
Abstract
Lithium-metal batteries with high energy/power densities have significant applications in electronics, electric vehicles, and stationary power plants. However, the unstable lithium-metal-anode/electrolyte interface has induced insufficient cycle life and safety issues. To improve the cycle life and safety, understanding the formation of the solid electrolyte interphase (SEI) and growth of lithium dendrites near the anode/electrolyte interface, regulating the electrodeposition/electrostripping processes of Li+, and developing multiple approaches for protecting the lithium-metal surface and SEI layer are crucial and necessary. This paper comprehensively reviews the research progress in SEI and lithium dendrite growth in terms of their classical electrochemical lithium plating/stripping processes, interface interaction/nucleation processes, anode geometric evolution, fundamental electrolyte reduction mechanisms, and effects on battery performance. Some important aspects, such as charge transfer, the local current distribution, solvation, desolvation, ion diffusion through the interface, inhibition of dendrites by the SEI, additives, models for dendrite formation, heterogeneous nucleation, asymmetric processes during stripping/plating, the host matrix, and in situ nucleation characterization, are also analyzed based on experimental observations and theoretical calculations. Several technical challenges in improving SEI properties and reducing lithium dendrite growth are analyzed. Furthermore, possible future research directions for overcoming the challenges are also proposed to facilitate further research and development toward practical applications.
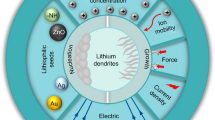
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
With the growing applications of portable electronics, electric vehicles, and smart grids, lithium (Li)-based metal batteries, including Li-ion batteries [1], Li-S batteries [2], and Li-air batteries [3], have been rapidly developed in recent years. To increase the mileage of applications, such as electric vehicles, power Li batteries must possess high energy densities. Li-ion batteries appear to have reached their energy density limit, and further increasing the energy density has become difficult. In this regard, Li-sulfur and Li-air batteries with both liquid- and solid-state electrolytes are expected to be the next-generation energy storage devices based on their extremely high theoretical energy densities. In such devices, lithium-metal anodes with a high theoretical specific capacity (3 860 mAh g−1) and a low reduction potential (− 3.04 V vs. SHE) are typically used [4,5,6,7,8]. Experimental results have shown that the energy density of lithium-metal-anode-based batteries is approximately 40%–50% higher than that of classical Li-ion batteries with graphite anodes [9].
Although lithium-metal-anode-based batteries have the advantage of high voltage, two key issues must be resolved before their practical applications. One is that lithium-metal batteries (LMBs) can exhibit rapid dendrite growth in the repeated charging/discharging process. This dendrite growth process punctures the separator, triggers short circuiting of the interior, and causes heat escape and even explosions, hindering the practical application of LMBs [10, 11]. The other issue is that Li dendrites can break away from the current collector to form dead Li, resulting in low Coulombic efficiency (CE) [12]. To resolve these issues, strategies for lithium-metal-anode protection have been developed, focusing on surface control of Li metal, including controlling the surface morphology, decorating the solid electrolyte interphase (SEI) layer, coating artificial protective films, and building three-dimensional lithiophilic frameworks to lower the sedimentary current density. In addition, using a high electrolyte concentration [13,14,15,16,17,18,19,20,21,22,23,24,25,26] and adding electrolyte additives [27,28,29] are reasonable means to prevent dendrite formation.
Regarding regulation and modulation, the electrochemical kinetics play a decisive role in the Li+ stripping/plating process. In this regard, many efforts have been made to investigate the electrochemical kinetics. For example, the Poisson–Nernst–Planck equation can describe the electrochemical mass transport and electrostatic potential across the cell [30], the deposition kinetics of lithium metal can also be analyzed by the Butler–Volmer equation [31], and the heterogeneous nucleation equation gives a growth model for Li embryos [32]. Understanding these kinetic processes can shed light on dendrite growth and the delamination from the current collector mentioned above.
In this Review, we comprehensively summarize the fundamental understanding of the electrochemical kinetics and dendrite growth of lithium-metal anodes in lithium-based batteries using both theoretical approaches, such as density functional theory (DFT) calculations, ab initio molecular dynamics (AIMD), molecular dynamics (MD), and finite element numerical simulations, and experimental validation. We also present a critical survey of the unique phenomena and kinetic mechanisms of Li+ stripping/plating in the electrochemical process. Details of the fundamental mechanisms are also analyzed in terms of electrolyte reduction, charge transfer, the local current distribution, solvation, desolvation, ion diffusion through the interface, inhibition of dendrites by the SEI, additives, asymmetric processes during stripping/plating, the host matrix, and in situ observation of nucleation, as outlined in Fig. 1. Note that extension of the concept of electrochemical kinetics can also play a key role in battery design and improvement of the electrochemical performance, such as the stability of lithium-metal-anode-based batteries.

Copyright © 2016, American Chemical Society. Reprinted with permission from Ref. [34]. Copyright © 2010, American Chemical Society. Reprinted with permission from Ref. [35]. Copyright © 2020, Elsevier. Reprinted with permission from Ref. [36]. Copyright © 2013, American Chemical Society. Reprinted with permission from Ref. [37]. Copyright © 2020, John Wiley and Sons. Reprinted with permission from Ref. [38]. Copyright © 2015, Springer Nature. Reprinted with permission from Ref. [39]. Copyright © 2020, Springer Nature. Reprinted with permission from Ref. [40]. Copyright © 2016, John Wiley and Sons. Reprinted with permission from Ref. [41]. Copyright © 2015, American Chemical Society
Schematic diagram of the Li+ diffusion process from the bulk electrolyte to the anode surface, which is divided into different parts to describe the multi-interface and multidimension issues. Reprinted with permission from Ref. [33].
2 Fundamentals of Electrode Nucleation/Growth Processes
2.1 Electrochemical Processes and Dendrite Growth
In general, understanding the mechanisms of electrode geometric evolution and interface reactions at the electrode/electrolyte interface remains a challenge. The cationic deposition process on the surface of the lithium-metal anode is inevitably involved in the design of a lithium-metal-anode battery. The electrode geometric evolution is a coupled dynamic process of mass transfer and local current density governed by Faraday’s law of electrolysis. The analysis of liquid phase mass transfer can provide insight into lithium dendrite formation and protection. The liquid phase mass transfer is simply divided into two stages: the transient state and the steady state. The transient state is dominated by fast electromigration. Due to the rapid formation of electric fields induced by the nonfaradaic current, the rate of mass transfer via electromigration is much higher than that via diffusion. Meanwhile, the variation range and area of the electrode surface concentration are constantly expanding. As identified in the steady state, the landmark phenomenon is concentration polarization. A dynamic balance can be formed, and the polarization region is no longer extended. In the steady state, mass transfer is the decisive step.
For further analysis, Fig. 2 shows a thin rectangular cell, where the two electrodes are made of Li metal, and the cell is filled with a dilute solution of Li+ salt. The motion of the Li+ salt can be described by the charge, the potential gradient, and diffusion constants (Di), which is a reasonable approximation for dilute solutions if the convection of the electrolyte is neglected. To accurately describe the ion and electron transport behavior, a numerical model can be used to solve the time-based evolution of the Poisson–Nernst–Planck system of equations (PNP equations) that describe the electrochemical mass transport and the electrostatic potential across the cell. The PNP equations take the following forms [42]:
where \(J_{i}\) is the mass flux of the ith species, \(D_{i}\) is the diffusivity of the species, \(Z_{i}\) and \(C_{i}\) are the charge and concentration of the ith species, \(x\) is the position, \(F\) is Faraday’s constant, \(R\) is the ideal gas constant, \(T\) is the thermodynamic temperature, and \(\varphi\) is the electrostatic potential.

Copyright © 1990, American Physical Society. b Profile of the distributions of V (electrostatic potential), Cc, and Ca (ion concentrations of cations and anions), which were obtained from numerical studies with a space-charge model. Reprinted with permission from Ref. [42]. Copyright © 1990, American Physical Society. c The different areas in the diagram can be used to determine whether the size of the embryo is suitable for different nucleation and growth stages. Reprinted with permission from Ref. [32]. Copyright © 2013, The Electrochemical Society. d Profile of a spherical cap-shaped nucleus (N) deposited on a smooth substrate (S) in a liquid electrolyte. a (= rsin θ) is the radius of the circular contact region between the cap-shaped nucleus and substrate. The height of the electrodeposit is h (= r(1 – cos θ)). \(\theta\) is the contact angle, h is the height of the embryo, and r is the radius of curvature. The volume of the cap is \(S_{{\text{V}}} r^{3}\), where \(S_{{\text{V}}} = \frac{\uppi }{3}\left( {2 + \cos \theta } \right)\left( {1 - \cos \theta } \right)^{2}\). The surface area of the cap is \(S_{{\text{A}}} r^{2}\), where \(S_{{\text{A}}} = 2{\uppi }\left( {1 - {\text{cos }}\theta } \right)\). Reprinted with permission from Ref. [32]. Copyright © 2013, The Electrochemical Society
a Scheme of the thin rectangular cell. Reprinted with permission from Ref. [42].
The reaction kinetics at the electrolyte/electrode interface can be described by the Butler–Volmer equation:
where \(\alpha_{{\text{A}}}\) and \(\alpha_{{\text{C}}}\) are the charge transfer coefficients of the anodic and cathodic reactions due to the overpotential \(\eta\). The exchange current density \(i_{0}\) depends on the composition of the reactants and products.
Several factors can affect the electrode kinetics at the electrode/electrolyte interface due to mass transfer and electron transfer. The affecting factors range from the interatomic interaction of crystal nucleation and side reactions to macroscopic electrode geometric evolution. During surface reactions, interface mass transfer and charge transport occur instantaneously, and dendrite growth and accumulation of dead Li can occur, leading to battery failure over time [43].
To date, several models, such as the space-charge model [42], SEI-induced model [4], and heterogeneous nucleation model [32], have been verified to describe Li dendrite formation (including nucleation and growth processes) from different aspects. However, these models cannot accurately describe the nucleation and growth behavior of lithium anodes [44]. Exploring the electrochemical behavior in LMBs via these nucleation models is still worthwhile.
The space-charge model was proposed in the 1990s. It is one of the widely accepted models providing guidance on the initial nucleation process of lithium dendrites [42]. In a dilute solution, the model describes that the deposition rate of Li+ is higher than that of the anions. Li+ depletion on the electrode surface results in a charge space, which ignores the convection of the electrolyte near the electrode, and only electromigration and diffusion are considered. Hence, the space-charge layer near the electrode/electrolyte interface can directly induce dendrite formation. As shown in Fig. 2a and b, the space-charge model simulates a thin rectangular cell filled with dilute solution. When Li+ is deposited on an electrode surface, Region 1 (the quasi-neutral region, \(z_{{\text{c}}} C_{{\text{c}}} \approx z_{{\text{a}}} C_{{\text{a}}}\)) and Region 2 (the space-charge region, \(C_{{\text{c}}} \ll C_{{\text{a}}}\)) are defined (\(z_{{\text{c}}} C_{{\text{c}}}\) and \(z_{{\text{a}}} C_{{\text{a}}}\) are the electrostatic charge densities of anions and cations). Region 1 occurs in the bulk electrolyte, and Region 2 occurs in a tiny area near the electrode/electrolyte interface. In addition, the two regions are dominated by different factors. In Region 1, the ion concentrations of Li+ and anions are mainly regulated and controlled by electromigration. In Region 2, the ion concentration of Li+ is mainly limited by surface diffusion. More specifically, some factors, such as increasing the electrolyte concentration, enhancing the mobility of anions, and reducing the applied electric field (plating at a low current density), could effectively reduce the thickness of the space-charge layer. Because the space-charge layer induces the formation of dendrites, there is a strong correlation between the two. Reducing the thickness of the space-charge layer can directly suppress the formation of dendrites.
However, the space-charge model does not take the reaction between Li metal and the electrolyte into account, and the SEI-induced model was proposed to mitigate the limitations. The presence of the SEI makes the deposition behavior of lithium metal more complex than that of other metals. To understand the role of the SEI in the nucleation process, we first need to describe the structure of the SEI. The SEI is generally considered to be an organic/inorganic composite mosaic structure produced by the reduction of lithium salt and the electrolyte by lithium metal. In the process of Li+ diffusion from the bulk electrolyte to the anode surface, Li+ ion diffusion through the SEI is the largest barrier. The activation energy barrier of Li+ diffusion through the SEI is higher than that through the bulk electrolyte. The Li+ concentration tends to be enriched near the holes of the SEI, which directly leads to a local nonuniform Li+ flux on the surface of the lithium-metal anode. In short, the porous SEI structure can lead to nonuniform Li deposition, resulting in rapid Li plating and dendrite growth. Additionally, during the subsequent stripping/plating processes, due to the fragile SEI, the sizeable volumetric expansion of the lithium-metal anode is not effectively buffered, and tremendous amounts of cracks on the SEI surface can be resulted. When these cracks react with the electrolyte and form a new thin SEI, the resulting electric field and Li+ ion flux are higher. Then, Li+ ions prefer to deposit on these cracks, leading to rapid growth of Li dendrites and stress concentration. Therefore, development of effective strategies for suppressing dendrite growth by stabilizing and strengthening the SEI layer is necessary.
Ely and García [32] employed the heterogeneous nucleation model, beginning from fundamental thermodynamic principles, unifying the concepts of electrochemistry and nucleation and growth theory, and explaining the effect of initial nucleation on Li+ deposition. Five regimes of behavior were identified: the nucleation suppression regime, long incubation time regime, short incubation time regime, early growth regime, and late growth regime. The size of the embryo directly determines whether the nucleation process can form a lithium nucleus. An embryo larger than or equal to the thermodynamic and kinetic critical radius is involved in an electrodeposited stable nucleus. Otherwise, the embryo tends to redissolve into the electrolyte again. In the long incubation time regime, the size of the embryo exceeds the critical radius after overcoming the critical energy barrier. Such embryos will remain and sequentially grow under fluctuations in the electric field and ionic field. Then, in the early growth and late growth regimes, the nuclei gradually grow to reach the same terminal size. Eventually, these embryos become the Li nuclei. Hence, the critical radius limits the initial Li nucleation process.
The growth of Li during electroplating in a flat way is virtually inaccessible. Electrolyte decomposition and SEI formation can occur during electroplating. Then, the Li+ ion diffuses through the SEI layer during the subsequent plating, which is different from the conventional electrodeposition process. Therefore, many embryos will appear at the electrode interface after a delay or incubation period because lithium electrodeposits can easily achieve heterogeneous nucleation conditions [32]. In addition, in situ characterization shows that the growth rate is constant after nucleation, which suggests a variety of competitive driving factors before the growth of the lithium nucleus.
Regarding the heterogeneous nucleation model, the volume change during stripping/plating processes is considered. For the surface of a lithium-metal anode without a host structure, the largest issue is the sharp volume change during stripping/plating processes. The change in volume is favorable for dendrite nucleation from the point of view of the heterogeneous nucleation model. However, it can also destroy the stability of the SEI and continuously consume active substances and the electrolyte, ultimately leading to failure of lithium-metal-anode-based batteries. Chazalviel [42] believed that cations could be rapidly consumed for lithium-metal anodes charged at a high rate in a binary electrolyte, and the cation concentration near the anode was expected to drop to zero [42]. After that, a strong electric field could induce absorption and deposition of a large amount of Li+ ions in a short time, causing dendrite growth. The Sand’s time (τ) can be used to describe the initial nucleation time of lithium dendrites:
where D is the diffusion constant, \(C_{0}\) is the initial concentration, \(J\) is the effective current density, and \(\mu_{{\text{a}}}\) and \(\mu_{{{\text{Li}}^{ + } }}\) are the anionic and Li+ mobilities, respectively.
Equation (4) indicates that the initial nucleation time of lithium dendrites (τ) is proportional to \(J^{ - 2}\). Furthermore, the velocity for the formation of dendrites can be determined by:
where \(\nu\) is the velocity for the formation of dendrites and E is the applied electric field.
Theoretical and numerical studies by Chazalviel [42] found that the dendrite growth rate was constant under a high current density and proportional to the applied electric field and the mobility of anions (\(\mu_{{\text{a}}}\)) for diffusion-limited systems. The critical values of high and low current densities can be expressed by Eq. (6):
where L is the interelectrode distance. Obviously, \(J^{*}\) is inversely proportional to the interelectrode distance (L) according to Eq. (6), indicating that the long electrode spacing of a pouch battery cell makes dendrite growth easier than the short electrode spacing of a coin battery cell.
Furthermore, in addition to at a high current density, dendrites can also grow at a low current density, induced by the nonuniform morphology of the electrode surface. In other words, under the condition of a low current density, uniformity of the electrode surface has an inhibitory effect on dendrite nucleation, and the anode surface also affects the initial nucleation time of lithium dendrites.
In the interpretation of dendrite nucleation evolution, heterogeneous nucleation can be expressed as Eq. (7):
The most basic and widely accepted model is the basis of studying the nucleation dynamics, as shown in Fig. 2c and d [32]. Mathematically, spherical cap-shaped deposition from a liquid electrolyte can be expressed by the total Gibbs free energy (Eq. 7) of transformation. In Eq. (7), \(\Delta G_{{\text{f}}}\) is the volume free energy of transformation, \(\gamma_{{{\text{NE}}}}\) is the interfacial free energy between the nucleus and electrolyte, \(\gamma_{{{\text{SN}}}}\) is the interfacial free energy between the substrate and the nucleus, \(\gamma_{{{\text{SE}}}}\) is the interfacial free energy between the substrate and electrolyte, z is the valence of the electrodepositing ion, F is Faraday’s constant, Ω is the molar volume, \(\eta\) is the overpotential, and \(r\) is the radius of curvature of the nucleus (Fig. 2d). \(S_{{\text{V}}} = \frac{\uppi }{3}\left( {2 + \cos \theta } \right)\left( {1 - \cos \theta } \right)^{2}\), and \(S_{{\text{A}}} = 2{\uppi }\left( {1 - {\text{cos }}\theta } \right)\), which account for the volumetric and area shape factors, where θ is the contact angle.
The critical radius to form a thermodynamically stable precipitate, i.e., \(\frac{{{\text{d}}G_{{\text{T}}} }}{{{\text{d}}r}} = 0\), can be expressed as Eq. (8):
The thermodynamically unstable embryos tend to redissolve in the electrolyte during the nucleation suppression regime, as shown in Fig. 2c. A statistically representative embryo larger than the thermodynamic and kinetic critical radius is required to successfully grow a thermodynamically stable electrodeposited nucleus. In regimes where only \(r > r_{{{\text{eq}}}}^{*}\) is satisfied, embryos can form, redissolve in the electrolyte and interact with each other. Different types of surfaces favored in the nucleation and growth of electrodeposits are demonstrated in Fig. 2c. Heterogeneous nucleation of electrodeposits at a large overpotential is favored on a substrate with a high degree of roughness. In addition, if its radii are larger than the critical kinetic nucleus, then the smooth anode surface in distributed particle form can serve as nucleation sites. However, if the substrate is very smooth or the radius of curvature of the substrate is very large, then no heterogeneous deposition can occur.
The initial nucleation phase can be considered heterogeneous nucleation behavior, which was demonstrated based on both thermodynamics and kinetics by Ely and García [32]. They treated the nucleation as having five regimes: the nucleation suppression regime, long incubation time regime, short incubation time regime, early growth regime, and late growth regime [32]. In the subsequent incubation regimes, the incubation of Li nuclei was entirely driven by thermal noise, local electrochemical reactions, and the Gibbs–Thomson interaction. If the applied overpotential was lower than the critical overpotential, then the long incubation regime would appear. Li nuclei with a broader size could be beneficial in this situation and grow for a long time. In the short incubation time regime, if the applied overpotential was higher than the critical overpotential, then only Li nuclei with a relatively narrow size could grow in a short time. Constant terminal velocities in the early growth regime and the late growth regime were found due to the thermodynamically and kinetically stable Li nucleus. According to the above analysis and calculation, dendrite formation can be inhibited by (1) reducing the roughness of the anode surface; (2) reducing the anode particle size to below the critical thermodynamic radius; (3) lowering the deposition potential to below the critical overpotential and cycle frequency to below the characteristic incubation frequency \(\left( {\frac{1}{\tau }} \right)\); and (4) adjusting the wettability of the Li deposition surface.
2.2 Interfacial Properties of the Anode/Electrolyte Interface
For Li stripping/plating processes, the kinetics of atom/electron diffusion to another phase seem important in understanding dendrite growth. At present, researchers have carried out multidimensional and multiexperimental studies on the mass transfer, electrode morphology evolution, and charge transfer occurring at the electrode interface. Thus, a novel understanding of real-world Li stripping/plating processes involving electrochemistry is required to correctly predict interatomic interactions in SEI and dendrite formation. Researchers have proposed several models for the SEI, as shown in Fig. 3, to fit the complex behavior of this interface. Concerning this, various characterizations and multiscale simulations have been carried out. Atomic- and molecular-level and macroscopic-level interfacial challenges are involved in the search for high-performance LMBs [52, 53].

Copyright © 1979, The Electrochemical Society. b XPS spectra of a Li metal surface. Reprinted with permission from Ref. [46]. Copyright © 1985, The Electrochemical Society. c Schematic diagram of the polyhetero-microphase SEI model. Reprinted with permission from Ref. [47]. Copyright © 1997, The Electrochemical Society. d Schematic presentation of the SEI formation process. Reprinted with permission from Ref. [48]. Copyright © 2000, Elsevier. e Schematic energy diagram of an electrolyte. Reprinted with permission from Ref. [34]. Copyright © 2011, Elsevier. f Schematic illustration of Li+ diffusion through a porous organic layer and a dense inorganic layer. Reprinted with permission from Ref. [49]. Copyright © 2012, American Chemical Society. g Schematic illustration of mosaic and multilayered structures formed on a Li surface. Reprinted with permission from Ref. [50]. Copyright © 2017, The American Association for the Advancement of Science. h Accelerator fluctuations with geometric deformation, and COMSOL simulation with THU. Reprinted with permission from Ref. [51]. Copyright © 2019, John Wiley and Sons
Presentation and recognition of key achievements in SEI and dendrite growth research history. a The effect of a partial short on the deposition process of the SEI. Reprinted with permission from Ref. [45].
At the atomic and molecular level, high chemical reactivity is induced by surface atoms with active atomic and electronic structures, enabling interactions with the electrolyte and solvents [54]. DFT calculations and in situ characterization techniques can enable the investigation and understanding of the adsorbed intermediates as well as the reaction mechanism. In recent years, some specific measurements have been carried out on new types of additives and concentrated electrolyte systems, revealing some novel mechanisms. Atomic and molecular interactions can modify the height and shape of the reaction and migration barriers, eventually affecting the nucleation kinetics, electrochemical polarization, and mass transport of Li+ ions in porous SEIs.
At the macroscopic level, several unique phenomena and the electrode geometric evolution can be captured with the development of in situ characterization technologies. Some reaction kinetics can be described with prominent nonlinear characteristics. The morphologies of the anode and SEI are prone to affect the stability and evolution. The initial conditions greatly affect the system and its changes with time based on the kinetic equation. In the following sections, interface phenomena at both the micro- and macrolevels will be introduced from several perspectives.
3 Interface Interactions
3.1 Electrolyte Reduction Mechanisms
Interface interactions have different features in their respective electrochemical processes, mainly in the SEI at the anode/electrolyte interface of lithium-metal-anode-based batteries. The SEI is the key factor of many processes, limiting the lifetime, performance, and safety. It can affect the inhomogeneous growth of the lithium-metal anode and diffusion of Li+ ions. Thermal runaway in battery failure can be caused primarily by SEI decomposition. The primary capacity fade can also be attributed to the consumption of lithium by the continuous growth of the SEI. Generally, an improvement in interfacial stability is required for high voltage. Thus, innovative design of stable electrolytes and the associated batteries requires a fundamental understanding of the SEI formation mechanism through both modeling and experimental measurements [48, 55, 56].
The initial formation of an SEI film is generally regarded as the process of electrolyte reduction on the lithium-metal-anode surface, which plays a significant role in the SEI composition [57]. At the same time, the formation of the SEI is directly related to the battery cycling process. In the practical battery charging process, the Li+ in the cathode diffuses toward the anode through separators and electrolytes, driven by the increase in the cathode potential and decrease in the anode potential (the cell voltage potential of the battery is increased). During the discharging process, the Li+ ions return to the cathode, where they insert into the cathode material (the cell voltage of the battery decreases) [58]. Considering that an electrode (an anode or cathode) itself has large numbers of ion and electron channels, it can be treated as an equipotential body in which the potential inside the electrode is equal everywhere. Therefore, the change in potential is mainly due to the electrolyte/electrode interface, which can be regarded as a parallel electric double layer (EDL) with resistance. There will be a potential gap between the electrode and electrolyte. Apparently, this potential gap is a trigger that activates the electrochemical reaction between the electrode and electrolyte, resulting in a “new interface” at the middle of the electrode and electrolyte. Hence, the “new interface” plays a key role in balancing the potential of the battery. The interface generated at the surface of the anode is named the SEI [45, 59], and that at the cathode is named the cathode electrolyte interphase (CEI), collectively known as the electrode electrolyte interphase (EEI) [60]. In practice, the SEI is a dominant factor that influences the Li+ diffusion on the anode surface. The reduction process can significantly affect the SEI components. Therefore, many mechanisms have been proposed to explain the process of electrolyte reduction, such as the one- and double-electron reduction mechanisms of electrolytes [61,62,63]. Furthermore, although the SEI chemical composition is diverse and disturbed by local variables, it has been studied by a multitude of experimental techniques, including photoelectron spectroscopy [64], Fourier transform infrared spectroscopy [65], redox shuttle analysis [66, 67], nuclear magnetic resonance (NMR) [68], atomic force microscopy [69], neutron reflectometry [70], capacity analysis during cycling [71], and state-of-charge (SoC) analysis [72, 73]. Some reactions may occur on the picosecond (ps) timescale, and directly capturing reactions at the electrode/electrolyte interface is challenging. Therefore, not only in situ characterization tests but also quantum chemistry (QC), DFT, and MD simulations have been widely used to reveal the initial electrolyte reduction and decomposition mechanisms [74]. Due to the complex structure and physical scale of the SEI, there are many deficiencies in our understanding of the SEI. As a result, researchers have varied in their research emphasis to meet different functional requirements and in their opinions on the SEI. For example, some researchers focused on the failure mechanisms and corresponding SEI regulation strategies [43], a new understanding of the LiF component in the SEI [75], and the formation and transport mechanisms of the SEI. These works have different perspectives and understandings. Therefore, understanding the SEI is a dynamic and tortuous process.
Goodenough et al. [34] used frontier orbital theory (FOT), as shown in Fig. 4a, to describe the nature of the anode and cathode (involving the electrolyte) of the battery. \({\mu }_{\mathrm{A}}\) and \({\mu }_{\mathrm{C}}\) are the chemical potentials of the anode and cathode, respectively. \({E}_{\mathrm{LUMO}}\) and \({E}_{\mathrm{HOMO}}\) represent the voltages corresponding to the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO). The electrolyte is affected by the LUMO and the HOMO. If \({\mu }_{\mathrm{A}}>{E}_{\mathrm{LUMO}}\), then the anode electrons tend to diffuse to the electrolyte LUMO, whereas the stability window is extended by the SEI on the negative electrode. In contrast, if \({\mu }_{\mathrm{C}}<{E}_{\mathrm{HOMO}}\), then the electrons of the electrolyte tend to transfer to the cathode surface to form a CEI film [41, 77]. The stability window is extended by surface films, denoted as the CEI and SEI on the electrodes.

Copyright © 2016, Elsevier. c Methods to compute the reduction voltage. Molecules/ions in the solution phase, solid phase, and gas phase are denoted by sol., s, and g, respectively. The subscripts sol and vap represent the vaporization and solvation processes, respectively. ∆G is the free energy, ΦM is the work function of the anode, and the subscript e indicates the ionization process. Reprinted with permission from Ref. [62]. Copyright © 2001, American Chemical Society
a Schematic open-circuit energy diagram of an electrolyte. Eg is the electrolyte electrochemical stability window. μA and μC are the redox potentials of the anode and cathode, respectively. b Several common solvents, additives, and desolvated ion reduction potentials versus Li/Li+, calculated from QC with the developed solvation model. Reprinted with permission from Ref. [76].
The dominant factors of SEI formation are usually attributed to the initial adsorption and formation of the inner Helmholtz plane (IHP) and the solvated coordination behavior [78]. The specific adsorption behavior is the formation of the initial interface on the anode surface, which is the foundation of the SEI and controls the initial state of the interface structure and the chemical composition. Solvated coordination behavior can impact the maintenance of the SEI film in cycling [59]. According to traditional electrochemical theory, the negative electrode tends to lose electrons due to the balance of the high free-electron concentrations when the electrode contacts with the electrolyte.
As shown in Fig. 4c, a thermodynamic chemical cycle for the lithium reaction can be expressed as Eq. (9):
where ∆Gsol is the solvation free energy of Li+, ΦM is the work function of the anode, ∆Ge is the ionization free energy, and ∆Gvap is the vaporization free energy. Using Eq. (9), the difference in the lithium electrode potential can be measured in multitudinous electrolyte systems, which depends only on the variation in G(Li+, sol), the free energy of Li+ in solution. Wang et al. [62] used this equation and found that the reduction potential of Li+(EC)4 (Li+ coordination with four ethylene carbonate (EC) molecules) was approximately − 2.2 V on the physical scale, which agrees with the experimental findings of Naji et al. [79] and Novak et al. [80] of a reduction potential for EC of 0.8 V versus Li/Li+ (corresponding to − 2.36 V on the physical scale). In addition, Arora et al. [81], in dealing with the capacity fade in lithium-ion batteries, obtained a reduction potential of − 3.045 V versus Li+/Li. Many cases have different reduction potentials, as demonstrated in Fig. 4b [76]. H-transfer reactions (intra- and intermolecular H atom transfer during electrolyte oxidation) are usually accompanied by electrolyte oxidation, which directly results in the oxidation potential being lower than the calculated HOMO value [76]. In addition, the LUMO of most electrolyte components is higher than the lithiated graphite (~ 0.1 eV) and lithium metal (0 eV) potentials. Therefore, if the chemical stability window of the electrolyte is not widened, then the reduction reaction of the electrolyte on the anode is inevitable. The SEI on the lithium-metal anode is more fragile than the CEI on the cathode due to the large volume expansion caused by Li+ deposition at the lithium anode surface. Considering their importance in battery performance and durability, extensive research from different perspectives has been performed on anode SEI films. Different anodes and/or electrolytes have diverse lithiation/delithiation characteristics, which lead to variations in the failure mechanisms of the SEI.
With the rapid development of supercomputers and highly sensitive spectroscopy, identification of the products and types of reactions on Li surfaces has become feasible for complex systems. For example, Leung et al. [82] utilized AIMD to track the initial decomposition process of liquid EC with different edge terminations. Two EC decomposition pathways were confirmed. Depending on which C–O bond was cleaved, CO or C2H4 gas could be generated. The multireaction pathways were further elucidated in later studies and confirmed by electron paramagnetic resonance spectroscopy measurements [83, 84]. The major solvent reduction products precipitating on lithium electrodes in ether, ester, and alkyl carbonate solutions were ROLi, RCOOLi, and ROCO2Li species, respectively. As claimed, the specific surface components formed on lithium-metal anodes could be identified, such as (CH2OCO2Li)2 and CH3CH(OCO2Li)CH2OCO2Li formed by reduction of EC and propylene carbonate (PC), CH3OLi and CH3OCO2Li formed by reduction of dimethyl carbonate (DMC), and HCOOLi formed by reduction of methyl formate (MF). Combining experimental and computational approaches, Liu et al. [85] found unambiguous answers in understanding how 1,2-dimethoxyethane (DME) and 1,3-dioxolane (DOL) reductively decompose on lithium through the interfacial chemistry and morphology. A series of possible SEI components, such as LiOCH2CH2OLi, LiOCH2O(CH2)3O(CH2)2OLi, (CH2CH2OCH2OLi)2, LiO(CH2)2O(CH2)3OLi, LiOCH2CH2OCH2CH2OLi, and (CH2OCH2CH2OLi)2, were proposed, all of which arise from reductive decomposition of the cyclic solvent DOL.
Using X-ray photoelectron spectroscopy (XPS), some perfluorinated anions in Li salt of the MFy− type (M = As, P, B) were identified to be reduced on Li to surface LiF and species of the LixMFy type by XPS [86]. The following reaction scheme for surface film formation on Li in this system can be outlined based on the above studies.
EC reduction
One-electron reduction [62, 63]
Two-electron reduction of 2 EC molecules [62]
Two-electron reduction of 1 EC molecule [62]
Two-electron reduction [82]
DMC reduction
One-electron reduction [48, 87]
Two-electron reduction of 1 DMC molecule [88, 89]
Two-electron reduction of 2 DMC molecules [88, 89]
DOL reduction [85]
PC reduction
One-electron reduction of PC [90]
Two-electron reduction of 2 PC molecules [90]
LiPF6 reduction [48, 87, 89, 91,92,93,94,95,96,97]
3.2 Charge Transfer in the Lithium-Metal-Anode Reaction
The homogenization of dendrites at the lithium-metal-anode/electrolyte interface is determined by the relationship between the morphology of the SEI and the deposition behavior of lithium atoms. Electron and Li+ transport during the process of Li deposition on the SEI can systematically achieve orientation of Li atoms during the deposition, which is affected by the subsurface atoms [98]. In addition, the origins of dendrite formation could be associated with the Li+ plating mechanism and the charge transfer mode during Li reduction or lithium-metal oxidation at the anode-electrolyte interface [99]. Charge transfer is usually manifested as the electron tunneling mode [100, 101].
The kinetics of electron and Li+ transport at the interface have been intensively investigated by analyzing the overpotential equation known as the Butler–Volmer Eq. (3), as previously described. In the Butler–Volmer equation, the exchange current density i0 can be expressed by the charge transfer resistance RCT via i0 = RT(FRCT)−1, measured in the linear regime of the electrode kinetics. If RCT is set equal to the interfacial impedance, then the charge transfer can be illustrated by the impedance method [102]. In addition to impedance, the reaction kinetics can also be expressed by the landscape of the excess chemical potential, \(\mu^{{{\text{ex}}}} \left( x \right)\), between local minima \(\mu_{1}^{{{\text{ex}}}}\) and \(\mu_{2}^{{{\text{ex}}}}\) with transitions over an activation barrier \(\mu_{\ddag }^{{{\text{ex}}}}\) (Fig. 5a). In this case, Eq. (25) can be used to express the reaction, where R is the reaction rate, \(k_{0}\) is the reaction rate constant, and \(k_{{\text{B}}}\) is the Boltzmann constant. In a balanced state, R will be equal to zero in equilibrium with \(\mu_{1}\) = \(\mu_{2}\) in Eq. (25), and the reaction rate (R) is consistent with nonequilibrium thermodynamics:

Copyright © 2013, American Chemical Society
a Schematic diagram of the excess chemical potential reflected by the reaction S1 → S2. b Process of cation adsorption from a liquid onto an electrode, where the transition state (TS) excludes multiple surface sites (s > 1) while shedding the primary solvation shell. c Solid diffusion on a lattice, where the TS excludes two sites. Reprinted with permission from Ref. [36].
In addition to classical kinetics methods, DFT and Monte Carlo (MC)/MD calculations have also been conducted to analyze electron transfer because the interfacial reactions and charge transfer at the lithium-metal-anode/electrolyte interface are difficult to track in experiments. Typically, the extreme reactivity of the Li metal surface can induce a strongly inhomogeneous electron distribution upon deposition of a cation on the surface. Once Li+ receives electrons and plates on the lithium-metal surface, different conditions can result in different Li plating layers. This charge inhomogeneity may promote uneven Li nucleation and growth, eventually resulting in dendritic behavior. Qin et al. [99] reported a simulation model describing both Li (100) and Li cations located in proximity to the surface. In this model, the solvent used was EC and DME, and the electrolyte was a LiPF6 salt; the Li cations were distributed over perfect, defect-containing, and Li2CO3-passivated Li (100) surfaces. The authors demonstrated that Li cations were easily reduced when bonded to DME rather than EC, and their preferred deposition site was the hollow site on both perfect and defective Li (100). Additionally, a compact Li2CO3 layer could inhibit the charge transfer from Li metal to Li cations, thus modifying Li plating. Bertolini et al. [103] utilized MD and observed the evolution of the Li (100) surface, including the formation of regions with varying Li density and oxidation states and the penetration of electrolyte molecules. Furthermore, electron transfer-driven decomposition could lead to the initial formation of SEI products, as shown in Fig. 6a. The formation of the structure and oxidation state of Li and some fragments were followed through the metal dissolution process. A porous SEI can be identified, and a change in the lithium concentration and oxidation state throughout this phase can be found by using simulation approaches. An increase in Li concentration closer to the dense phase is suggested (Fig. 6a). The entire porous phase is pervaded by Li ions and reacted species coexisting with intact electrolyte molecules. The Li distribution is more uneven in the nest phase than in the disperse phase. In the nest phase, Li atoms are organized as nanochannels and nanovoids. With time, clusters are formed in this region (Fig. 6a), and their size can grow over time. The clusters formed in the disperse phase have a high concentration of interconnected Li atoms. The clusters also have a high concentration of other species, such as O- and F-based compounds with Li ions and poly(carbon monosulfide). In the electrolyte containing Li triflate (LiTf), clusters are formed after a time scale of 1.2 ns. Both in the nest and disperse phases, Li has a very different structure from that in the bulk body-centered cubic (bcc) phase. In the Li–Li radial pair distribution function (RPDF) (Fig. 6b and c), the nest and disperse phases present a first peak at approximately 3.2 and 3.5 Å (1 Å = 1 × 10−10 m), respectively, for each simulated electrolyte. In the disperse phase, Li is mainly organized in pairs with a liquid-like structure, which can be formed and dissolved over time, while in the nest phase, Li has an amorphous structure, organized as a solid. As shown in the RPDF, a lower density of dissolved Li atoms can be obtained in pure DME than in other electrolytes for both the nest (Fig. 6b) and disperse (Fig. 6c) phases. Norio et al. [104] developed a hybrid MC/MD reaction method to study the atomistic picture of the SEI film structure (Fig. 6d). The mass density of dimers in the EC-based SEI film (Fig. 6e) was larger than that in the PC-based film (Fig. 6f), indicating a denser network among the organic salts in the EC-based SEI film. From another point of view, the PC-based SEI film does not have a dominant peak but rather a broad distribution of dimers, indicating that flaws exist in the network formation of the PC-based SEI film. As observed, electron tunneling can only work in the initial part of SEI growth. In this mode, electron and Li+ transport processes in the SEI were addressed by an atomistic simulation method [103, 104]. As claimed, electron tunneling allows the migration of electrons over SEI layers as thin as 2–3 nm [105], while the thickness of the SEI usually exceeds 10 nm [106]. This suggested that electron tunneling plays a role only in the initial part of the first-cycle SEI growth.

a Model simulation of the 1.0 M (1 M = 1 mol L−1) LiTf/1,4-dioxane (DX) cell at 2 ns. (I) Front-side view of the cell showing all the atoms. (II) Front-side view of the cell showing only lithium atoms, while the other atoms are hidden. (III) Side view of the cell showing only lithium atoms. The molecules (shown on the right side) are (A) DOL, (B) dimethyltrifluoroacetamide (DMTFA), (C) EC, (D) fluoroethylene carbonate (FEC), (E) DME, (F) DX, and (G) LiTf. Color code: Li (purple), O (red), C (gray), H (white), F (cyan), and S (yellow). Li–Li RPDF in the b nest phase and c disperse phase tested in several electrolytes after 500 ps of simulation. Reprinted with permission from Ref. [103]. Copyright © 2018, American Chemical Society. d Configuration of the cell in the 1.1 M LiPF6/EC or PC simulation, where the origin of the Z-axis is taken as the position of the contact point between the graphite and electrolyte. Color code: Li (blue), C (cyan), O (red), H (white), P (orange), and F (green). Surface number densities \(\rho_{{\text{n}}}^{{\text{S}}}\) of several solvent molecules and reaction products during the MC/MD cycles, that is, SEI film formation simulations, in e EC-based and f PC-based electrolytes. Reprinted with permission from Ref. [104]. Copyright © 2014, American Chemical Society
3.3 Local Current Distribution over the Lithium-Metal Anode
In general, the electrode reaction and growth of the dendrites are determined by the local current density. Hence, understanding and controlling the electrode current distribution to limit dendrite formation is imperative for safe use of a lithium-metal anode. The local current density is expressed in a similar way to electron transport at the interface, which is described by the Butler–Volmer equation. The direction of these studies can be guided by theoretical studies on the relationship among the current distribution, concentration gradient, and dendrite nucleation. To model dendrite formation and growth, the concentration and potential fields in the electrolyte domain need to be considered, and the normal velocity of the lithium interface needs to be calculated. Mass conservation is typically governed by the PNP equations and electroneutrality. Electroneutrality is expressed as the total charge number of cations in the solution being equal to the total charge number of anions:
where the subscript i represents the species, which can be the PF6− anion or other lithium ion salt anions. For each species, ci is the concentration, and \({z}_{i}\) is the charge number. The electroneutrality gives c− = cLi.
The current density, i = \(F\sum\nolimits_{i} {Z_{i} {\varvec{N}}_{i} }\), where \({\varvec{N}}_{i}\) is the flux of species, is governed by Eq. (27):
The total lithium current density is given by it = iLi + iSEI, where the reaction current density, iLi, is described by the Butler–Volmer equation and the SEI formation current density iSEI is determined by the Tafel-type kinetics [107].
Liu and Lu [107] quantitatively analyzed the change in SEI thickness. Consider a small SEI line element and denote its length at time t by l. After a time interval (dt), the length of the element becomes l + dl, while its thickness becomes h + dh. Denote the SEI growth rate from the electrochemical reaction by qSEI, which is the increase in SEI thickness per unit time due to the reaction. Mass conservation gives the following expression:
Numerous experimental studies have been carried out to address the issue of electrode current distribution and dendrite growth in lithium batteries. The increase in the current density in adjacent dendrites or globules is the most prominent driving force for the nucleation process, thus far escaping experimental scrutiny. Therefore, the average current density is mostly reported in relevant reports on lithium-metal-anode-based batteries [108]. For example, Liu et al. [107] found that dendrite growth could show two distinct stages: fast acceleration during SEI penetration and stable growth after reaching an equilibrium SEI thickness at the dendrite tip. Under low current density conditions, the growth rate of dendrites could be scaled up by increasing the current density without much geometrical shape change. The dendrite morphology can be significantly changed by a high current density, forming a needle shape, as shown in Fig. 7a. The larger SEI thickness at the bottom corners of the dendrite can cause lithium to grow slower here than in the flat area, so slight piercing of the substrate surface with a V-shape can be observed at the bottom of the dendrite, as shown in Fig. 7b. This makes dendrite break away from the substrate easier. A simulation with the same parameters but without the SEI is shown in Fig. 7c. Compared to Fig. 7a, clear differences can be seen in the morphology and height, which are rounder and shorter without any V-shape in the bottom corner.

Copyright © 2017, The Electrochemical Society. The growth of the lithium globule was divided into four stages according to the local current density. The local current density is plotted according to the four stages of the growth of the lithium embryo. d In the initial stage, a perturbation at the anode/electrolyte interface results in a higher current density at the tip of the globule than that of the entire region. This current density is measured between time points 0 and 8.27 C cm−2. e With the growth of the embryo, the current is delocalized away from the tip of the globule. Measurement points: 8.27–16.53 C cm−2. f The delocalization behavior spreads. Measurement points: 16.53–35.82 C cm−2. g The globule has grown large enough that the current density concentration is significantly reduced. Measurement points: 35.82–54.72 C cm−2. Reprinted with permission from Ref. [108]. Copyright © 2016, The Electrochemical Society
a Profile of the lithium surface morphology (blue line) and SEI layer thickness (red layer) evolution process with time. b The profile of a V-shaped dendrite was formed at the left corner at t = 50 s. The substrate appears to be slightly pierced by V-shaped dendrites due to the thick SEI, resulting in slow growth at the corner. c Profile of the lithium surface morphology at t = 50 s without inducing an SEI in the simulation. The whole curve is smooth without a V-shape at the bottom corner. Reprinted with permission from Ref. [107].
The local current distribution of the lithium-metal anode with local mechanical stresses is a crucial parameter to reveal the details of the evolution of electrochemical deposition. Time-resolved hard X-ray microtomography was employed by Harry et al. [108] to monitor the internal structure of a symmetric lithium-polymer cell during galvanostatic polarization. The study demonstrated local current density evolution due to local mechanical stress from lithium metal nucleation and growth (Fig. 7d–g). In Fig. 7d, the tip of the protruding lithium globule directly leads to a high local current density. As the lithium globule grows, the local concentrated current density spreads from the protrusion tip to the perimeter, which is the current density delocalization effect. Eventually, when the radius of the globule is sufficiently large, the current density of the protruding globule is only slightly higher than that of the surrounding lithium metal. The current delocalization effect shown in Fig. 7e and f is hypothesized to be caused by the mechanical stiffness of the polymer electrolyte.
Anion depletion under an applied current density results in a local space charge near the electrode, which eventually causes highly branched growth of the metal. As identified, the dendrite growth rate is directly related to the applied current density [109]. The local geometric fluctuations of the substrate or the SEI layer could greatly influence the current and ion distributions, causing dendrite initiation and growth at even small current densities [110]. Based on this understanding, several dendrite prevention avenues have been investigated through studies of the local current density: (1) increasing the surface area of the anode to reduce the effective current density, i.e., using a lithium powder anode [111,112,113,114,115] and a high surface area current collector [114, 116,117,118,119]; and (2) exploiting a self-healing electrostatic mechanism based on repelling the incoming Li+ from the tip to the valley of the potential dendritic surfaces [112, 120,121,122,123].
3.4 Solvation Structure of the Lithium-Metal Anode
In general, the electrolyte provides an electric current between the positive pole (cathode) and the negative pole (anode), and the current is carried only by the moving ions for charge transfer. Dissociated salts in polar solvents can produce most of the mobile ions in the cell, except in the molten state (ionic liquid) or in the decoupled ceramic or glass state [124]. For conventional lithium-ion batteries, lithium ions are mainly provided by lithium salts, such as LiPF6, lithium bis(fluorosulfonyl)imide (LiFSI) and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI). The solvation sheath is closely related to the formation of the SEI. Therefore, some reports describe the formation of the SEI from the competitive reactions of the solvation sheath structure [125], and some describe the solvation structure arising from control of the interface to achieve a high-performance SEI structure [126]. At present, there are still some controversies about the influence of anions on the interface. However, in an organic electrolyte environment, the solvation of anions is highly system-specific because the Coulomb interaction between anions and solvents is weak. In most commonly used nonaqueous solvents, anions are much less solvated than cations [127]. Hence, the solvation structure with Li ions and solvent molecules constitutes the actual ionic species that are mobile in the electrolyte.
Recognition of the solvation structure of lithium ions is necessary to understand the interface interaction process of the solvation structure. Bernal and Fowler [124] proposed that the solvent molecules in the solvent were induced to surround and redirect the ion dipoles, thereby changing the coordination state of the solvent in dilute aqueous solution. From a solvation structure point of view, the most important finding thus far is the change in coordination of both solvent and salt from a dilute solution to a concentrated solution. On the basis of the strength of the association between the solvent molecules and the ions, the solvation sheath structure can be divided into three parts according to Borodin et al. [35] (Fig. 8): “salt-in-solvent”, “salt-solvate”, and “solvent-in-salt”. In the primary solvation sheath region, the center ion forms the strongest association with solvent molecules and maintains the sheath structure during motion, as shown in Fig. 8a. In the “second” solvation sheath region, solvent molecules break the balance and weakly associate with the center ion. Apart from these two regions, the solvent molecules can remain undisturbed and away from the center ion. As the concentration of the solution increases, a heterogeneous structure may occur in the electrolyte. Yu et al. [128] investigated the lithium bis(trifluoromethane)sulfonamide concentrated aqueous electrolyte, where heterogeneous structural features and different dynamic behaviors were observed between negative and positive ion domains with different Li+ mobilities, using experimentally corroborated MD simulations. With increasing salt concentration, the original solvation sheath structure (including the “primary” and “second” solvation sheaths) gradually disappears, and anions are then added into the “primary” solvation sheath, as shown in Fig. 8b. At extremely high salt concentrations, the anions and cations are close to each other in the same solvation sheath, as shown in Fig. 8c.

Copyright © 2020, Elsevier. d Schematic diagram of the solution structures in a concentrated electrolyte. Reprinted with permission from Ref. [129]. Copyright © 2019, Springer Nature
Schematic representation of three salt concentration systems in an electrolyte. a Li+ ions in a dilute electrolyte with three parts: the primary solvation sheath, secondary sheath and free solvent. b With increasing Li salt concentration, the structure of the secondary solvation sheath is disrupted in the absence of free solvent, and part of the primary sheath structure is squeezed in by anions. c The solvation sheath shares its limited solvent molecules, which may result in the presence of domains in the electrolyte. Reprinted with permission from Ref. [35].
For the quantitative ratio of solvent to lithium salt in a dilute electrolyte solution [approximately 1 M (1 M = 1 mol L−1) and a solvent/salt molar ratio > 10], the most common coordination state of lithium ions is being surrounded by three or four solvent molecules, which are named solvent-separated ion pairs (SSIPs) [130]. The solvent molecules mainly remain free, and decomposition of solvent molecules is induced, forming an SEI film. However, for the case of a concentrated electrolyte (> 3 M), the Li+ coordination number was indicated to be less than 1–2 as the solvent molecules were reduced. At the same time, more salt anions entered the electrolyte to coordinate with Li+, taking the place of the solvent molecule, thus forming contact ion pairs (CIPs) and cation–anion aggregates (AGGs), as shown in Fig. 8d. In addition, a higher Li+ salt concentration could degrade the coordination ability of solvents. Highly concentrated electrolytes have been confirmed to enable stable cycling of lithium-metal anodes due to their unique solvation structure and the lack of free solvent molecules [131]. The decomposition/reaction of salt anions instead of solvent molecules could gradually dominate SEI film formation with increasing Li salt concentration. In brief, when the Li+ concentration is increased from 1 M to more than 3 M, the number of free solvent molecules is reduced, and the coordination form is transformed from SSIPs to CIPs and AGGs, especially AGGs, which can be dominant in the case of higher concentrations to yield unusual functions.
The solvation structure is closely correlated with the electrochemical behavior of Li+ ions. The Debye–Hückel model assumes that salts are completely dissociated but not fully independent of the solution system when ignoring direct solvent-salt interactions [132]. Modern ionics science recognizes that the solvation structure is a vital factor for the electrochemical behavior of Li+. The solvation structure is becoming more flexible as a result of different Li+ salt concentrations, types of organic solvents, and additives. Generally, all kinds of organic solvents render coordination by the electrostatic force between high electronegativity atoms (N, O, and so on) and Li+, known as a “complexant”. The study of the solvation structure has attracted attention because of the exfoliation and solvent cointercalation phenomena of the graphite electrodes commonly used in most conventional lithium-ion batteries. Besenhard et al. [133]. proposed a 3D process describing the solvation sheath cointercalation path. The key to this model is to review the previously neglected specific solvation structure movement at the interface and decomposition process of the cation solvation sheath at the microscopic level. Subsequent work demonstrated this model. Wagner et al. [134] utilized in situ X-ray diffraction (XRD) to demonstrate that during the initial lithiation process, due to the solvation sheath cointercalation, the spacing between the graphene sheets expanded, and the potential was 200–300 mV higher than the reduction potential of the known solvent. They believed that the primary solvation sheath was more likely to cointercalate solvent molecules into graphitic electrodes. Xu et al. [135, 136] further suggested that the eventual interface chemistry on graphitic anodes was actually determined by the solvation sheath composition of Li+ in typical nonaqueous electrolytes. Obviously, the constitution of the solvation sheath could directly cause a change in the reduction potential at the interface. Therefore, further exploration of lithium ion complex solvents can directly affect the understanding of the interface interaction. High concentrations of lithium salt are also a double-edged sword, which can cause the electrolyte viscosity to be too high. This leads to poor ionic conductivity and poor wetting capability, which hinder practical applications. To alleviate these defects and reduce the cost, diluting a high concentration electrode liquid with nonpolar solvents to make it a localized high concentration electrolyte has become a hot research field [137, 138]. In addition, weakly solvating electrolytes could achieve an anion-derived, inorganic-rich SEI [139]. Many achievements have been realized by adjusting the solvation sheath of Li ions from the solvation structure, for example, improvement of the uniformity of the SEI, extension of the potential window [140, 141], prevention of electrode corrosion and dissolution [142,143,144], acceleration of electrode reactions [140, 145], increase of the Li+ transference number [146], and decrease of the volatility and flammability [141, 147, 148].
3.5 Desolvation of the Li+ Sheath
In consideration of the cation transport process at the electrolyte interface, researchers have distinguished the contributions of the so-called ion transfer barrier from two parts: (1) desolvation of Li+ before it deposits on the anode surface and (2) subsequent diffusion of bare Li+ through the SEI. Understanding the barrier between these two parts should clarify how lithium ions diffuse from the electrolyte environment to the negative electrode. In general, the diffusion process of the sheath structure is divided into three stages: (1) the sheath structure must diffuse from the electrolyte to being near the SEI interface; (2) the Li+ sheath structure must be desolvated near the SEI surface; and (3) the Li+ ion changes its solvation structure in the electrolyte to adjust the coordination state. Since the final state of the sheath structure is not bare, ions passing through the SEI may be amorphous [149]. The structure of the Li+ solvation sheath is very often accompanied by a change in solvent species [150, 151]. Since the SEI components are often derived from electrolyte reduction, the two phases of the SEI and electrolyte have different chemical environments. Hence, this process is often known as desolvation or, a more accurate description, resolvation. Actually, Li+ diffusing from the electrolyte into the SEI can pass through the defects (interstitials or vacancies) in the SEI film structure, as shown in Fig. 9a and b [152,153,154]. Shi et al. [49] further proposed a knock-off mechanism, showing that Li+ interstitials diffused through Li2CO3 by replacing another Li+ in the lattice position. The final step was Li+ passing through the SEI film to the anode surface in different ways, including diffusion along the grain boundaries, knock-off transport and defect migration, as shown in Fig. 9c–f [49, 155, 156]. Many researchers judged that the second and third stages could control the kinetics of battery chemistry (corresponding to the two ion transport barriers), and efforts have been made to judge the rate-determining step in the kinetics of the lithium-ion battery chemistry.

Copyright © 2018, American Chemistry Society. The transition state structure lies in the upper left inset, the lower figure shows the energy profile, and the upper right inset shows a schematic diagram of the Lii+ diffusion pathway from site Ai along the [010] direction following the c knock-off and d direct-hopping mechanisms with threefold coordination in the transition structure. Reprinted with permission from Ref. [49]. Copyright © 2012, American Chemistry Society. The migration barrier and diffusion direction of Li diffusion through the LiF/Li2O grain boundary are divided into e Path 1, f Path 2, and g Path 3. Reprinted with permission from Ref. [155]. Copyright © 2019, American Chemistry Society
Schematic diagrams of knock-off mechanisms obtained by AIMD. Schematic depiction as a function of the distance from a Li+ atom and b a phosphorus atom from the PF6− anion to an amorphous Li2EDC SEI film. The values of all coordinates in the bulk electrolyte or SEI film are normalized. Reprinted with permission from Ref. [149].
Desolvation of the Li+ solvation sheath is proposed when Li+ diffuses through the electrolyte/SEI interface, as shown in Fig. 10c, which can be fitted by DFT or MD simulations. The activation energy is used to quantitatively describe the barrier of desolvation of the Li+ solvation sheath because it can reflect the basic kinetics of the interfacial process. In fact, the apparent activation barriers could also be estimated through electrochemical impedance spectroscopy (EIS) data [157, 158]. As observed, the activation energy was not affected by the Li+ activity or the effective surface area [159]. Abe et al. [160,161,162] and Yamada et al. [163] reported that the slowest process occurred when the Li+ solvation sheath was trying to enter graphene before Li obtained electrons from the anode, where the primary solvation sheath had to be stripped. Xu et al. [164] proved that the Li+ solvation sheath stripping process gradually occurred in the outer pore diffusion stage, as shown in Fig. 10a.

Copyright © 2010, American Chemical Society. b Integrated ion densities at the SEI/electrolyte interface as a function of SEI thickness and composition. Solid red lines: total density up to 6 Å into the electrolyte region; black dotted lines: division of the density into an ion-adsorbed region; blue dashed lines: 6 Å from this region into the bulk electrolyte region near the interface. Reprinted with permission from Ref. [169]. Copyright © 2013, American Chemical Society. c Li+ activation energy barriers in different electrolyte composition systems. The reference lines for the activation energy at the bottom are obtained from 1.0 M LiPF6/tetrahydrofuran (THF) (red dotted line) and 1.0 M LiPF6/PEG222 (green dotted line) with LTO. Reprinted with permission from Ref. [164]. Copyright © 2010, American Chemical Society. d Schematic depiction of the Li+ desolvation process near the negative interface. Reprinted with permission from Ref. [171]. Copyright © 2019, American Chemistry Society
a Schematic diagram showing the process and activation energy barrier of Li+ diffusion from the bulk electrolyte to the graphite/electrolyte interface. Reprinted with permission from Ref. [164].
Many substantial computational efforts have been dedicated to studies of Li desolvation energetics at the SEI/electrolyte interface [149, 165,166,167]. The SEI-free Li4Ti5O12 (LTO) anode was successfully utilized in measuring the Li+ desolvation energy barrier [approximately (52 ± 3) kJ mol−1 at different EC concentrations], while the overall value of the activation energy of the SEI-covered graphite anode was approximately 60–70 kJ mol−1, as shown in Fig. 10c. Therefore, Li+ required approximately 20 kJ mol−1 energy to pass through the SEI. A similar conclusion was also proven by Pacific Northwest National Lab (PNNL) [168]. The Li+ desolvation process was the major kinetic barrier because of the high activation energies for charge transfer (0.5–0.7 eV). The high activation energies were experimentally determined at the low-temperature operation limit. In addition, the activation energy of the desolvation step was indicated to be constant over a wide range of EC:DMC concentrations [164]. Xu et al. [136] concluded that a similar activation energy to that of Yamada et al. [159, 163] was found in the LiClO4/EC:DMC system of different EC concentrations, where EC had little effect on the activation energy. A later MD simulation by Borodin and Bedrov [166] suggested that the activation energies of Li+ desolvation were approximately 40–44 and 39–41 kJ mol−1 for dilithium butylene decarbonate (Li2BDC)/EC:DMC(3:7)-LiPF6 and dilithium ethylene decarbonate (Li2EDC)/EC:DMC(3:7)-LiPF6, respectively. Jorn et al. [169] utilized AIMD simulations and found that with increasing SEI thickness and LiF, more Li+ was adsorbed on the SEI surface, suggesting that a large amount of Li+ could be easily desolvated, as shown in Fig. 10b. In terms of the composition of the SEI, the diffusion energy barriers of the inorganic and organic SEI components are also different. The energy barrier of the inorganic component is significantly lower than that of the organic component, which can also prove that desolvation is the key step [170]. Meanwhile, Jorn et al. [169] showed that the path for Li+ desolvation is a two-step mechanism, consisting of crossing two energy barriers and then adsorption. Ming et al. [171] presented a new interfacial model for desolvation. Desolvation was identified as a key factor that could affect anode stability. The solvent interaction with Li+ (e.g., strength and conformation) was the key factor for stability in different electrolytes, which was different from the prevailing SEI formation effect.
Recently, Nasara et al. [172] studied different electrolyte systems for a Li4Ti5O12 (LTO) anode at a lowered cutoff potential and found that increasing Li+ concentration and activity (\(\alpha_{{{\text{Li}}^{ + } }}\)) contributed less to the Li+ desolvation activation energy barrier. The activation energies from multiple scenarios suggested that the Li+ desolvation process was dependent on the element composition of the SEI film and the solvent molecule rather than the physical thickness of the SEI film [167]. There is much evidence to support the hypothesis that desolvation is the rate-determining step. Many simulation methods have been used to make further advances in the exploration of such complex reactions and diffusion processes at the interface. In addition, the Li+ transport through the SEI should be given more attention. Currently, there still appears to be no method for resolving the Li+ desolvation and Li migration through the SEI [173,174,175,176,177,178].
3.6 Li+ Diffusion Through the SEI/Electrolyte Interface
To understand Li+ diffusion at the electrode/electrolyte interface, the properties and their effect on battery performance should be deeply studied [179]. 6Li isotope labeling experiments were reported by using a combination of time-of-flight secondary-ion-mass spectrometry (TOF–SIMS) and NMR spectroscopy to directly observe and prove the exchange process between the electrolyte and SEI [180, 181]. The authors demonstrated the existence of an SEI and observed three stages for Li+ to travel from the electrolyte to the anode [168]. In addition, the time scale for Li+ diffusion through the SEI was indicated to be seconds or minutes.
Many studies have been reported on inorganic and organic components as diffusion barriers of the SEI [75]. Diffusion barriers can be limiting factors due to their topological and defect distribution in the mosaic structure of SEI films. Hence, DFT, MD, and multiscale modeling need to complement each other to accurately measure the diffusion process. Shi et al. [49] proposed Li+ diffusion in an SEI film with a two-layer/two-mechanism model. The model demonstrated that the outer porous layer (organic) allowed Li+, salt anions, and even solvent molecules to diffuse, but the dense inner layer (inorganic) could only facilitate Li+ interstitials to diffuse through Li2CO3 via replacement of another Li+ in the lattice position. The structure of the SEI was divided into organic components and inorganic components. The inorganic component had a larger resistance for Li+ diffusion through the SEI.
To quantitatively analyze the main diffusion barrier in the SEI, some researchers have focused on key components (including Li2CO3, Li2O, LiF or Li alkyl carbonate) [91, 182, 183] of the SEI to demonstrate a single crystal affecting Li+. Lu and Harris [180] demonstrated that Li2CO3 was the main component of the SEI in their experiments. Iddir and Curtiss [152] explored the Li+ migration energy in a bulk monoclinic Li2CO3 crystal and found that Li+ migration along the [010] channel could obtain a lower energy barrier and that hopping via this channel was less likely to occur. Chen et al. [184] further demonstrated that vacancies could contribute less to Li+ diffusion along the [010] plane and that vacancies substantially contributed more to Li+ diffusion across the [010] plane. Therefore, the effect of Li+ diffusion for a variety of concentrations of vacancies and interstitials in different crystal orientations was not consistent. The predominant diffusion mechanism for Li2CO3 was vacancy-interstitial diffusion. Chen et al. [184] systematically calculated Li+ diffusion in three structures (Li2CO3, Li2O, and LiF) using the nudged elastic band (NEB) method. LiF was demonstrated to have the slowest Li+ diffusion compared to Li2CO3 and Li2O because Li2CO3 (0.227 eV) and Li2O (0.152 eV) had lower energy barriers than LiF (0.729 eV). TOF–SIMS measurements showed that the anions and electrolyte could only infiltrate the outer porous layer to approximately 5 nm in the SEI, whereas Li+ infiltrated more into the dense inner layer to approximately 20 nm [180]. Pan et al. [185] and Yildirim et al. [153] studied the LiF diffusion mechanism via the nearest-neighbor Schottky pair and expounded that the ionic conductivity of LiF was highly dependent on the electrode it contacted. When it contacted a negative (graphite, silicon) electrode, the ionic conductivity of LiF was dramatically lower by 18 to 20 orders of magnitude compared to LiC2O3 [49, 154]. Moreover, Benitez et al.[186]. systematically observed the diffusion mechanisms under an applied electric field in LiF (knock-off), Li2O (vacancy-assisted) and Li2CO3 (combined vacancy-interstitial diffusion mechanism), as shown in Fig. 11. Benitez et al. [186] utilized the large-scale atomic/molecular massively parallel simulator (LAMMPS) to calculate the Li+ diffusion mechanism with two divided parts of chain movement (knock-off mechanism), as shown in Fig. 11a. The first chain movement (including purple, orange, and yellow balls) was initiated by Li+ (green ball). For the second chain movement, Li+ (red ball) displaces the blue ball (initial position) to start the chain movement. As shown in Fig. 11b, Li+ diffusion in Li2O relies on the vacancies, and Li+ ions move from their own site to the next empty lattice site in the direction of the applied field in sequence. The most special case is Li+ ion diffusion in Li2CO3 in Fig. 11c, which is mainly manifested as combined vacancy-interstitial diffusion. Recently, Lu and Liao [187] reported that the Li+ diffusion energy barrier in Li2O was considerable consistent, directly leading to Li2O cluster exfoliation.

Copyright © 2017, The Electrochemical Society
Schematic diagrams of the initial (left) and final (right) positions of Li+ ions. An electric field was applied to show the diffusion processes in a LiF, b Li2O and c Li2CO3. The diffusion mechanism of LiF was knock-off, which involves diffusion by two chain movements (the green line and orange line) of Li+ ions. The diffusion mechanism of Li2O relies on vacancies. Li+ diffusion in Li2CO3 is achieved through combined vacancies and interstitials. Li+ ions (multiple color balls), F (cyan), O (red), and C (brown). Reprinted with permission from Ref. [186].
In addition to the pristine SEI film, introducing other phases or taking advantage of the space-charge layer effect can also enhance the diffusion carrier concentration. Doping a second phase can increase the charge carrier concentration of the SEI film because the main inorganic components of the SEI (LiF, Li2CO3, and Li2O) can form a defect formation energy higher than the diffusion energy barrier. Shi et al. [154] utilized BO33− to replace CO32− in Li2CO3 crystals and created more Li+ interstitials that could increase the ionic conductivity. Doping common metallic cations such as Ca2+, Mg2+, Al3+ and even La3+ into LiF or Li2CO3 could also increase the Li+ vacancy concentration [185, 188]. Liang [189] introduced insulating Al2O3 nanoparticles into LiF to increase the Li+ carrier concentration, which could increase the space-charge layer effect to increase the ionic conductivity. Furthermore, heterogeneous structures and grain boundaries could also contribute to the ionic conductivity [156, 190]. Pan et al. [190] made a prediction based on the different values of Li diffusion carriers in Li2CO3 and LiF and the combined interfacial defect reaction energy. Zhang et al. [156] demonstrated the formation of a space-charge layer across the Li2CO3 and LiF interfaces and observed a dramatically increased defect concentration in Li2CO3 and LiF. In contrast, Li and Maier [191] found that introducing Al2O3 nanoparticles into the SEI could lead to fewer ionic carriers (decreased ionic conductivity) near the interface.
3.7 SEI Modification and Dendrite Inhibition
Since the lithium-metal-anode surface is initially smooth bulk, the main factor affecting surface nucleation is the state of the SEI film. Optimization of lithium-metal surface nucleation should deal with the SEI film. Typically, an in situ SEI film with an organic/inorganic composite structure can be formed from the reaction between the anode surface and electrolyte, controlled by the electrolyte composition, anode potential, temperature, and additives. The mosaic model is the most commonly accepted description of the SEI structure and describes the nonuniform surface of the anode. Peled et al. [47] directly proved Li transport via SEI grain boundaries. The heterogeneous structure could affect the SEI formation on the surface of lithium metal in terms of morphology and the difference in transport dynamics caused by local morphology. Figure 12 shows the mechanism of Li dendrite growth [4]. First, the SEI is formed on the Li surface cracks due to the volume expansion caused by Li+ plating (Step 1); second, Li dendrites are produced from the cracks as the plating progresses (Step 2); third, Li dendrites transform into isolated Li metal due to Li stripping, and the isolated Li is an ignorable part of the so-called dead Li, which deepens the degree of SEI fracture (Step 3); and last, upon continuous cycling of Steps 1–3, accumulated dead Li, a thick SEI and a porous lithium electrode are formed (Step 4). These four steps are spontaneous processes, leading to degradation of the anode performance. As identified, the stress accumulated in the lithium metal deposition process could cause Li dendrites to be generated at the thinner positions of the SEI. Wang et al. [192] proposed that the SEI passivates the lithium-metal surface, preventing stress relaxation through uniform diffusional creep with Li diffusion to the film surface. Inorganic components of the SEI, such as Li2CO3, were close to and adhered to Li metal. These components had a high strength. The lithium-metal/SEI interface was vacancy poor. Consequently, lithium atoms were harder to deposit nearby. Therefore, stress relief could be realized by local creep of the defect surface. This method was based on the stress release of lithium metal and was also involved in the mechanical strength of the SEI. As identified, the mechanical strength is also a key factor for the SEI to suppress dendrites. Many researchers have designed artificial SEIs [193], including organic [194,195,196,197,198,199,200] and inorganic [201, 202] components and their hybrids [203,204,205], to enhance the mechanical strength of the SEI. In addition, artificial SEIs can be classified into different types based on the manufacturing method, such as solvent casting, wet chemistry, chemical vapor deposition (CVD), physical vapor deposition (PVD), and atomic layer deposition [206]. Tao et al. [207] described in detail the advantages and disadvantages of artificial SEIs produced by different deposition methods in the practical application of zinc metal anodes. A thicker coating layer and increased ion diffusion are issues. Therefore, Xu et al.[206]. suggested that establishing a structure–property relationship between the surface properties and lithium stability is the key to success.

Copyright © 2017, Springer Nature
Schematic diagram of the formation of Li dendrites. Reprinted with permission from Ref. [4].
As observed, during long cycling of the lithium-metal anode, a fresh SEI that breaks and grows again appears on the surface, and its film thickness is uneven. Li+ ions easily pass through the thin locations, which have a high ionic conductivity rate for preferred deposition [6]. This means that the uneven SEI surface has inconsistent nucleation time on the time scale. The different lithium ionic conductivities directly lead to the existence of inconsistent nucleation and growth states on the spatial scale at the interface. An SEI with high ionic conductivity easily forms higher nuclei on the lithium metal surface. Meanwhile, when the SEI is composed of multiple components with different ionic conductivities, the spatially varying ionic conductivity will cause large differences in the lithium plating/stripping volume at different sites and the subsequent SEI. Therefore, high surface energy and low transport barrier of Li+ in the SEI are key to homogeneous nucleation.
Increasing the ionic conductivity of Li+ ions at the SEI is one of the key factors for inhibiting dendrites. According to the space-charge model [42], the formation of dendrites is closely related to the existence of a Li+ depletion layer. Hence, the diffusion rate of Li+ is closely related to the formation of dendrites. Normally, Li+ diffuses more slowly in the SEI than in the electrolyte, so the ionic conductivity of the SEI is several orders of magnitude lower than that of the bulk electrolyte. In this case, the inorganic component in the SEI usually corresponds to the rate-determining step of Li+ diffusion. More specifically, the inorganic components in the SEI are mainly composed of LiF and Li2O. Pan et al. [185] used DFT calculations to show that Li+ ion diffusion in LiF occurred via Li vacancy hopping. Vacancy-mediated diffusion mechanisms (including knock-off, direct exchange, and hopping) were identified by MD simulations of Li2CO3, LiF, and Li2O. The authors predicted that the ionic conductivity of LiF was at least three orders of magnitude lower than that of Li2CO3 and Li2O, and similar conclusions were also reported by Yildirim et al. [153]. Soto et al. [208] demonstrated that the SEI based on Na+ favored Li+ transport. This suggests that the ionic conductivity can be improved by switching the cation Li+ to Na+ in a premade SEI. In addition, the low adsorption energy of Li+ on LiF could lead to a low in-plane diffusion barrier for Li adatoms, which was considered to be beneficial to restraining Li dendrite growth. In addition to conventional SEI components, SEI components with higher ionic conductivity have also been used to regulate the formation of a dense lithium deposition morphology and inhibit lithium dendrites. For example, LiTeS3 [209], LiI [210], Li3N [211], LiPS4 [212], and Li2S/Li2Se [213], can have higher ionic conductivities, which could greatly improve the ionic conductivity. Rangasamy et al. [214] used LiI and LiPS4 to constitute Li7P2S8I, which exhibited an ionic conductivity of 6.3 × 10−4 S cm−1 at room temperature, a 400% improvement over the ionic conductivity of \(\upbeta\)-Li3PS4 (LPS) and a more than three orders of magnitude improvement over that of LiI.
The beneficial effect of SEI formation is to prevent further reaction between the Li metal anode and electrolyte. As identified, the SEI can be continuously destroyed by stripping/plating processes. The reduction in CE comes from the continuous consumption of the electrolyte and active material Li during the stripping/plating processes. In this case, the SEI as a protective layer can suppress the growth of dendrites. With respect to this, its size, composition and mechanical properties are of great significance for uniform deposition and high performance of the lithium-metal anode. However, the SEI thickness, granularity, chemical composition, and distribution are usually not well controlled. The organic components in the SEI are derived from the reaction between lithium metal and solvent, while the inorganic components are mainly derived from the decomposition of lithium salts [90]. In summary, an SEI film on lithium metal is composed of an electrolyte and additive, among which the electrolyte can be divided into solvent and salt. Therefore, rational selection of organic solvents, lithium salts and functional additives can enhance the SEI layer on lithium metal and heighten its performance.
In summary, SEI suppression of dendrites is generally strongly dependent on the regulation of the Li+ diffusion rate and the multiple physical properties of the SEI. SEI modification should focus on achieving a fast Li+ diffusion rate, stress release, mechanical strength, and structural uniformity (thickness, granularity, chemical composition, and distribution) of the SEI.
3.8 Mechanism of Additives
The additives for lithium-metal-anode-based batteries consist of salt additives and organic molecule additives. The desired lithium salt should also be conducive to the formation of a thin and stable SEI film that generally has the characteristics to meet the basic requirements: (1) wide voltage windows; (2) high ionic conductivity; (3) high chemical stability; and (4) electrochemical compatibility with the anode and cathode. Salt additives can be divided into transport-type lithium salts and auxiliary-type salts. At present, the commonly used lithium salts include lithium hexafluorophosphate (LiPF6), lithium bis(oxalato)borate (LiBOB), lithium difluorooxalatoborate (LiDFOB), lithium tetrafluoroborate (LiBF4), lithium trifluoromethanesulfonate (LiCF3SO3), lithium perchlorate (LiClO4), lithium hexafluoroarsenate (LiAsF6), LiTFSI, and LiFSI. As the most commonly used electrolyte salt, LiPF6 is widely used in lithium-ion and lithium-metal batteries. LiPF6 is generally believed to help form a highly resistive SEI in EC-ethyl methyl carbonate (EMC). This SEI has a large number of resistive decomposition products of LiF and Li2CO3 [216]. As a moisture-sensitive lithium salt, LiPF6 could also produce HF after reacting with water-induced impurities formed in the production process, which could corrode the battery system. Ding et al. [215] investigated different types of Li salts, as shown in Fig. 13a. Compared to LiPF6, LiBOB has the best Li morphology under SEM. LiBOB also has the second-best CE after LiAsF6, as shown in Table 1. When LiDFOB, LiBF4, LiCF3SO3 or LiClO4 serves as the Li salt, Li cannot cover the surface of the Cu collector well. LiCF3SO3 has less influence on the morphology and interface stability of lithium dendrites. LiCF3SO3, LiTFSI and LiFSI have high dissociation constants, high voltage stability and good thermal stability. LiAsF6 has the highest CE because it produces AsF6, which can coat the surface of lithium metal to prevent side reactions, as shown in Table 1 [215]. However, LiAsF6 has a highly toxic element. In the LMB system, LiAsF6 may not be used as a conventional lithium salt. LiClO4 faces similar conditions as LiAsF6. Perchlorates are highly oxidizing and are not widely accepted, although their reactivity with lithium-metal anodes is lower than those of other lithium salts. In summary, a single Li salt is currently defective, and binary or polysalt electrolyte additives have been developed to balance the ionic conductivity and viscosity of the electrolytes to evenly deposit Li+ in lithium-metal-anode batteries [217,218,219,220,221,222,223,224,225,226,227]. Zhang et al. [216] added 0.05 M LiPF6 to the bicarbonate/carbonate solvent electrolyte, and the battery could maintain a capacity of 97.1% after 500 cycles at a medium–high load of 1.75 mAh cm−2. Nuli et al. [228] reported that a binary salt electrolyte consisting of LiTFSI/LiFSI could result in a dendrite-free lithium coating with a CE of 99% during cycling.

Copyright © 2013, Springer Nature. b The upper two pictures are electrostatic potential maps based on the electron densities of g-butyrolactone and LiNO3 and of FEC and LiNO3. O, red; Li, purple; C, gray; H, white; N, blue; and F, green. The left end (red color) of the scale bar below the map reflects a lower Coulombic potential, and the right end (blue color) reflects a higher Coulombic potential. The bottom two pictures are SEM images of the Li plating morphology on the copper working electrode with LiNO3 and without LiNO3. Reprinted with permission from Ref. [37]. Copyright © 2020, John Wiley and Sons
a Scanning electron microscopy (SEM) images of the lithium-metal-anode surface deposited in electrolytes of LiBOB, LiPF6, LiAsF6, LiTFSI, LiI, LiDFOB, LiBF4, LiCF3SO3, and LiClO4 in PC solvent. Reprinted with permission from Ref. [215].
As recognized, auxiliary-type salts mainly contribute to the formation of the SEI instead of ion conduction, and they can interact with electrolytes or anodes through their own properties. For example, lithium nitrate (LiNO3), as well as the sulfurized species in the SEI layer, was investigated because it can significantly ameliorate the correlative properties of the SEI in lithium-sulfur batteries [131]. The solvent molecules could coordinate with NO3− and Li+ in the electrolyte due to the large electrostatic potential, as shown in Fig. 13b. LiNO3 plays a major role in stable lithium-metal anodes through the synergistic effect with lithium polysulfides (LPSs) [229]. However, the detailed mechanism of the improvement is still not fully understood. There is some evidence that nitrate ions could react with LPSs and metallic lithium as an oxidizer, resulting in the formation of a dual-layer SEI rich in LiNxOy and polysulfide (e.g., Li2SO4 and Li2S2O3) [229]. The interaction between LPSs and LiNO3 was investigated by using theoretical reactive MD simulations [230]. As observed, once LiNO3 was added, a multilayer SEI emerged with a homogeneous LixNOy cluster. The LPSs were captured by the dipole–dipole interaction in the presence of LixNOy to form lithium bonds [231]. As a result, the anode could be significantly shielded from directly contacting LPSs in the presence of the multilayer SEI. Zhang et al. [232] revealed the connection of NO3− anions and Li+ solvation in a 2 M LiFSI-DME electrolyte. The presence of LiNO3 was observed to be beneficial to complete decomposition of FSI− and generation of a dense SEI for enhancing the ion–dipole interaction between Li+ and solvent molecules so that the FSI− anions were mediated and polarized. In addition, there are also some reports about the contribution of other metal cations (Mg2+ [233,234,235], Mn2+ [236], Cu2+ [236], Sn2+ [237], Zn2+ [238], In3+ [238,239,240], and Bi3+ [238]) as auxiliary-type additives to stable alkali metal anodes. Ye et al. [241] employed AlCl3 as an electrolyte additive to achieve dendrite-free Li deposition. The electrolyte in the production process introduces trace water, which could react with Al3+ to produce Al(OH)3 nanoparticles. Al(OH)3 nanoparticles could react with excess Al3+ (electrolyte) and form positively charged colloidal particles (PCCPs). Subsequently, the as-generated and Al2O3 particles deposit on the lithium anode surface and form a stable and dense Al-containing SEI layer. Using this method, the CE of such a lithium-metal anode was increased to 99% at 0.5 mA cm−2 in 1.0 M LiPF6 (EC/DMC/DEC) electrolyte. The Zhang group adjusted the Li embryos by using Cs+ and Rb+ [120, 242, 243]. According to the Nernst equation, if the concentration of Cs+ or Rb+ is lower than 0.05 M, then the effective reduction potential of Cs+ or Rb+ is lower than that of Li+ at 1.0 M (− 3.04 V). Therefore, upon deposition, Cs+/Rb+ could be adsorbed on the Li surface and not form metal-alloy layers on it. If an uneven Li surface is formed, then protuberances with high charge densities will adsorb Cs+/Rb+ near the tips, resulting in an electrostatic shield. The positively charged shield could repel Li+. Thus, Li+ is forced to deposit in adjacent areas, resulting in smoothing.
In addition to salts, there are also a wide variety of organic electrolyte additives. In the electrochemical aspect, organic electrolyte additives can be divided into three main categories: SEI formation agents, kinetics acceleration agents, and kinetics suppression agents [244, 245]. Currently, fluoride-related compounds are widely accepted as SEI formation additives (FEC). The main purpose is to produce a LiF-rich SEI. FEC is a primary and familiar fluorinated solvent used to construct fluorinated SEIs in various fluorinated cyclic and acyclic carbonates. The LiF-rich SEI has the effect of forming uniform surface morphology and ion flux on the SEI film. Cui et al. [50] used cryogenic electron microscopy (cryo-EM) and revealed that the fluorine-containing SEI nanostructures formed in the FEC electrolyte were multilayered SEIs rather than the mosaic SEIs in the electrolyte without FEC. More importantly, when using FEC additives, observation by high-resolution cryo-EM imaging and fast Fourier transform (FFT) patterns showed that the lattices of these Li2O crystalline components were fairly aligned in the same direction. As the lithium nucleation dynamics was controlled by the SEI states, grain boundaries of uniform arrangement and regional uniform ion fluxes could contribute to the absence of significant differences in the time and size of lithium nucleation. Other SEI formation additives include nitrides, phosphides, and sulfides, and Li3N-rich [246,247,248], Li3P-rich [249], and Li2S-rich [250,251,252] SEIs are formed, respectively.
As identified, kinetics suppression agents and kinetics acceleration agents can suppress dendrites and affect the cell performance in the electrochemical process. Kinetics suppression agents can coordinate with Li+ or adsorb on the lithium-metal-anode surface to reduce activity and suppress desolvation of the Li+ solvation sheath, slowing down the electrochemical kinetics. Dai et al. [253] reported a hexadecyltrimethylammonium chloride (CTAC) additive, a kind of surfactant, to achieve a smooth surface. Upon CTAC addition to the electrolyte with an uneven surface, electrostatic interactions made CTA+ adsorb around the protuberances. Additionally, the protuberances had a higher surface energy and could absorb surfactant molecules. Thus, in the subsequent nucleation process, solvent Li+ could not diffuse through the absorbed layer. Hence, Li metal was forced to adjacent areas, further leading to a smooth surface. With addition of this surfactant, Li‖Li cells could stably cycle over 300 h at a high current density of 4.0 mAh cm−2. Wang et al. [254] introduced polyethylene glycol (PEG) and 12-crown-4 as electrolyte additives to inhibit dendrite growth and increase the cycling stability of the lithium-metal anode, as shown in Fig. 14a–d. Li+ was found to favor complexation with four O atoms in the sense of the coordination energy. For comparison, the absorption energy and complexation energy of the 12-crown-4 molecule with four-coordination sites (Fig. 14c) and the PEG-200 molecule with four-coordination sites (Fig. 14b) were calculated and compared with those of the DME solvent molecule with two-coordination sites (Fig. 14a). Using DFT calculations, PEG-200 molecules and 12-crown-4 were found to be more favorable for absorption on lithium-metal surfaces than DME solvent molecules, which can be attributed to ether molecules. The favorable complexation for PEG-200 and 12-crown-4 molecules could be confirmed. The ΔG values for the complexation of Li+ with PEG-200, 12-crown-4 and DME are − 115.375, − 117.281 and − 113.597 kcal mol−1 (1 kcal mol−1 = 4 185.851 J mol−1), respectively (Fig. 14a–c). Complexation with four-coordination-site ethers is more stable than that with two-coordination-site ethers. Notably, after complexation with Li+, the four oxygen atoms in PEG-200 or 12-crown-4 are arranged in a coplanar manner. However, for DME, the four oxygen atoms lie in a crossed manner, which can increase the system energy. Different arrangements in Li+ complexation were further investigated by NMR, as shown in Fig. 14d. When PEG-200 or 12-crown-4 is added to the electrolyte, the 7Li spectrum shows a small increase, which can be attributed to the strong complexation between the four-coordination-site ether and Li+. The four-coordination-site ether exhibits a deshielding effect in the NMR spectrum of 7Li, where the electronegativity of oxygen remarkably makes oxygen a deshielding element. The coplanar arrangement of oxygen further decreases the electron density of Li+ perpendicular to the plane. Therefore, the structures of different ethers are associated with their behaviors in the electrochemical process by complexation and absorption. In electrochemical deposition, kinetics suppression agents with stable complexation and absorption can reduce and homogenize the ionic activity of Li+ near the lithium-metal anode to render dendrite-free Li deposition easy.

Copyright © 2020, John Wiley and Sons. e Accelerator fluctuations with geometric deformation. f COMSOL simulation with THU; blue represents the electrolyte, and white represents the electrode. Reprinted with permission from Ref. [51]. Copyright © 2019, John Wiley and Sons
DFT calculations and NMR analysis. Optimized geometries for the binding of Li+ to a two DME molecules, b one PEG-200 molecule, and c one 12-crown-4 molecule. d 7Li NMR spectra of bare electrolyte and electrolyte with PEG-200 or 12-crown-4 additive (1 ppm = 10−6). Reprinted with permission from Ref. [37].
A kinetics acceleration agent denotes a common electroplating additive to fill tiny scratches, pits, and filaments by electroplating to smooth the clad layer, accompanied by improvement of the mechanical properties, such as hardness and ductility, and is also called an accelerator in the conventional electroplating industry [255, 256]. An appropriate accelerator should adsorb on the lithium-metal-anode surface and accelerate the electroplating procedure. As observed, the adsorption was shifted with the evolution of the microscopic surface, increasing the local current density in areas with different roughness and concave-convex extents, which was described as the curvature enhanced accelerator coverage (CEAC) mechanism [257]. However, new accelerators in LMBs are rarely found due to the laborious process. Wang et al. [51] leveled a plated lithium film via step growth through the CEAC mechanism by simply adding thiourea (THU, 0.02 M) into the electrolyte. Unlike the traditional electrolyte additive or protecting layer, THU displayed a catalytic role in electroplating of lithium metal, acting as an accelerator rather than a suppressor. The geometric deformation characteristic of the anode was simulated by a numerical method. For the trivial deposition process, the Butler–Volmer (B–V) equation was introduced for the cathodic reaction. However, with THU adsorbed on the surface, the B–V equation takes the surface concentration into consideration, which is given by:
where i is the anodic current density, i0 is the exchange current density, \({{C}}^{\prime}\) is the coefficient of the Li+ concentration, α is the transfer coefficient of the anode, F is the Faraday constant, R is the molar gas constant, \(k\) is the coefficient of the THU concentration, and \(k^{\prime}\) is the promoting transfer coefficient of THU. By comparing the geometric deformation characteristics of the anode, trivial deposition tended to occur on the dendrites, where the dendrites grew in a propagative manner. Notably, the situation was different with THU. Upon THU addition, it was adsorbed and enriched on the concavities rather than the convexities after geometric deformation of the anode, as shown in Figure 14f. The THU content on the surface (θ) increased with shrinkage, which accelerated the filling rate in the concave pits. Hence, Li+ tended to be reduced and deposited on the concave regions due to the acceleration of THU, as shown in Fig. 14e. With prolonged electroplating time, a dendrite-free lithium deposition layer was formed. The concave surface shrunk due to geometric deformation. In this way, a superfilling phenomenon could occur on the lithium-metal surface.
4 Electrode Geometric Evolution
4.1 Geometric Evolution of the Lithium-Metal Anode
Comprehensively understanding the morphological evolution of the stripping/plating process is very important in the design of lithium-metal-anode batteries. Most of the previous theoretical models on dendrite formation in stripping/plating were based on linear stability analysis. For example, the pioneering studies by Mullins/Sekerka [260, 261], Ahmad/Viswanathan [262, 263], and Tikekar et al. [264] revealed the impact of some key physical parameters: elastic properties, molar volume, etc. Notably, the stability to small perturbations was also studied in terms of potential chemical changes by Mullins–Sekerka linear stability analysis [262, 264,265,266,267]. A further understanding of dendrite initiation by taking other influencing parameters into account, such as the growth dimensions and kinetics of the growth process, seems needed.
In the graphite electrode, the intercalation reaction and deposition reaction always occur simultaneously, meaning that the regulated characterization methods have difficulty identifying the contributions of these reactions. Normally, the intercalation reaction preferentially occurs in the reaction path compared to the Li deposition reaction in the graphite electrode. Because the intercalation reaction potential is in the range from 200 to 65 mV versus Li/Li+ in the graphite electrode, the Li deposition potential is below 0 V versus Li/Li+ [268]. The intercalation reaction potential is clearly more positive than the deposition potential, suggesting that Li deposition is not a competitive reaction to Li insertion thermodynamically [269]. Based on this, lithium metal acting as a negative electrode is typically employed to study Li+ deposition, which is easier to quantify than a graphite electrode.
The asymmetry of electrode evolution can be established based on the difference in the dynamic hindrance between the stripping and deposition potentials simultaneously, including the asymmetric overpotential, asymmetric kinetics and asymmetric morphology evolution. The discovery of the asymmetry between the cathode and anode originates from the seemingly contradictory fact that in the constant current experiment of a Li‖Li symmetric battery, a sharp overpotential “peak” is usually observed during the first few cycles, and the overpotential changes into a flatter plateau after several cycles. More specifically, there are some fundamental variabilities in the macroscopic and microscopic morphology evolution during stripping and deposition. As observed, in the first cycle of the Li‖Li symmetric battery, there was a difference in the shape of the overpotential between the first half-cycle and the second half-cycle; second, the shape in the first few cycles was more complicated than that in the last few cycles, meaning that the charging and discharging overpotential plateaus were more volatile than the first [33]. In contrast, the charging and discharging overpotential plateaus in the last few cycles were flatter and less volatile. Therefore, there were two asymmetry phenomena in the overpotential: (1) under the same periodic cycle, the overpotential in the first half-cycle and the second half-cycle was asymmetric; and (2) under different cycle periods, there was an asymmetry in the overpotential profiles during stripping and deposition. Asymmetric overpotentials were generated in the system. The asymmetry phenomenon was essentially due to the different crucial steps at the electrode interface in the dynamic process. The overpotentials of lithium deposition and stripping processes were considered to be determined by the properties of the electrode/electrolyte interface.
In addition to the overpotential, an asymmetric process can be identified in the morphology evolution. The initial electrode state with a smoother surface can be initially considered to be a symmetric condition. However, at the beginning of the cycling, the symmetry of the initial state is directly destroyed due to the natural asymmetry of the deposition and stripping, as shown in Fig. 15b. The first cycle is the most apparent period for this asymmetry. During continuous stripping/plating, the surface of the electrode becomes more uneven. With increasing porous surface area, clearly displaying the asymmetry between cathode deposition and anode stripping on the surface becomes difficult. This also reveals that the overpotential profiles are flatter in the subsequent cycles than those in previous cycles.

Copyright © 2016, American Chemistry Society. b Cross-sectional operando microscopy images of the lithium-metal-anode surface in earlier cycles and later cycles. Reprinted with permission from Ref. [258]. Copyright © 2017, Royal Society of Chemistry. c Snapshots of the phase parameter (upper), Li+ concentration (middle), and electric potential (lower) during the electrodeposition process. Reprinted with permission from Ref. [259]. Copyright © 2014, AIP Publishing. d Changes in cell polarization (top) correlated with schematic representations of the morphology and energy barrier diagrams (bottom). Reprinted with permission from Ref. [33]. Copyright © 2016, American Chemistry Society
a Schematic representation of the cathode deposition processes divided into lithium nucleation and growth and the anode stripping processes divided into dendrite stripping, bulk stripping and pit stripping. Reprinted with permission from Ref. [33].
The factors that directly affect the asymmetry phenomenon at the electrode/electrolyte interface during battery cycling are as follows: (1) the difference in energy barriers of plating and stripping, which can be caused by different surface morphologies; and (2) the difference in the Li+ transport at the electrode/electrolyte interface, which primarily goes through the SEI film. The asymmetric process is the change in dynamics due to the surface morphology and chemical atmosphere, which ultimately affects the Butler–Volmer equation. Hence, the study of the asymmetry phenomenon can reveal the specific change in dynamics in stripping/plating at the electrode/electrolyte interface and provide a reference for the design of LMBs.
The morphology evolution leading to the asymmetry phenomenon can also be considered a nonlinear process [270]. For a smooth electrode, the smooth surface of the electrode was destroyed in the stripping process, and dendrites were formed as the deposition process proceeded. After several cycles, the area of the smooth surface accounted for a lower percentage, while the area of the porous surface accounted for a higher percentage. The entire electrode surface could be occupied by porous or nonuniform structures (sharp edges, cracks, and holes on the nanometer to micrometer scale), which was proven by Dees et al.’s research on morphological transitions in a lithium-metal anode in a 1.2 M LiPF6/EC:EMC (30:70 in weight ratio) electrolyte [271]. They showed that the porous and smooth morphologies had completely different overpotential profiles.
Wood et al. [33] studied the processes of lithium-metal stripping and deposition via numerical simulation analysis. The stripping process was divided into three stages: dendrite stripping, bulk stripping and pit stripping. The deposition process was divided into two stages: nucleation and growth. According to the observation by in situ atomic force microscopy in alkyl carbonate solution, as shown in Fig. 15a, the deposition and stripping on the electrode could result in completely different morphologies [272]. In more detail, for two bulk Li/Li electrodes assembled into a symmetric battery with the same initial state (one serves as the anode and the other as the cathode), the two electrodes will have completely different surface morphologies after a half-cycle. Obviously, the surface morphologies of the cathode and anode are naturally asymmetric. Fan et al. [273] further demonstrated that the deposition mode of lithium metal evolved from a granular growth mode to a fibrous growth mode, which essentially came from the increase in current density/areal capacity. Widening of the difference in morphology evolution was indicated to be possibly due to the current density/areal capacity. The ratio of effective current to the surface area for the dendrite morphology is different from that on the electrode surface with only pits. Liu et al. [274] studied the morphology evolution of lithium-metal after a half-cycle. Gaps of 15–20 and 13–16 mV were found for the end voltages between S-Li (stripped-Li, the morphology was pits) and P-Li (plated-Li, the morphology was dendrites) during stripping and plating, respectively. Although the polarity was transformed, the morphologies of the two were still not consistent. Because the reaction at each electrode should be governed by B–V kinetics, a modified form of the current–overpotential relationship was employed [33]:
where \(\gamma\) is a parameter that accounts for the roughness of the electrode surface, \(k\) is an effective heterogeneous rate constant that depends on the morphology of the electrode, \(\beta\) is a weighting factor, and \(\eta\) is the overpotential of the electrode. The roughness parameter \(\gamma\) is the ratio between the total surface area of the electrode (including deposits) and the 2D projected surface area.
Many factors could conceivably influence the change in overpotential during the evolution of Li+ deposition. Liang and Chen [259] studied a nonlinear phase field model that included three parts that could affect the overpotential: the ion concentration in the electrolyte, electrochemical reaction at the interface, and stable overpotential of lithium-metal bulk. According to this model, as shown in Fig. 15c, the local inhomogeneities of deposits are expected to be able to induce local variations in current density and Li concentration. Similar results by Chen et al. [275] showed that the electrode surface morphology could be related to the Li+ concentration during the charging process. However, the electrode surface morphology, in turn, could result in a concentration gradient as well as in an electric potential gradient near the electrode surface. At the same time, due to the electrochemical reaction, the amount of Li+ deposition increased with time, and a part of the electrode surface eventually formed a continuous dense layer. In contrast, the growth of the dense layer was relatively slow compared to that of the deposition layer due to the uneven current density. The deposition behavior of lithium-metal on Cu foil and Cu foam was compared by Fan et al. [273]. As the current density/areal capacity increased, the granular growth mode of Li deposition changed to a fibrous growth mode. The change in lithiophilic and electrode nucleation sites could lead to the asymmetric process. Most of the above reports focus on the impact of applied voltage and initial electrode morphology on dendritic patterns during charging.
Due to the passivation characteristics, the SEI can change the ionic conductivity transmission during stripping/plating processes, which directly affects the evolution of the surface morphology of lithium metal [6]. As observed, the electrode surface had different Li+ diffusion velocities during stripping/plating processes. In the deposition process, the dendrites continuously grew, penetrated the previously formed SEI film and produced a newly formed SEI film. However, the surface composition and thickness of the previously formed SEI were inconsistent with those of the newly formed SEI, which had a lower surface resistance [276]. Physically, the kinetic barrier of ionic conductivity transmission for the newly formed SEI was lower. The cathode was dominated by a thin SEI with a lower transmission kinetic barrier, whereas the anode was dominated by a thick SEI with a larger kinetic barrier. Thus, the asymmetry phenomenon of Li+ transport between the anode and cathode could result.
Therefore, in situ optical measurements can be used to directly observe details of Li dendrite nucleation and growth on the lithium anode surface and the Li+ ionic conductivity in the SEI film during battery cycling. Furthermore, the local change in Li+ ionic conductivity within the SEI film can undoubtedly directly affect the morphology of dendrites during the deposition process.
4.2 Multiscale Host Matrix for Nucleation in Lithium-Metal Deposition
A three-dimensional (3D) host matrix for uniform deposition during battery charging is considered to be effective from the perspective of the space-charge model, which is the most widely accepted lithium dendrite nucleation model. For the 3D host matrix, a larger surface area can significantly reduce the effective current density (\(J\)), meaning that the initial nucleation time of lithium dendrites (τ) can be increased, which is beneficial to uniform deposition of the lithium surface. Meanwhile, experimental results showed that the time of dendrite occurrence was proportional to \(J^{ - 2}\), and the time for passing through the cell was directly proportional to \(J^{ - 1}\), which was consistent with the model conclusions [277]. Using different materials or modified 3D host matrices, more properties may be improved, such as mechanical flexibility, lithium affinity, and surface energy [4, 38, 114, 116,117,118,119, 278,279,280,281,282,283,284,285,286,287,288,289,290,291,292,293,294,295,296,297,298,299,300]
Yang et al. [38] proposed a 3D porous Cu current collector with excellent plating/stripping efficiency (98.5%) performance and more material expansion. The 3D porous Cu current collector was treated as being composed of submicrometer Cu fibers with nanosized protuberances on the 2D Cu surface, which could act as charge centers and nucleus modulation sites, as shown in Fig. 16a. In addition, such a 3D porous structure also had some favorable electrochemical properties. The areal capacity density of lithium metal deposited in such a 3D porous structure was up to 3.1 mAh cm−2. A Li‖Li symmetric cell with 3D porous Cu collectors showed a stable voltage hysteresis over 600 h without a short circuit. Li et al. [282] employed different sized copper meshes (pore diameters from 60 to 170 µm) embedded in lithium metal using mechanical pressure to precisely control the areal capacity density of the collector. This method could effectively avoid uneven local mass distribution of the 3D collector and achieve uniform distribution of nucleation sites on the surface of the collector bulk. A further advance was to improve the lithiophilicity of the collector and the wettability of the deposited coating. The disadvantage of the 3D host matrix was that it could consume more electrolyte to form an SEI, which could cause low CE. Meanwhile, the 3D host matrix also showed poor performance in terms of overpotential and uniform lithium capture ability because of the absence of lithiophilic materials. Many researchers have proposed using lithiophilic materials, such as Li-C@ZnO [280, 301], Li-PI@ZnO [279], Li-C@Si [278], and C-SiO2@Au [302], to solve these issues. Yang et al. [285] further demonstrated that Cu foam@graphene coating (GC) could improve the cycling stability of the LMB over a wide current density range. Such Cu foam@GC could reduce the local current density and avoid excess consumption of electrolyte because of its moderate surface area of 1.12 m2 g−1.

Copyright © 2015, Springer Nature. b Simulated current densities of the micropatterned lithium metal surface and its 2D cross-section image. Reprinted with permission from Ref. [297]. Copyright © 2018, Elsevier
a Schematic of the proposed electrochemical deposition processes of lithium metal on a planar current collector and a 3D current collector. Reprinted with permission from Ref. [38].
As identified, a micropatterned anode similar to the 3D collector can achieve an increased surface area of the electrode. The difference is that micropatterns are often fabricated in a simple way. As identified, micropatterns can be pressed on the surface of a lithium-metal anode by a mold to achieve a regular uniform microarray [293,294,295,296,297,298,299, 303]. The arrays of micropatterns could guide uniform Li deposition to achieve better cycling performance than bare lithium metal. Therefore, from the perspective of industrialization, micropatterned anodes have significant advantages in terms of processing costs. Since Li+ was deposited at the bottom of the array holes, the shapes and area geometries of the 3D porous arrays, such as needles [299], square holes [295], pyramids [297, 298], and cylinders [296], could be obtained, which directly affected the local current distribution. At present, micropatterned anode analysis is mainly conducted through finite element analysis, as shown in Fig. 16b, such as MATLAB and COMSOL. Nevertheless, more research into the details of this field is needed.
Qualitative simulation research found that the surface uniformity of the electrode structure could reach parts per million (PPM) and effectively inhibit dendrite growth [304]. Although a uniform surface is key to dendrite suppression and the protuberances impact dendrite growth, many nanoscale protuberances, such as charge centers and nucleation sites, still need to be introduced into the nanostructure layer. In addition, the introduction of lithophilic modification can effectively guide the deposition of Li+ on the microstructure and compensate for the electrostatic interaction between the protuberances and ions. At present, achieving uniformity at the PPM level in cost-effective large-scale industrial production is still difficult. Therefore, controlling the uniform roughness of the graded submicron structure and specific surface area of lithium-metal anodes is very important. Different strategies can be used to balance the advantages and disadvantages of the graded microstructure.
4.3 In Situ Observation of Nucleation and Electrode Geometric Evolution
Several in situ characterization techniques have been developed to observe the reaction processes to fundamentally understand the mechanisms. For example, in modern manufacturing processes, integrated circuits require continuous miniaturization, and in situ ec-S/TEM can be initially employed to investigate Cu electrodeposition for integrated circuits [277]. With the rapid development of the battery field, in situ characterization techniques seem more important for mechanism exploration and performance optimization. The method of in situ ec-S/TEM can serve this purpose. Microfabricated electrochemical liquid cells can be specifically designed for such S/TEM, as shown in Fig. 17a. Although some helpful information can be obtained, some fundamental issues still need to be further studied. For example, in the electrodeposition of Cu on a Au substrate, which is a well-studied system [311,312,313,314,315,316], the results obtained from current transient analysis and those obtained from post-growth microscopy seem inconsistent [317,318,319]. Hence, the kinetics of nucleation evolution related to ion transfer, electron transfer, ion concentration distribution, and electrode morphology should be further studied. To investigate these processes and the electrode geometric evolution, in situ characterization techniques with high spatial resolution that enable detailed site-specific observation of the electrochemical processes and nucleation evolution at the electrode/electrolyte interface occurring in a natural electrolyte environment in real time have been developed. These techniques typically utilize closed microfabrication electrochemical cells, which combine the capability of quantitative electrochemical measurements with multiple characterization measurement systems such as high-spatial- and temporal-resolution imaging, spectroscopy, and diffraction. Many reports have been devoted to in situ characterization of the nucleation and growth stages of lithium crystals. A lithium-metal particle can be a single crystal with a bcc structure [306]. Since the Li (110) surface has the lowest surface energy among all index surfaces [320, 321], Li embryos can be preferentially terminated on (110) surfaces, as shown in Fig. 17c. The surface energy has been proven to significantly affect the growth of the crystal plane in the lithium dendrite nucleation stage. The direction of Li whisker growth along the electric field has been widely demonstrated in early in situ characterization [322, 323]. Li whiskers prefer to grow along crystal planes with high surface energy. However, He et al. [306] used an AFM cantilever coupled with in situ ETEM and demonstrated that whiskers grew without a preponderant orientation. The classical theory of surface crystal growth is also suitable for Li nucleation [324]. According to high-resolution in situ ETEM characterization, the surface energy obviously plays a more important role in Li nucleation than in the growth process. In fact, there are still many factors that directly affect Li nucleation and growth. Kushima et al.[305]. provided more details about the Li nucleation and growth processes via ETEM, and two competing mechanisms of surface growth of dense Eden-like clusters and root growth of whiskers were identified. There is voltage-dependent competition between lithium electrodeposition and SEI formation reactions, as shown in Fig. 17b. As shown, the diameter of the Li embryo is proportional to the square root of time. With the growth of the Li embryo, the SEI layer passivates the embryo surface and gradually reduces the lithium deposition rate because Li+ diffusion is limited by the SEI. Ishikawa et al. [307] indicated that the Li deposition substrate crystallographic planes could also affect the size of Li embryos, as observed by in situ SEM, as shown in Fig. 17d. Different crystal surfaces of substrates have different adsorption capacities for Li atoms, which can lead to different adsorption ion concentrations, through which the size of Li embryos can be directly determined. Li et al. [325] modified the surface structure of the lithium-metal anode and studied the dominant nucleation sites and ion concentration gradients in the electrolyte. Using in situ optical microscopy, they observed that the anode surface with nucleation sites was significantly smoother than that without nucleation sites during cycling.

Copyright © 2020, Springer Nature. b Three models of the morphology of lithium deposits based on in situ STEM. Reprinted with permission from Ref. [305]. Copyright © 2017, Elsevier. c Utilization of in situ atomic force microscopy (AFM)-environmental transmission electron microscopy (ETEM) to observe the Li nucleation process during electrochemical deposition of Li+ in a CO2 environment. Blue dotted lines highlight the nucleus. The red arrow indicates that the Li embryo grew over time at the electrode/electrolyte interface. Red dotted lines emphasize the side surface and shape of the Li whisker. Reprinted with permission from Ref. [306]. Copyright © 2019, Springer Nature. d SEM images of Li precipitates with various size distributions on various index Cu. Reprinted with permission from Ref. [307]. Copyright © 2017, American Chemistry Society
a Assembly diagram of an in situ electrochemical scanning/transmission electron microscopy (ec-S/TEM) holder, and SEM images of electrochemical spacer microchips. Reprinted with permission from Ref. [39].
There are still many other factors influencing the lithium nucleation process, as observed by in situ characterization techniques. A key factor is the size of the lithium nucleus, which is still one of the important factors in relation to dendrite formation. Mehdi et al. [326] utilized scanning transmission electron microscopy (STEM) to directly observe that the electrode surface in an electrolyte with an additive could have a larger grain size than that without an additive. The larger grain sizes were indicative of more uniform Li deposition, which was directly connected to better performance in terms of CE and an overall decrease in the formation of Li dendrites. Without an additive, the round-trip efficiency of Li stripping/plating processes was reduced, with poor electronic/ionic conductivity. The cell with additives could give a favorable nucleus, benefiting from the high ionic conductivity of the inorganic component LiF, which led to early dominant growth of some nucleation sites [326]. The concentration gradient of the electrolyte may affect not only the size of the lithium nucleus but also the dendrite formation directly, suggesting that it can affect the growth stage. The ion concentration gradient at the electrode surface can be chemically mapped by in situ STEM imaging rather than TEM. White et al. [308] observed an increased ion concentration gradient during dendrite growth, as shown in Fig. 18a. Therefore, the lower ion concentration gradient is helpful for suppressing the formation of dendrites and conducive to the formation of larger Li nuclei, as confirmed by in situ STEM [306, 320, 321, 324]. Recently, Yan et al. [327] demonstrated that increasing the deposition temperature could result in a large nucleus size, a low nucleation density, and compact growth, as observed by a series of ex situ and in situ microscopic analyses.

Copyright © 2012, American Chemical Society. b Experimental equipment for operando X-ray imaging with a V-slot Li electrode holder, and comparison diagram of lithium deposition before/after 4 h in a Li-Cu cell. Reprinted with permission from Ref. [309]. Copyright © 2019, American Chemical Society. c Li-Cu cell static in situ NMR spectra, and visualization of lithium deposited for 1 cycle. Reprinted with permission from Ref. [310]. Copyright © 2020, Elsevier
a STEM beam scanned from top to bottom to obtain the time evolution of the Pb2+ ionic concentration. During the acquisition of image a0 (b0), the potential V on the electrode induces polarity switching, and plating (stripping) begins, which creates white (black) regions on the electrode boundary. Images a1 and b1 depict the same events with 8 × 8 spatial binning and rescaling of the intensities to enhance the contrast. Data (a2 and b2) extracted from the boxed regions in a1 and b1 are fitted to the diffusion equation with increasing t downward and x to the right. The best-fitting region (a3 and b3) returns D = 1.3 × 10–11 and 1.5 × 10−11 m2 s–1, respectively. Reprinted with permission from Ref. [308].
From a more macroscopic perspective, in situ characterization showed that the morphologies of the lithium-metal anode can be significantly different before and after cycling, where the anode surface is smooth before and porous after cycling. Other new methods are being developed for studying electrode evolution with cycling. As observed, the formation of dendrites can directly lead to a change in morphology during electrode evolution. Yu et al. [309] demonstrated the evolution process of electrodes via in situ operando X-ray imaging. Plated Li first grew and uniformly filled the whole space under the separator during plating. With further plating, Li plating predominantly occurred near the separator, leading to a much denser layer of plated Li near the separator than in the distal regions, as shown in Fig. 18b. For the lithium-metal anode, pits formed on the anode surface during the stripping processes. Pits were preferred deposition sites where the next lithium dendrites formed in plating. “Dead lithium” was formed on the anode surface during continuous cycling. To quantitatively measure the amount of dead lithium on the porous anode surface, Hsieh et al. [310] employed in situ NMR spectroscopy to reveal lithium metal microstructures upon plating and stripping, as shown in Fig. 18c. The spectrum exhibits a single peak at (245 ± 1) ppm before cycling, which represents the pristine smooth lithium foil anode. Upon plating, a signal at (263 ± 1) ppm began to grow, reaching the maximum peak intensity after full plating, which represents a nonuniform surface morphology.
Although in situ characterization is instrumental, the current reported results are mainly limited by the dendrite growth accuracy. Observing how Li+ is adsorbed on the anode surface is difficult. In addition, whether rearrangement occurs and how rearrangement develops in the early stage of dendrite nucleation are still unclear. There is still a vast growing research area about the early stage of dendrite nucleation. Undoubtedly, in situ characterization methods often play an indispensable role in the exploration of the nucleation process of dendrite formation.
5 Conclusion and Prospects
Lithium-metal-anode-based batteries have tremendous application potential for portable electronics, electric cars, and smart grids. However, the unstable lithium/electrolyte interface restricts their subsequent development in terms of battery durability and safety. In dealing with the degradation of lithium-metal anodes, this review aims to clarify the basic understanding of the electrochemical processes, key advances in the electrochemical understanding and multiple approaches to control Li stripping/plating with respect to the formation of the SEI and dendrite growth. The critical and in-depth comprehension of the interfacial challenges, nucleation and electrode geometric evolution is emphasized. The key advances in and novel electrochemical strategies for Li stripping/plating are summarized as follows: (1) electrolyte reduction mechanisms; (2) charge transfer processes; (3) local current distribution; (4) solvation and desolvation of the Li+ sheath; (5) SEI modification and inhibition of dendrite growth; (6) mechanisms of additives; (7) asymmetric processes in stripping/plating; and (8) in situ characterization. The following specific challenges and concrete analyses need to be addressed.
-
(1)
The understanding of SEI formation in terms of the reaction kinetics and mechanisms, composition, and role in battery performance is insufficient. The SEI is closely related to LMB performance and is an inseparable process during battery operation. As identified, there are a large number of single/two-electron reduction paths in electrolyte reduction. Therefore, in an actual electrode-liquid system with different solvent types, the formed SEI has a large number of complex components, and the formation is an extremely fast reaction process (ps timescale). In this regard, modeling the whole reaction process in detail is difficult. In this case, QC/AIMD/DFT calculations and in situ microscopic characterization methods may be employed to obtain a fundamental understanding of the mechanisms.
-
(2)
The specific kinetic processes of charge transport, especially electron transfer, at the anode/electrolyte interface require more interpretation. Mainstream research on charge transfer involves the tunneling mode, combining Marcus theory and the B–V equation while lacking a detailed description of the kinetic processes during electron transfer. In particular, the close contact between electrons and Li+ at the electrode interface in the specific kinetic process is not described in detail.
-
(3)
A deeper understanding of the fluctuating local current density distribution in terms of dendrite nucleation and growth is lacking. Reducing the local current density is a practical and effective method to suppress dendrites. However, the limited research ignores the local current inhomogeneity caused by the local geometric distribution. To achieve global current density reduction, a uniform geometric structure should be considered for the current collectors. The precise difference between the formation of dendrites at low current and at larger current has not been thoroughly explored.
-
(4)
Results on the rate-determining step in Li+ ion diffusion from the bulk electrolyte to the anode surface are inconclusive. In general, the diffusion process of the sheath structure can be divided into three stages: (1) the sheath structure must diffuse from the electrolyte to near the SEI interface; (2) the Li+ sheath structure must be desolvated near the SEI surface; and (3) Li+ ions change their solvation structure in the electrolyte to adjust the coordination state. Distinguishing which is the rate-determining step is difficult in the current report. Utilizing a variety of sheath structures to adjust interface redox reactions is the most effective tool for exploring the rate-determining step and achieving electrochemical interface regulation.
-
(5)
There are difficulties in unifying the various mechanical properties and chemical components of SEIs to obtain a general consensus for standard lithium batteries. Many reports quantitatively discuss how the mechanical properties and varieties of chemical components of SEIs correspond to battery performance. The mainstream belief is generally that the structure of the SEI film is a complex mosaic structure, including the organic components near the electrolyte and the inorganic components near the lithium-metal anode. However, the simulation of SEIs is generally built via single chemical components at present. Thus, accurately simulating the complex working conditions of SEIs is difficult. The understanding of the SEI is not perfect, and the corresponding structure–activity relationship is not yet clear. Meanwhile, distinguishing how coupled properties affect the battery performance is difficult.
-
(6)
Whether a variety of additives will produce a synergistic effect is unknown. The additives for LMBs consist of salt additives and organic molecule additives. According to the functional classification, they can be divided into single functional additives (SEI formation additives or self-healing electrostatic shielding additives) or multifunctional additives. Research on the interaction between inhibiting additives and accelerating additives is lacking. At present, there is no effective framework providing guidance on multiple additives to achieve multiple functions. In practical terms, the theory on additives can be applied to further suppress dendrites and improve the cell efficiency.
-
(7)
The understanding of the disappearance of asymmetric processes after long cycling, which seems like presentational symmetrization, is insufficient. Intuitively, the asymmetric process corresponds to a difference in the shape of the overpotential between the first half-cycle and the second half-cycle. After long cycling, the smooth morphology of electrodes is altered to a porous morphology. Meanwhile, the compositions of the electrolyte and SEI are also changed compared to the initial states. All of these factors may make the overall asymmetric process more symmetrical. Discriminating which of these factors contributes more is difficult.
-
(8)
In situ characterization still has many shortcomings. For example, the current distribution and electric field gradient in an in situ electrochemical cell are different from those in coin batteries or commercial batteries. In situ electrochemical cells (such as S/TEM spacer microchips) are limited by their electrolyte volume, in which the width is always less than a micron. The width of the electrodes directly affects the electric field distribution in the electrolyte. The influence of cell size on the driving force of the nucleation process and local electrochemical reactions could still be substantial. Hence, in situ characterization has difficulty directly reflecting the same process in coin batteries or commercial batteries at present.
However, the above challenges and issues are a new world that we do not yet know. Every coin has two sides. These issues give us more possibilities to explore. To overcome the challenges discussed above for practical application, several future research directions may be proposed as follows.
-
(1)
QC/AIMD/DFT calculations have a brilliant future in SEI modeling. With the continuous optimization of computing software and the improvement in the computing capability, full-scale modeling of SEIs has been gradually implemented. QC/AIMD/DFT calculations with larger fluxes are more appropriate for performing SEI fitting under larger models. Building the mosaic model with organic/inorganic components will provide us with more details for exploring changes at the anode/electrolyte interface of LMBs.
-
(2)
Based on Marcus theory and the B–V equation, new theories can be developed, and multiscale characterization of the electrode transfer kinetics process at the anode/electrolyte interface can be performed. The current understanding of electrode transfer limits our understanding of interface issues. The development of new theories and multiscale characterization will be helpful for solving the dendrite deposition and low CE issues of lithium-metal electrodes in the future.
-
(3)
The influence of geometric uniformity on local current density can be investigated to further understand the deposition behavior. Researchers generally utilize powder electrodes and 3D current collectors to achieve a low current density. The current is affected by the uneven geometric distribution, leading to an inconsistent current density, which has a significant impact on dendrite nucleation and growth. Thus, the core issue lies in quantitatively analyzing the subtle current difference in the dominant nucleation and growth of lithium dendrites.
-
(4)
The desolvation process in EC-based ester electrolytes with local high concentrations can be investigated. The composition of the solvation sheath structure of Li+ is totally different between dilute and high concentration solution systems. Due to the different solvent types, the activation energy barrier between desolvation and ion diffusion near the SEI needs to be explored in detail.
-
(5)
The inhibition, acceleration and hybrid mechanism of additives can be in-depth studied to achieve "self-repair" of ion transport at the anode/electrolyte interface. Since most additives follow the acceleration or inhibition mechanism, the investigation of this mechanism can explore a screening principle for additives.
-
(6)
Research on desolvation and microscopic nucleation may provide a new understanding of asymmetric dynamics. Investigating the appearance of the asymmetry phenomenon at the beginning of cycling and the disappearance of the asymmetry phenomenon after long cycling of the battery can reveal the relationship between Li+ transport and dendrite nucleation and growth.
-
(7)
Some in situ characterization methods with low input energy density that correspond to large-scale characterization at the same time can be developed. A high-energy electron beam is often employed for in situ experiments. The results of the high-energy electron beam cause unexpected interactions, such as radiolysis, electrolyte degradation and redox reactions. Therefore, low-energy in situ characterization methods can guarantee the accuracy of experimental results.
In this review, we collect the issues of the SEI, anode/electrolyte interface film and anode surface deposition process of Li+ transport from the electrolyte to anode surface. Meanwhile, factors such as the solvation structure, Li+ concentration, anode/SEI components and surface geometry are explained. In view of the interfacial complexity of LMBs, only small parts of them have been solved under specific conditions. More phenomena are pending and not yet comprehended. In addition, the electrochemical processes are not the only factor in improving the performance of LMBs. Some studies have occasionally presented a series of contradictory explanations and illustrations of similar phenomena. For LMBs and these electrochemical processes, the blueprint for the future lies in meticulous treatment of the interface processes to determine the characteristics of the SEI and provide an adequate explanation of nucleation under different environments, taking this research to new heights. This rational and critical thinking would eventually pay considerable dividends in battery development.
References
Zhang, H.L., Zhao, H.B., Khan, M.A., et al.: Recent progress in advanced electrode materials, separators and electrolytes for lithium batteries. J. Mater. Chem. A 6, 20564–20620 (2018). https://doi.org/10.1039/c8ta05336g
Umeshbabu, E., Zheng, B.Z., Yang, Y.: Recent progress in all-solid-state lithium-sulfur batteries using high Li-ion conductive solid electrolytes. Electrochem. Energy Rev. 2, 199–230 (2019). https://doi.org/10.1007/s41918-019-00029-3
Ma, Z., Yuan, X.X., Li, L., et al.: A review of cathode materials and structures for rechargeable lithium-air batteries. Energy Environ. Sci. 8, 2144–2198 (2015). https://doi.org/10.1039/c5ee00838g
Lin, D., Liu, Y., Cui, Y.: Reviving the lithium metal anode for high-energy batteries. Nat. Nanotechnol. 12, 194–206 (2017). https://doi.org/10.1038/nnano.2017.16
Albertus, P., Babinec, S., Litzelman, S., et al.: Status and challenges in enabling the lithium metal electrode for high-energy and low-cost rechargeable batteries. Nat. Energy 3, 16–21 (2018). https://doi.org/10.1038/s41560-017-0047-2
Cheng, X.B., Zhang, R., Zhao, C.Z., et al.: Toward safe lithium metal anode in rechargeable batteries: a review. Chem. Rev. 117, 10403–10473 (2017). https://doi.org/10.1021/acs.chemrev.7b00115
Xin, S., You, Y., Wang, S.F., et al.: Solid-state lithium metal batteries promoted by nanotechnology: progress and prospects. ACS Energy Lett. 2, 1385–1394 (2017). https://doi.org/10.1021/acsenergylett.7b00175
Zhao, X.D., Kong, X.L., Liu, Z.L., et al.: The cutting-edge phosphorus-rich metal phosphides for energy storage and conversion. Nano Today 40, 101245 (2021). https://doi.org/10.1016/j.nantod.2021.101245
Wang, H.S., Liu, Y.Y., Li, Y.Z., et al.: Lithium metal anode materials design: interphase and host. Electrochem. Energy Rev. 2, 509–517 (2019). https://doi.org/10.1007/s41918-019-00054-2
Feng, X.N., Ouyang, M.G., Liu, X., et al.: Thermal runaway mechanism of lithium ion battery for electric vehicles: a review. Energy Storage Mater. 10, 246–267 (2018). https://doi.org/10.1016/j.ensm.2017.05.013
Liu, H.Q., Wei, Z.B., He, W.D., et al.: Thermal issues about Li-ion batteries and recent progress in battery thermal management systems: a review. Energy Convers. Manag. 150, 304–330 (2017). https://doi.org/10.1016/j.enconman.2017.08.016
Li, S., Jiang, M.W., Xie, Y., et al.: Developing high-performance lithium metal anode in liquid electrolytes: challenges and progress. Adv. Mater. 30, 1706375 (2018). https://doi.org/10.1002/adma.201706375
Amine, R., Liu, J.Z., Acznik, I., et al.: Regulating the hidden solvation-ion-exchange in concentrated electrolytes for stable and safe lithium metal batteries. Adv. Energy Mater. 10, 2000901 (2020). https://doi.org/10.1002/aenm.202000901
Chen, S.J., Xiang, Y.X., Zheng, G.R., et al.: High-efficiency lithium metal anode enabled by a concentrated/fluorinated ester electrolyte. ACS Appl. Mater. Interfaces 12, 27794–27802 (2020). https://doi.org/10.1021/acsami.0c06930
Drvarič Talian, S., Bobnar, J., Moškon, J., et al.: Effect of high concentration of polysulfides on Li stripping and deposition. Electrochim. Acta 354, 136696 (2020). https://doi.org/10.1016/j.electacta.2020.136696
Généreux, S., Gariépy, V., Rochefort, D.: On the relevance of reporting water content in highly concentrated electrolytes: the LiTFSI-acetonitrile case. J. Electrochem. Soc. 167, 120536 (2020). https://doi.org/10.1149/1945-7111/abb34c
Glaser, R., Wu, F.X., Register, E., et al.: Tuning low concentration electrolytes for high rate performance in lithium-sulfur batteries. J. Electrochem. Soc. 167, 100512 (2020). https://doi.org/10.1149/1945-7111/ab7183
Hou, J.X., Lu, L.G., Wang, L., et al.: Thermal runaway of lithium-ion batteries employing LiN(SO2F)2-based concentrated electrolytes. Nat. Commun. 11, 5100 (2020). https://doi.org/10.1038/s41467-020-18868-w
Jiang, L.H., Liang, C., Li, H., et al.: Safer triethyl-phosphate-based electrolyte enables nonflammable and high-temperature endurance for a lithium ion battery. ACS Appl. Energy Mater. 3, 1719–1729 (2020). https://doi.org/10.1021/acsaem.9b02188
Kremer, L.S., Danner, T., Hein, S., et al.: Influence of the electrolyte salt concentration on the rate capability of ultra-thick NCM 622 electrodes. Batter. Supercaps 3, 1172–1182 (2020). https://doi.org/10.1002/batt.202000098
Lee, S., Park, K., Koo, B., et al.: Safe, stable cycling of lithium metal batteries with low-viscosity, fire-retardant locally concentrated ionic liquid electrolytes. Adv. Funct. Mater. 30, 2003132 (2020). https://doi.org/10.1002/adfm.202003132
Lin, S.S., Hua, H.M., Li, Z.S., et al.: Functional localized high-concentration ether-based electrolyte for stabilizing high-voltage lithium-metal battery. ACS Appl. Mater. Interfaces 12, 33710–33718 (2020). https://doi.org/10.1021/acsami.0c07904
Peng, Z., Cao, X., Gao, P.Y., et al.: High-power lithium metal batteries enabled by high-concentration acetonitrile-based electrolytes with vinylene carbonate additive. Adv. Funct. Mater. 30, 2001285 (2020). https://doi.org/10.1002/adfm.202001285
Wang, Z.C., Sun, Y.Y., Mao, Y.Y., et al.: Highly concentrated dual-anion electrolyte for non-flammable high-voltage Li-metal batteries. Energy Storage Mater. 30, 228–237 (2020). https://doi.org/10.1016/j.ensm.2020.05.020
Zhang, M.M., Hao, H.X., Zhou, D.X., et al.: Understanding the microscopic structure of a “water-in-salt” lithium ion battery electrolyte probed with ultrafast IR spectroscopy. J. Phys. Chem. C 124, 8594–8604 (2020). https://doi.org/10.1021/acs.jpcc.0c00937
Zhou, Y.F., Su, M., Yu, X.F., et al.: Real-time mass spectrometric characterization of the solid-electrolyte interphase of a lithium-ion battery. Nat. Nanotechnol. 15, 224–230 (2020). https://doi.org/10.1038/s41565-019-0618-4
Nagy, K.S., Kazemiabnavi, S., Thornton, K., et al.: Thermodynamic overpotentials and nucleation rates for electrodeposition on metal anodes. ACS Appl. Mater. Interfaces 11, 7954–7964 (2019). https://doi.org/10.1021/acsami.8b19787
Goodman, J.K.S., Kohl, P.A.: Effect of alkali and alkaline earth metal salts on suppression of lithium dendrites. J. Electrochem. Soc. 161, D418–D424 (2014). https://doi.org/10.1149/2.0301409jes
Choudhury, S., Tu, Z.Y., Stalin, S., et al.: Electroless formation of hybrid lithium anodes for fast interfacial ion transport. Angew. Chem. Int. Ed. 56, 13070–13077 (2017). https://doi.org/10.1002/anie.201707754
Liu, J.L., Eisenberg, B.: Molecular mean-field theory of ionic solutions: a Poisson-Nernst-Planck-Bkerman model. Entropy 22, 550 (2020). https://doi.org/10.3390/e22050550
Peled, E., Menkin, S.: Review—SEI: past, present and future. J. Electrochem. Soc. 164, A1703–A1719 (2017). https://doi.org/10.1149/2.1441707jes
Ely, D.R., García, R.E.: Heterogeneous nucleation and growth of lithium electrodeposits on negative electrodes. J. Electrochem. Soc. 160, A662–A668 (2013). https://doi.org/10.1149/1.057304jes
Wood, K.N., Kazyak, E., Chadwick, A.F., et al.: Dendrites and pits: untangling the complex behavior of lithium metal anodes through operando video microscopy. ACS Cent. Sci. 2, 790–801 (2016). https://doi.org/10.1021/acscentsci.6b00260
Goodenough, J.B., Kim, Y.: Challenges for rechargeable batteries. J. Power Sources 196, 6688–6694 (2011). https://doi.org/10.1016/j.jpowsour.2010.11.074
Borodin, O., Self, J., Persson, K.A., et al.: Uncharted waters: super-concentrated electrolytes. Joule 4, 69–100 (2020). https://doi.org/10.1016/j.joule.2019.12.007
Bazant, M.Z.: Theory of chemical kinetics and charge transfer based on nonequilibrium thermodynamics. Acc. Chem. Res. 46, 1144–1160 (2013). https://doi.org/10.1021/ar300145c
Jie, Y.L., Liu, X.J., Lei, Z.W., et al.: Enabling high-voltage lithium metal batteries by manipulating solvation structure in ester electrolyte. Angew. Chem. Int. Ed. 59, 3505–3510 (2020). https://doi.org/10.1002/anie.201914250
Yang, C.P., Yin, Y.X., Zhang, S.F., et al.: Accommodating lithium into 3D current collectors with a submicron skeleton towards long-life lithium metal anodes. Nat. Commun. 6, 8058 (2015). https://doi.org/10.1038/ncomms9058
Unocic, R.R., Jungjohann, K.L., Mehdi, B.L., et al.: In situ electrochemical scanning/transmission electron microscopy of electrode-electrolyte interfaces. MRS Bull. 45, 738–745 (2020). https://doi.org/10.1557/mrs.2020.226
Cheng, X.B., Hou, T.Z., Zhang, R., et al.: Dendrite-free lithium deposition induced by uniformly distributed lithium ions for efficient lithium metal batteries. Adv. Mater. 28, 2888–2895 (2016). https://doi.org/10.1002/adma.201506124
Gauthier, M., Carney, T.J., Grimaud, A., et al.: Electrode-electrolyte interface in Li-ion batteries: current understanding and new insights. J. Phys. Chem. Lett. 6, 4653–4672 (2015). https://doi.org/10.1021/acs.jpclett.5b01727
Chazalviel, J.N.: Electrochemical aspects of the generation of ramified metallic electrodeposits. Phys. Rev. A 42, 7355–7367 (1990). https://doi.org/10.1103/physreva.42.7355
Ding, J.F., Xu, R., Yan, C., et al.: A review on the failure and regulation of solid electrolyte interphase in lithium batteries. J. Energy Chem. 59, 306–319 (2021). https://doi.org/10.1016/j.jechem.2020.11.016
Chen, X.R., Zhao, B.C., Yan, C., et al.: Review on Li deposition in working batteries: from nucleation to early growth. Adv. Mater. 33, 2004128 (2021). https://doi.org/10.1002/adma.202004128
Peled, E.: The electrochemical behavior of alkali and alkaline earth metals in nonaqueous battery systems: the solid electrolyte interphase model. J. Electrochem. Soc. 126, 2047–2051 (1979). https://doi.org/10.1149/1.2128859
Nazri, G., Muller, R.H.: Composition of surface layers on Li electrodes in PC, LiClO4 of very low water content. J. Electrochem. Soc. 132, 2050–2054 (1985). https://doi.org/10.1149/1.211428810.1149/1.2114288
Peled, E., Golodnitsky, D., Ardel, G.: Advanced model for solid electrolyte interphase electrodes in liquid and polymer electrolytes. J. Electrochem. Soc. 144, L208–L210 (1997). https://doi.org/10.1149/1.1837858
Aurbach, D.: Review of selected electrode-solution interactions which determine the performance of Li and Li ion batteries. J. Power Sources 89, 206–218 (2000). https://doi.org/10.1016/S0378-7753(00)00431-6
Shi, S.Q., Lu, P., Liu, Z.Y., et al.: Direct calculation of Li-ion transport in the solid electrolyte interphase. J. Am. Chem. Soc. 134, 15476–15487 (2012). https://doi.org/10.1021/ja305366r
Li, Y., Li, Y., Pei, A., et al.: Atomic structure of sensitive battery materials and interfaces revealed by cryo-electron microscopy. Science 358, 506–510 (2017). https://doi.org/10.1126/science.aam6014
Wang, Q., Yang, C.K., Yang, J.J., et al.: Dendrite-free lithium deposition via a superfilling mechanism for high-performance Li-metal batteries. Adv. Mater. 31, 1903248 (2019). https://doi.org/10.1002/adma.201903248
Bentley, C.L., Kang, M., Unwin, P.R.: Nanoscale surface structure-activity in electrochemistry and electrocatalysis. J. Am. Chem. Soc. 141, 2179–2193 (2019). https://doi.org/10.1021/jacs.8b09828
Sigman, M.S., Harper, K.C., Bess, E.N., et al.: The development of multidimensional analysis tools for asymmetric catalysis and beyond. Acc. Chem. Res. 49, 1292–1301 (2016). https://doi.org/10.1021/acs.accounts.6b00194
Wagle, D.V., Zhao, H., Baker, G.A.: Deep eutectic solvents: sustainable media for nanoscale and functional materials. Acc Chem Res 47, 2299–2308 (2014). https://doi.org/10.1021/ar5000488
Wang, A.P., Kadam, S., Li, H., et al.: Review on modeling of the anode solid electrolyte interphase (SEI) for lithium-ion batteries. Npj Comput. Mater. 4, 15 (2018). https://doi.org/10.1038/s41524-018-0064-0
Palacín, M.R., de Guibert, A.: Why do batteries fail? Science 351, 1253292 (2016). https://doi.org/10.1126/science.1253292
Ramos-Sanchez, G., Soto, F.A., Martinez De La Hoz, J.M., et al.: Computational studies of interfacial reactions at anode materials: initial stages of the solid-electrolyte-interphase layer formation. J. Electrochem. Energy Convers. Storage 13, 031002 (2016). https://doi.org/10.1115/1.4034412
Aurbach, D., Ein-Eli, Y., Chusid, O., et al.: The correlation between the surface chemistry and the performance of Li-carbon intercalation anodes for rechargeable ‘rocking-chair’ type batteries. J. Electrochem. Soc. 141, 603–611 (1994). https://doi.org/10.1149/1.2054777
Yan, C., Xu, R., Xiao, Y., et al.: Toward critical electrode/electrolyte interfaces in rechargeable batteries. Adv. Funct. Mater. 30, 1909887 (2020). https://doi.org/10.1002/adfm.201909887
Cheng, X.B., Yan, C., Zhang, X.Q., et al.: Electronic and ionic channels in working interfaces of lithium metal anodes. ACS Energy Lett. 3, 1564–1570 (2018). https://doi.org/10.1021/acsenergylett.8b00526
Aurbach, D., Daroux, M.L., Faguy, P.W., et al.: Identification of surface films formed on lithium in propylene carbonate solutions. J. Electrochem. Soc. 134, 1611–1620 (1987). https://doi.org/10.1149/1.210072210.1149/1.2100722
Wang, Y.X., Nakamura, S., Ue, M., et al.: Theoretical studies to understand surface chemistry on carbon anodes for lithium-ion batteries: reduction mechanisms of ethylene carbonate. J. Am. Chem. Soc. 123, 11708–11718 (2001). https://doi.org/10.1021/ja0164529
Borodin, O., Olguin, M., Spear, C.E., et al.: Towards high throughput screening of electrochemical stability of battery electrolytes. Nanotechnology 26, 354003 (2015). https://doi.org/10.1088/0957-4484/26/35/354003
Maibach, J., Lindgren, F., Eriksson, H., et al.: Electric potential gradient at the buried interface between lithium-ion battery electrodes and the SEI observed using photoelectron spectroscopy. J. Phys. Chem. Lett. 7, 1775–1780 (2016). https://doi.org/10.1021/acs.jpclett.6b00391
Chapman, N., Borodin, O., Yoon, T., et al.: Spectroscopic and density functional theory characterization of common lithium salt solvates in carbonate electrolytes for lithium batteries. J. Phys. Chem. C 121, 2135–2148 (2017). https://doi.org/10.1021/acs.jpcc.6b1223410.1021/acs.jpcc.6b12234
Tang, M., Miyazaki, K., Abe, T., et al.: Effect of graphite orientation and lithium salt on electronic passivation of highly oriented pyrolytic graphite. J. Electrochem. Soc. 159, A634–A641 (2012). https://doi.org/10.1149/2.073205jes
Kranz, T., Kranz, S., Miß, V., et al.: Interrelation between redox molecule transport and Li+ ion transport across a model solid electrolyte interphase grown on a glassy carbon electrode. J. Electrochem. Soc. 164, A3777–A3784 (2017). https://doi.org/10.1149/2.1171714jes
Michan, A.L., Leskes, M., Grey, C.P.: Voltage dependent solid electrolyte interphase formation in silicon electrodes: monitoring the formation of organic decomposition products. Chem. Mater. 28, 385–398 (2016). https://doi.org/10.1021/acs.chemmater.5b04408
Kumar, R., Lu, P., Xiao, X.C., et al.: Strain-induced lithium losses in the solid electrolyte interphase on silicon electrodes. ACS Appl. Mater. Interfaces 9, 28406–28417 (2017). https://doi.org/10.1021/acsami.7b06647
Steinhauer, M., Stich, M., Kurniawan, M., et al.: In situ studies of solid electrolyte interphase (SEI) formation on crystalline carbon surfaces by neutron reflectometry and atomic force microscopy. ACS Appl. Mater. Interfaces 9, 35794–35801 (2017). https://doi.org/10.1021/acsami.7b09181
Attia, P.M., Das, S., Harris, S.J., et al.: Electrochemical kinetics of SEI growth on carbon black: Part I. Experiments. J. Electrochem. Soc. 166, E97–E106 (2019). https://doi.org/10.1149/2.0231904jes
Keil, P., Schuster, S.F., Wilhelm, J., et al.: Calendar aging of lithium-ion batteries. J. Electrochem. Soc. 163, A1872–A1880 (2016). https://doi.org/10.1149/2.0411609jes
Keil, P., Jossen, A.: Calendar aging of NCA lithium-ion batteries investigated by differential voltage analysis and coulomb tracking. J. Electrochem. Soc. 164, A6066–A6074 (2016). https://doi.org/10.1149/2.0091701jes
Chu, Y.L., Shen, Y.B., Guo, F., et al.: Advanced characterizations of solid electrolyte interphases in lithium-ion batteries. Electrochem. Energy Rev. 3, 187–219 (2020). https://doi.org/10.1007/s41918-019-00058-y
Tan, J., Matz, J., Dong, P., et al.: A growing appreciation for the role of LiF in the solid electrolyte interphase. Adv. Energy Mater. 11, 2100046 (2021). https://doi.org/10.1002/aenm.202100046
Delp, S.A., Borodin, O., Olguin, M., et al.: Importance of reduction and oxidation stability of high voltage electrolytes and additives. Electrochim. Acta 209, 498–510 (2016). https://doi.org/10.1016/j.electacta.2016.05.100
Hou, J.B., Yang, M., Wang, D.Y., et al.: Fundamentals and challenges of lithium ion batteries at temperatures between –40 and 60 °C. Adv. Energy Mater. 10, 2070079 (2020). https://doi.org/10.1002/aenm.202070079
Yan, C., Li, H.R., Chen, X., et al.: Regulating the inner Helmholtz plane for stable solid electrolyte interphase on lithium metal anodes. J. Am. Chem. Soc. 141, 9422–9429 (2019). https://doi.org/10.1021/jacs.9b05029
Naji, A., Ghanbaja, J., Humbert, B., et al.: Electroreduction of graphite in LiClO4-ethylene carbonate electrolyte. Characterization of the passivating layer by transmission electron microscopy and Fourier-transform infrared spectroscopy. J. Power Sources 63, 33–39 (1996). https://doi.org/10.1016/S0378-7753(96)02439-1
Novák, P., Joho, F., Imhof, R., et al.: In situ investigation of the interaction between graphite and electrolyte solutions. J. Power Sources 81(82), 212–216 (1999). https://doi.org/10.1016/S0378-7753(99)00119-6
Arora, P., White, R.E., Doyle, M.: Capacity fade mechanisms and side reactions in lithium-ion batteries. J. Electrochem. Soc. 145, 3647–3667 (1998). https://doi.org/10.1149/1.1838857
Leung, K., Budzien, J.L.: Ab initio molecular dynamics simulations of the initial stages of solid-electrolyte interphase formation on lithium ion battery graphitic anodes. Phys. Chem. Chem. Phys. 12, 6583–6586 (2010). https://doi.org/10.1039/b925853a
Onuki, M., Kinoshita, S., Sakata, Y., et al.: Identification of the source of evolved gas in Li-ion batteries using 13C-labeled solvents. J. Electrochem. Soc. 155, A794–A797 (2008). https://doi.org/10.1149/1.2897970
Yu, J.M., Balbuena, P.B., Budzien, J., et al.: Hybrid DFT functional-based static and molecular dynamics studies of excess electron in liquid ethylene carbonate. J. Electrochem. Soc. 158, A400–A410 (2011). https://doi.org/10.1149/1.3545977
Liu, Q., Cresce, A., Schroeder, M., et al.: Insight on lithium metal anode interphasial chemistry: reduction mechanism of cyclic ether solvent and SEI film formation. Energy Storage Mater. 17, 366–373 (2019). https://doi.org/10.1016/j.ensm.2018.09.024
Simmen, F., Foelske-Schmitz, A., Verma, P., et al.: Surface layer formation on Li1+xMn2O4−δ thin film electrodes during electrochemical cycling. Electrochim. Acta 56, 8539–8544 (2011). https://doi.org/10.1016/j.electacta.2011.07.046
Eriksson, T., Andersson, A.M., Bishop, A.G., et al.: Surface analysis of LiMn2O4 electrodes in carbonate-based electrolytes. J. Electrochem. Soc. 149, A69 (2002). https://doi.org/10.1149/1.142639810.1149/1.1426398
Leroy, S., Martinez, H., Dedryvère, R., et al.: Influence of the lithium salt nature over the surface film formation on a graphite electrode in Li-ion batteries: an XPS study. Appl. Surf. Sci. 253, 4895–4905 (2007). https://doi.org/10.1016/j.apsusc.2006.10.071
Leroy, S., Blanchard, F., Dedryvère, R., et al.: Surface film formation on a graphite electrode in Li-ion batteries: AFM and XPS study. Surf. Interface Anal. 37, 773–781 (2005). https://doi.org/10.1002/sia.2072
Aurbach, D., Zaban, A., Gofer, Y., et al.: Recent studies of the lithium-liquid electrolyte interface electrochemical, morphological and spectral studies of a few important systems. J. Power Sources 54, 76–84 (1995). https://doi.org/10.1016/0378-7753(94)02044-4
Xu, K.: Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem Inform 35, 4303–4417 (2004). https://doi.org/10.1002/chin.200450271
Aurbach, D., Zaban, A., Ein-Eli, Y., et al.: Recent studies on the correlation between surface chemistry, morphology, three-dimensional structures and performance of Li and Li-C intercalation anodes in several important electrolyte systems. J. Power Sources 68, 91–98 (1997). https://doi.org/10.1016/S0378-7753(97)02575-5
Kawamura, T., Kimura, A., Egashira, M., et al.: Thermal stability of alkyl carbonate mixed-solvent electrolytes for lithium ion cells. J. Power Sources 104, 260–264 (2002). https://doi.org/10.1016/S0378-7753(01)00960-0
Harris, S.J., Lu, P.: Effects of inhomogeneities—nanoscale to mesoscale—on the durability of Li-ion batteries. J. Phys. Chem. C 117, 6481–6492 (2013). https://doi.org/10.1021/jp311431z
Eshkenazi, V., Peled, E., Burstein, L., et al.: XPS analysis of the SEI formed on carbonaceous materials. Solid State Ion. 170, 83–91 (2004). https://doi.org/10.1016/S0167-2738(03)00107-3
Sloop, S.E., Pugh, J.K., Wang, S., et al.: Chemical reactivity of PF5 and LiPF6 in ethylene carbonate/dimethyl carbonate solutions. Electrochem. Solid-State Lett. 4, A42 (2001). https://doi.org/10.1149/1.1353158
Aurbach, D., Markovsky, B., Shechter, A., et al.: A comparative study of synthetic graphite and Li electrodes in electrolyte solutions based on ethylene carbonate-dimethyl carbonate mixtures. J. Electrochem. Soc. 143, 3809–3820 (1996). https://doi.org/10.1149/1.1837300
Ren, Y., Qi, Z.H., Zhang, C., et al.: The charge transfer of intercalated Li atoms around islands on Li-halide (F, Br, Cl) surface of SEIs: a first principles calculation. Comput. Mater. Sci. 176, 109535 (2020). https://doi.org/10.1016/j.commatsci.2020.109535
Qin, X.P., Shao, M.H., Balbuena, P.B.: Elucidating mechanisms of Li plating on Li anodes of lithium-based batteries. Electrochim. Acta 284, 485–494 (2018). https://doi.org/10.1016/j.electacta.2018.07.159
Tang, M., Lu, S.D., Newman, J.: Experimental and theoretical investigation of solid-electrolyte-interphase formation mechanisms on glassy carbon. J. Electrochem. Soc. 159, A1775–A1785 (2012). https://doi.org/10.1149/2.025211jes
Li, D.J., Danilov, D., Zhang, Z.R., et al.: Modeling the SEI-formation on graphite electrodes in LiFePO4 batteries. J. Electrochem. Soc. 162, A858–A869 (2015). https://doi.org/10.1149/2.0161506jes
Krauskopf, T., Mogwitz, B., Hartmann, H., et al.: The fast charge transfer kinetics of the lithium metal anode on the garnet-type solid electrolyte Li6.25Al0.25La3Zr2O12. Adv. Energy Mater. 10, 2000945 (2020). https://doi.org/10.1002/aenm.202000945
Bertolini, S., Balbuena, P.B.: Buildup of the solid electrolyte interphase on lithium-metal anodes: reactive molecular dynamics study. J. Phys. Chem. C 122, 10783–10791 (2018). https://doi.org/10.1021/acs.jpcc.8b03046
Takenaka, N., Suzuki, Y., Sakai, H., et al.: On electrolyte-dependent formation of solid electrolyte interphase film in lithium-ion batteries: strong sensitivity to small structural difference of electrolyte molecules. J. Phys. Chem. C 118, 10874–10882 (2014). https://doi.org/10.1021/jp5018696
Lin, Y.X., Liu, Z., Leung, K., et al.: Connecting the irreversible capacity loss in Li-ion batteries with the electronic insulating properties of solid electrolyte interphase (SEI) components. J. Power Sources 309, 221–230 (2016). https://doi.org/10.1016/j.jpowsour.2016.01.078
Nie, M.Y., Abraham, D.P., Chen, Y.J., et al.: Silicon solid electrolyte interphase (SEI) of lithium ion battery characterized by microscopy and spectroscopy. J. Phys. Chem. C 117, 13403–13412 (2013). https://doi.org/10.1021/jp404155y
Liu, G.Y., Lu, W.: A model of concurrent lithium dendrite growth, SEI growth, SEI penetration and regrowth. J. Electrochem. Soc. 164, A1826–A1833 (2017). https://doi.org/10.1149/2.0381709jes
Harry, K.J., Higa, K., Srinivasan, V., et al.: Influence of electrolyte modulus on the local current density at a dendrite tip on a lithium metal electrode. J. Electrochem. Soc. 163, A2216–A2224 (2016). https://doi.org/10.1149/2.0191610jes
Yang, H., Fey, E.O., Trimm, B.D., et al.: Effects of pulse plating on lithium electrodeposition, morphology and cycling efficiency. J. Power Sources 272, 900–908 (2014). https://doi.org/10.1016/j.jpowsour.2014.09.026
Rosso, M., Gobron, T., Brissot, C., et al.: Onset of dendritic growth in lithium/polymer cells. J. Power Sources 97(98), 804–806 (2001). https://doi.org/10.1016/S0378-7753(01)00734-0
Heine, J., Krüger, S., Hartnig, C., et al.: Coated lithium powder (CLiP) electrodes for lithium-metal batteries. Adv. Energy Mater. 4, 1300815 (2014). https://doi.org/10.1002/aenm.201300815
Jin, D., Oh, J., Friesen, A., et al.: Self-healing wide and thin Li metal anodes prepared using calendared Li metal powder for improving cycle life and rate capability. ACS Appl. Mater. Interfaces 10, 16521–16530 (2018). https://doi.org/10.1021/acsami.8b02740
Pu, K.C., Qu, X.L., Zhang, X., et al.: Nanoscaled lithium powders with protection of ionic liquid for highly stable rechargeable lithium metal batteries. Adv. Sci. 6, 1901776 (2019). https://doi.org/10.1002/advs.201901776
Jin, D., Roh, Y., Jo, T., et al.: Submicron interlayer for stabilizing thin Li metal powder electrode. Chem. Eng. J. 406, 126834 (2021). https://doi.org/10.1016/j.cej.2020
Jin, D., Bae, H.S., Hong, J., et al.: Scaffold-structured polymer binders for long-term cycle performance of stabilized lithium-powder electrodes. Electrochim. Acta 364, 136878 (2020). https://doi.org/10.1016/j.electacta.2020.136878
Lu, L.L., Ge, J., Yang, J.N., et al.: Free-standing copper nanowire network current collector for improving lithium anode performance. Nano Lett. 16, 4431–4437 (2016). https://doi.org/10.1021/acs.nanolett.6b01581
Wang, J.S., Liu, P., Sherman, E., et al.: Formulation and characterization of ultra-thick electrodes for high energy lithium-ion batteries employing tailored metal foams. J. Power Sources 196, 8714–8718 (2011). https://doi.org/10.1016/j.jpowsour.2011.06.071
An, Y.L., Fei, H.F., Zeng, G.F., et al.: Vacuum distillation derived 3D porous current collector for stable lithium-metal batteries. Nano Energy 47, 503–511 (2018). https://doi.org/10.1016/j.nanoen.2018.03.036
Yun, Q.B., He, Y.B., Lv, W., et al.: Chemical dealloying derived 3D porous current collector for Li metal anodes. Adv. Mater. 28, 6932–6939 (2016). https://doi.org/10.1002/adma.201601409
Ding, F., Xu, W., Graff, G.L., et al.: Dendrite-free lithium deposition via self-healing electrostatic shield mechanism. J. Am. Chem. Soc. 135, 4450–4456 (2013). https://doi.org/10.1021/ja312241y
Yang, X.F., Sun, Q., Zhao, C.T., et al.: Self-healing electrostatic shield enabling uniform lithium deposition in all-solid-state lithium batteries. Energy Storage Mater. 22, 194–199 (2019). https://doi.org/10.1016/j.ensm.2019.07.015
Munaoka, T., Yan, X.Z., Lopez, J., et al.: Ionically conductive self-healing binder for low cost Si microparticles anodes in Li-ion batteries. Adv. Energy Mater. 8, 1703138 (2018). https://doi.org/10.1002/aenm.201703138
Nan, Y., Li, S.M., Zhu, M.Q., et al.: Endowing the lithium metal surface with self-healing property via an in situ gas-solid reaction for high-performance lithium metal batteries. ACS Appl. Mater. Interfaces 11, 28878–28884 (2019). https://doi.org/10.1021/acsami.9b07942
Bernal, J.D., Fowler, R.H.: A theory of water and ionic solution, with particular reference to hydrogen and hydroxyl ions. J. Chem. Phys. 1, 515–548 (1933). https://doi.org/10.1063/1.1749327
Xu, R., Yan, C., Huang, J.Q.: Competitive solid-electrolyte interphase formation on working lithium anodes. Trends Chem. 3, 5–14 (2021). https://doi.org/10.1016/j.trechm.2020.10.008
Xu, R., Shen, X., Ma, X.X., et al.: Identifying the critical anion-cation coordination to regulate the electric double layer for an efficient lithium-metal anode interface. Angew. Chem. Int. Ed. 60, 4215–4220 (2021). https://doi.org/10.1002/anie.202100788
von Wald Cresce, A., Gobet, M., Borodin, O., et al.: Anion solvation in carbonate-based electrolytes. J. Phys. Chem. C 119, 27255–27264 (2015). https://doi.org/10.1021/acs.jpcc.5b08895
Yu, Z., Curtiss, L.A., Winans, R.E., et al.: Asymmetric composition of ionic aggregates and the origin of high correlated transference number in water-in-salt electrolytes. J. Phys. Chem. Lett. 11, 1276–1281 (2020). https://doi.org/10.1021/acs.jpclett.9b03495
Yamada, Y., Wang, J.H., Ko, S., et al.: Advances and issues in developing salt-concentrated battery electrolytes. Nat. Energy 4, 269–280 (2019). https://doi.org/10.1038/s41560-019-0336-z
von Wald Cresce, A., Borodin, O., Xu, K.: Correlating Li+ solvation sheath structure with interphasial chemistry on graphite. J. Phys. Chem. C 116, 26111–26117 (2012). https://doi.org/10.1021/jp303610t
Jie, Y.L., Liu, X.J., Lei, Z.W., et al.: Enabling high-voltage lithium metal batteries by manipulating solvation structure in ester electrolyte. Angew. Chem. 132, 3533–3538 (2020). https://doi.org/10.1002/ange.201914250
Pitzer, K.S.: Ion interaction approach: theory and data correlation. In: Pitzer, K.S. (ed.) Activity Coefficients in Electrolyte Solutions, pp. 75–153. CRC Press, Boca Raton (2018). https://doi.org/10.1201/9781351069472
Besenhard, J.O., Winter, M., Yang, J., et al.: Filming mechanism of lithium-carbon anodes in organic and inorganic electrolytes. J. Power Sources 54, 228–231 (1995). https://doi.org/10.1016/0378-7753(94)02073-C
Wagner, M.R., Albering, J.H., Moeller, K.C., et al.: XRD evidence for the electrochemical formation of Li+(PC)yCn− in PC-based electrolytes. Electrochem. Commun. 7, 947–952 (2005). https://doi.org/10.1016/j.elecom.2005.06.009
Kang, X.: “Charge-transfer” process at graphite/electrolyte interface and the solvation sheath structure of Li+ in nonaqueous electrolytes. J. Electrochem. Soc. 154, A162 (2007). https://doi.org/10.1149/1.240986610.1149/1.2409866
Xu, K., Lam, Y., Zhang, S.S., et al.: Solvation sheath of Li+ in nonaqueous electrolytes and its implication of graphite/electrolyte interface chemistry. J. Phys. Chem. C 111, 7411–7421 (2007). https://doi.org/10.1021/jp068691u
Chen, S.R., Zheng, J.M., Mei, D.H., et al.: High-voltage lithium-metal batteries enabled by localized high-concentration electrolytes. Adv. Mater. 30, 1870144 (2018). https://doi.org/10.1002/adma.201870144
Ding, J.F., Xu, R., Yao, N., et al.: Non-solvating and low-dielectricity cosolvent for anion-derived solid electrolyte interphases in lithium metal batteries. Angew. Chem. Int. Ed. 60, 11442–11447 (2021). https://doi.org/10.1002/anie.202101627
Yao, Y.X., Chen, X., Yan, C., et al.: Regulating interfacial chemistry in lithium-ion batteries by a weakly solvating electrolyte. Angew. Chem. Int. Ed. 60, 4090–4097 (2021). https://doi.org/10.1002/anie.202011482
Yamada, Y., Furukawa, K., Sodeyama, K., et al.: Unusual stability of acetonitrile-based superconcentrated electrolytes for fast-charging lithium-ion batteries. J. Am. Chem. Soc. 136, 5039–5046 (2014). https://doi.org/10.1021/ja412807w
Yoshida, K., Nakamura, M., Kazue, Y., et al.: Oxidative-stability enhancement and charge transport mechanism in glyme-lithium salt equimolar complexes. J. Am. Chem. Soc. 133, 13121–13129 (2011). https://doi.org/10.1021/ja203983r
Yamada, Y., Chiang, C.H., Sodeyama, K., et al.: Corrosion prevention mechanism of aluminum metal in superconcentrated electrolytes. ChemElectroChem 2, 1687–1694 (2015). https://doi.org/10.1002/celc.201500426
Matsumoto, K., Inoue, K., Nakahara, K., et al.: Suppression of aluminum corrosion by using high concentration LiTFSI electrolyte. J. Power Sources 231, 234–238 (2013). https://doi.org/10.1016/j.jpowsour.2012.12.028
McOwen, D.W., Seo, D.M., Borodin, O., et al.: Concentrated electrolytes: decrypting electrolyte properties and reassessing Al corrosion mechanisms. Energy Environ. Sci. 7, 416–426 (2014). https://doi.org/10.1039/c3ee42351d
Yamada, Y., Yaegashi, M., Abe, T., et al.: A superconcentrated ether electrolyte for fast-charging Li-ion batteries. Chem. Commun. 49, 11194 (2013). https://doi.org/10.1039/c3cc46665e
Suo, L.M., Hu, Y.S., Li, H., et al.: A new class of solvent-in-salt electrolyte for high-energy rechargeable metallic lithium batteries. Nat. Commun. 4, 1481 (2013). https://doi.org/10.1038/ncomms2513
Wang, J.H., Yamada, Y., Sodeyama, K., et al.: Superconcentrated electrolytes for a high-voltage lithium-ion battery. Nat. Commun. 7, 12032 (2016). https://doi.org/10.1038/ncomms12032
Wang, J.H., Yamada, Y., Sodeyama, K., et al.: Fire-extinguishing organic electrolytes for safe batteries. Nat. Energy 3, 22–29 (2018). https://doi.org/10.1038/s41560-017-0033-8
Raguette, L., Jorn, R.: Ion solvation and dynamics at solid electrolyte interphases: a long way from bulk? J. Phys. Chem. C 122, 3219–3232 (2018). https://doi.org/10.1021/acs.jpcc.7b11472
Zheng, D., Qu, D.Y., Yang, X.Q., et al.: Preferential solvation of lithium cations and impacts on oxygen reduction in lithium-air batteries. ACS Appl. Mater. Interfaces 7, 19923–19929 (2015). https://doi.org/10.1021/acsami.5b04005
Uchida, S., Ishikawa, M.: Lithium bis(fluorosulfonyl)imide based low ethylene carbonate content electrolyte with unusual solvation state. J. Power Sources 359, 480–486 (2017). https://doi.org/10.1016/j.jpowsour.2017.05.090
Iddir, H., Curtiss, L.A.: Li ion diffusion mechanisms in bulk monoclinic Li2CO3 crystals from density functional studies. J. Phys. Chem. C 114, 20903–20906 (2010). https://doi.org/10.1021/jp1086569
Yildirim, H., Kinaci, A., Chan, M.K.Y., et al.: First-principles analysis of defect thermodynamics and ion transport in inorganic SEI compounds: LiF and NaF. ACS Appl. Mater. Interfaces 7, 18985–18996 (2015). https://doi.org/10.1021/acsami.5b02904
Shi, S.Q., Qi, Y., Li, H., et al.: Defect thermodynamics and diffusion mechanisms in Li2CO3 and implications for the solid electrolyte interphase in Li-ion batteries. J. Phys. Chem. C 117, 8579–8593 (2013). https://doi.org/10.1021/jp310591u
Ramasubramanian, A., Yurkiv, V., Foroozan, T., et al.: Lithium diffusion mechanism through solid-electrolyte interphase in rechargeable lithium batteries. J. Phys. Chem. C 123, 10237–10245 (2019). https://doi.org/10.1021/acs.jpcc.9b00436
Zhang, Q.L., Pan, J., Lu, P., et al.: Synergetic effects of inorganic components in solid electrolyte interphase on high cycle efficiency of lithium ion batteries. Nano Lett. 16, 2011–2016 (2016). https://doi.org/10.1021/acs.nanolett.5b05283
Nikitina, V.A., Vassiliev, S.Y., Stevenson, K.J.: Metal-ion coupled electron transfer kinetics in intercalation-based transition metal oxides. Adv. Energy Mater. 10, 1903933 (2020). https://doi.org/10.1002/aenm.201903933
Chekushkin, P.M., Merenkov, I.S., Smirnov, V.S., et al.: The physical origin of the activation barrier in Li-ion intercalation processes: the overestimated role of desolvation. Electrochim. Acta 372, 137843 (2021). https://doi.org/10.1016/j.electacta.2021.137843
Yamada, Y., Iriyama, Y., Abe, T., et al.: Kinetics of lithium ion transfer at the interface between graphite and liquid electrolytes: effects of solvent and surface film. Langmuir 25, 12766–12770 (2009). https://doi.org/10.1021/la901829v
Abe, T., Ohtsuka, M., Sagane, F., et al.: Lithium ion transfer at the interface between lithium-ion-conductive solid crystalline electrolyte and polymer electrolyte. J. Electrochem. Soc. 151, A1950 (2004). https://doi.org/10.1149/1.1804813
Abe, T., Fukuda, H., Iriyama, Y., et al.: Solvated Li-ion transfer at interface between graphite and electrolyte. J. Electrochem. Soc. 151, A1120 (2004). https://doi.org/10.1149/1.1763141
Abe, T., Sagane, F., Ohtsuka, M., et al.: Lithium-ion transfer at the interface between lithium-ion conductive ceramic electrolyte and liquid electrolyte-a key to enhancing the rate capability of lithium-ion batteries. J. Electrochem. Soc. 152, A2151 (2005). https://doi.org/10.1149/1.2042907
Yamada, Y., Sagane, F., Iriyama, Y., et al.: Kinetics of lithium-ion transfer at the interface between Li0.35La0.55TiO3 and binary electrolytes. J. Phys. Chem. C 113, 14528–14532 (2009). https://doi.org/10.1021/jp9043539
Xu, K., von Cresce, A., Lee, U.: Differentiating contributions to “ion transfer” barrier from interphasial resistance and Li+ desolvation at electrolyte/graphite interface. Langmuir 26, 11538–11543 (2010). https://doi.org/10.1021/la1009994
Li, Y.S., Qi, Y.: Energy landscape of the charge transfer reaction at the complex Li/SEI/electrolyte interface. Energy Environ. Sci. 12, 1286–1295 (2019). https://doi.org/10.1039/c8ee03586e
Borodin, O., Bedrov, D.: Interfacial structure and dynamics of the lithium alkyl dicarbonate SEI components in contact with the lithium battery electrolyte. J. Phys. Chem. C 118, 18362–18371 (2014). https://doi.org/10.1021/jp504598n
Jorn, R., Raguette, L., Peart, S.: Investigating the mechanism of lithium transport at solid electrolyte interphases. J. Phys. Chem. C 124, 16261–16270 (2020). https://doi.org/10.1021/acs.jpcc.0c03018
Li, Q.Y., Lu, D.P., Zheng, J.M., et al.: Li+-desolvation dictating lithium-ion battery’s low-temperature performances. ACS Appl. Mater. Interfaces 9, 42761–42768 (2017). https://doi.org/10.1021/acsami.7b13887
Jorn, R., Kumar, R., Abraham, D.P., et al.: Atomistic modeling of the electrode-electrolyte interface in Li-ion energy storage systems: electrolyte structuring. J. Phys. Chem. C 117, 3747–3761 (2013). https://doi.org/10.1021/jp3102282
Xu, R., Yan, C., Xiao, Y., et al.: The reduction of interfacial transfer barrier of Li ions enabled by inorganics-rich solid-electrolyte interphase. Energy Storage Mater. 28, 401–406 (2020). https://doi.org/10.1016/j.ensm.2019.12.020
Ming, J., Cao, Z., Li, Q., et al.: Molecular-scale interfacial model for predicting electrode performance in rechargeable batteries. ACS Energy Lett. 4, 1584–1593 (2019). https://doi.org/10.1021/acsenergylett.9b00822
Nasara, R.N., Ma, W., Kondo, Y., et al.: Charge-transfer kinetics of the solid-electrolyte interphase on Li4Ti5O12 thin-film electrodes. Chemsuschem 13, 4041–4050 (2020). https://doi.org/10.1002/cssc.202001086
Jow, T.R., Delp, S.A., Allen, J.L., et al.: Factors limiting Li+ charge transfer kinetics in Li-ion batteries. J. Electrochem. Soc. 165, A361–A367 (2018). https://doi.org/10.1149/2.1221802jes
Li, Q.Y., Jiao, S.H., Luo, L.L., et al.: Wide-temperature electrolytes for lithium-ion batteries. ACS Appl. Mater. Interfaces 9, 18826–18835 (2017). https://doi.org/10.1021/acsami.7b04099
Hu, J.Z., Jaegers, N.R., Chen, Y., et al.: Adsorption and thermal decomposition of electrolytes on nanometer magnesium oxide: an in situ 13C MAS NMR study. ACS Appl. Mater. Interfaces 11, 38689–38696 (2019). https://doi.org/10.1021/acsami.9b11888
Ohba, N., Ogata, S., Asahi, R.: Hybrid quantum-classical simulation of Li ion dynamics and the decomposition reaction of electrolyte liquid at a negative-electrode/electrolyte interface. J. Phys. Chem. C 123, 9673–9679 (2019). https://doi.org/10.1021/acs.jpcc.8b11737
Markevich, E., Salitra, G., Rosenman, A., et al.: The effect of a solid electrolyte interphase on the mechanism of operation of lithium–sulfur batteries. J. Mater. Chem. A 3, 19873–19883 (2015). https://doi.org/10.1039/c5ta04613k
Marino, C., Boulaoued, A., Fullenwarth, J., et al.: Solvation and dynamics of lithium ions in carbonate-based electrolytes during cycling followed by operando infrared spectroscopy: the example of NiSb2, a typical negative conversion-type electrode material for lithium batteries. J. Phys. Chem. C 121, 26598–26606 (2017). https://doi.org/10.1021/acs.jpcc.7b06685
Huang, W., Boyle, D.T., Li, Y., et al.: Nanostructural and electrochemical evolution of the solid-electrolyte interphase on CuO nanowires revealed by cryogenic-electron microscopy and impedance spectroscopy. ACS Nano 13, 737–744 (2019). https://doi.org/10.1021/acsnano.8b08012
Lu, P., Harris, S.J.: Lithium transport within the solid electrolyte interphase. Electrochem. Commun. 13, 1035–1037 (2011). https://doi.org/10.1016/j.elecom.2011.06.026
Ilott, A., Jerschow, A.: Probing solid-electrolyte interphase (SEI) growth and ion permeability at undriven electrolyte-metal interfaces using 7Li NMR. J. Phys. Chem. C 122, 12598–12604 (2018). https://doi.org/10.1021/acs.jpcc.8b01958
Markevich, E., Fridman, K., Sharabi, R., et al.: Amorphous columnar silicon anodes for advanced high voltage lithium ion full cells: dominant factors governing cycling performance. J. Electrochem. Soc. 160, A1824–A1833 (2013). https://doi.org/10.1149/2.085310jes
Xu, K.: Electrolytes and interphases in Li-ion batteries and beyond. Chem. Rev. 114, 11503–11618 (2014). https://doi.org/10.1021/cr500003w
Chen, Y.C., Ouyang, C.Y., Song, L.J., et al.: Electrical and lithium ion dynamics in three main components of solid electrolyte interphase from density functional theory study. J. Phys. Chem. C 115, 7044–7049 (2011). https://doi.org/10.1021/jp112202s
Pan, J., Cheng, Y.T., Qi, Y.: General method to predict voltage-dependent ionic conduction in a solid electrolyte coating on electrodes. Phys. Rev. B 91, 134116 (2015). https://doi.org/10.1103/physrevb.91.134116
Benitez, L., Seminario, J.M.: Ion diffusivity through the solid electrolyte interphase in lithium-ion batteries. J. Electrochem. Soc. 164, E3159–E3170 (2017). https://doi.org/10.1149/2.0181711jes
Lu, X., Liao, X.Q.: Oxidization of fluid-like Li metal with inherent Li-Li2O interface from simulation insights. J. Mater. 6, 692–701 (2020). https://doi.org/10.1016/j.jmat.2020.05.007
Li, Y.S., Leung, K., Qi, Y.: Computational exploration of the Li-electrode|electrolyte interface in the presence of a nanometer thick solid-electrolyte interphase layer. Acc. Chem. Res. 49, 2363–2370 (2016). https://doi.org/10.1021/acs.accounts.6b00363
Liang, C.C.: Conduction characteristics of the lithium iodide-aluminum oxide solid electrolytes. J. Electrochem. Soc. 120, 1289 (1973). https://doi.org/10.1149/1.2403248
Pan, J., Zhang, Q.L., Xiao, X.C., et al.: Design of nanostructured heterogeneous solid ionic coatings through a multiscale defect model. ACS Appl. Mater. Interfaces 8, 5687–5693 (2016). https://doi.org/10.1021/acsami.5b12030
Li, C.L., Maier, J.: Ionic space charge effects in lithium fluoride thin films. Solid State Ion. 225, 408–411 (2012). https://doi.org/10.1016/j.ssi.2012.02.036
Wang, X., Zeng, W., Hong, L., et al.: Stress-driven lithium dendrite growth mechanism and dendrite mitigation by electroplating on soft substrates. Nat. Energy 3, 227–235 (2018). https://doi.org/10.1038/s41560-018-0104-5
Wang, L., Zhou, Z.Y., Yan, X., et al.: Engineering of lithium-metal anodes towards a safe and stable battery. Energy Storage Mater. 14, 22–48 (2018). https://doi.org/10.1016/j.ensm.2018.02.014
Luo, J., Fang, C.C., Wu, N.L.: High polarity poly(vinylidene difluoride) thin coating for dendrite-free and high-performance lithium metal anodes. Adv. Energy Mater. 8, 1701482 (2018). https://doi.org/10.1002/aenm.201701482
Liu, K., Pei, A., Lee, H.R., et al.: Lithium metal anodes with an adaptive “solid-liquid” interfacial protective layer. J. Am. Chem. Soc. 139, 4815–4820 (2017). https://doi.org/10.1021/jacs.6b13314
Gao, Y., Zhao, Y.M., Li, Y.C., et al.: Interfacial chemistry regulation via a skin-grafting strategy enables high-performance lithium-metal batteries. J. Am. Chem. Soc. 139, 15288–15291 (2017). https://doi.org/10.1021/jacs.7b06437
Li, N.W., Shi, Y., Yin, Y.X., et al.: A flexible solid electrolyte interphase layer for long-life lithium metal anodes. Angew. Chem. Int. Ed. 57, 1505–1509 (2018). https://doi.org/10.1002/anie.201713193
Gao, Z.G., Zhang, S.J., Huang, Z.G., et al.: Protection of Li metal anode by surface-coating of PVDF thin film to enhance the cycling performance of Li batteries. Chin. Chem. Lett. 30, 525–528 (2019). https://doi.org/10.1016/j.cclet.2018.05.016
Zhu, M., Wu, J.X., Wang, Y., et al.: Recent advances in gel polymer electrolyte for high-performance lithium batteries. J. Energy Chem. 37, 126–142 (2019). https://doi.org/10.1016/j.jechem.2018.12.013
Yang, Q.L., Li, W.L., Dong, C., et al.: PIM-1 as an artificial solid electrolyte interphase for stable lithium metal anode in high-performance batteries. J. Energy Chem. 42, 83–90 (2020). https://doi.org/10.1016/j.jechem.2019.06.012
Peng, Z., Wang, S.W., Zhou, J.J., et al.: Volumetric variation confinement: surface protective structure for high cyclic stability of lithium metal electrodes. J. Mater. Chem. A 4, 2427–2432 (2016). https://doi.org/10.1039/c5ta10050j
Han, X.G., Gong, Y.H., Fu, K., et al.: Negating interfacial impedance in garnet-based solid-state Li metal batteries. Nat. Mater. 16, 572–579 (2017). https://doi.org/10.1038/nmat4821
Zhou, W.D., Wang, S.F., Li, Y.T., et al.: Plating a dendrite-free lithium anode with a polymer/ceramic/polymer sandwich electrolyte. J. Am. Chem. Soc. 138, 9385–9388 (2016). https://doi.org/10.1021/jacs.6b05341
Duan, H., Yin, Y.X., Shi, Y., et al.: Dendrite-free Li-metal battery enabled by a thin asymmetric solid electrolyte with engineered layers. J. Am. Chem. Soc. 140, 82–85 (2018). https://doi.org/10.1021/jacs.7b10864
Yan, C., Cheng, X.B., Tian, Y., et al.: Dual-layered film protected lithium metal anode to enable dendrite-free lithium deposition. Adv. Mater. 30, 1870181 (2018). https://doi.org/10.1002/adma.201870181
Xu, R., Cheng, X.B., Yan, C., et al.: Artificial interphases for highly stable lithium metal anode. Matter 1, 317–344 (2019). https://doi.org/10.1016/j.matt.2019.05.016
Tao, F., Liu, Y., Ren, X.Y., et al.: Different surface modification methods and coating materials of zinc metal anode. J. Energy Chem. 66, 397–412 (2022). https://doi.org/10.1016/j.jechem.2021.08.022
Soto, F.A., Yan, P.F., Engelhard, M.H., et al.: Tuning the solid electrolyte interphase for selective Li- and Na-ion storage in hard carbon. Adv. Mater. 29, 1606860 (2017). https://doi.org/10.1002/adma.20160686010.1002/adma.201606860
Nanda, S., Bhargav, A., Manthiram, A.: Anode-free, lean-electrolyte lithium-sulfur batteries enabled by tellurium-stabilized lithium deposition. Joule 4, 1121–1135 (2020). https://doi.org/10.1016/j.joule.2020.03.020
Han, F.D., Yue, J., Zhu, X.Y., et al.: Suppressing Li dendrite formation in Li2S-P2S5 solid electrolyte by LiI incorporation. Adv. Energy Mater. 8, 1703644 (2018). https://doi.org/10.1002/aenm.201703644
Dong, Q.Y., Hong, B., Fan, H.L., et al.: Inducing the formation of in situ Li3N-rich SEI via nanocomposite plating of Mg3N2 with lithium enables high-performance 3D lithium-metal batteries. ACS Appl. Mater. Interfaces 12, 627–636 (2020). https://doi.org/10.1021/acsami.9b16156
Fan, X.L., Ji, X., Han, F.D., et al.: Fluorinated solid electrolyte interphase enables highly reversible solid-state Li metal battery. Sci. Adv. 4, eaau9245 (2018). https://doi.org/10.1126/sciadv.aau9245
Liu, F.F., Wang, L.F., Zhang, Z.W., et al.: A mixed lithium-ion conductive Li2S/Li2Se protection layer for stable lithium metal anode. Adv. Funct. Mater. 30, 2001607 (2020). https://doi.org/10.1002/adfm.202001607
Rangasamy, E., Liu, Z.C., Gobet, M., et al.: An iodide-based Li7P2S8I superionic conductor. J. Am. Chem. Soc. 137, 1384–1387 (2015). https://doi.org/10.1021/ja508723m
Ding, F., Xu, W., Chen, X.L., et al.: Effects of carbonate solvents and lithium salts on morphology and coulombic efficiency of lithium electrode. J. Electrochem. Soc. 160, A1894–A1901 (2013). https://doi.org/10.1149/2.100310jes
Zheng, J.M., Engelhard, M.H., Mei, D.H., et al.: Electrolyte additive enabled fast charging and stable cycling lithium metal batteries. Nat. Energy 2, 17012 (2017). https://doi.org/10.1038/nenergy.2017.12
Ding, M.S., von Cresce, A., Xu, K.: Conductivity, viscosity, and their correlation of a super-concentrated aqueous electrolyte. J. Phys. Chem. C 121, 2149–2153 (2017). https://doi.org/10.1021/acs.jpcc.6b12636
Huang, F.F., Ma, G.Q., Wen, Z.Y., et al.: Enhancing metallic lithium battery performance by tuning the electrolyte solution structure. J. Mater. Chem. A 6, 1612–1620 (2018). https://doi.org/10.1039/c7ta08274f
Giffin, G.A., Moretti, A., Jeong, S., et al.: Connection between lithium coordination and lithium diffusion in [Pyr12O1][FTFSI] ionic liquid electrolytes. Chemsuschem 11, 1981–1989 (2018). https://doi.org/10.1002/cssc.201702288
Peng, H., Nguyen, A.V.: A link between viscosity and cation-anion contact pairs: adventure on the concept of structure-making/breaking for concentrated salt solutions. J. Mol. Liq. 263, 109–117 (2018). https://doi.org/10.1016/j.molliq.2018.04.145
Kondou, S., Thomas, M.L., Mandai, T., et al.: Ionic transport in highly concentrated lithium bis(fluorosulfonyl)amide electrolytes with keto ester solvents: structural implications for ion hopping conduction in liquid electrolytes. Phys. Chem. Chem. Phys. 21, 5097–5105 (2019). https://doi.org/10.1039/c9cp00425d
Dong, Y., Zhang, N., Li, C.X., et al.: Fire-retardant phosphate-based electrolytes for high-performance lithium metal batteries. ACS Appl. Energy Mater. 2, 2708–2716 (2019). https://doi.org/10.1021/acsaem.9b00027
Dong, X.L., Lin, Y.X., Li, P.L., et al.: High-energy rechargeable metallic lithium battery at –70 °C enabled by a cosolvent electrolyte. Angew. Chem. Int. Ed. 58, 5623–5627 (2019). https://doi.org/10.1002/anie.201900266
Jafta, C.J., Sun, X.G., Veith, G.M., et al.: Probing microstructure and electrolyte concentration dependent cell chemistry via operando small angle neutron scattering. Energy Environ. Sci. 12, 1866–1877 (2019). https://doi.org/10.1039/c8ee02703j
Heist, A., Lee, S.H.: Improved stability and rate capability of ionic liquid electrolyte with high concentration of LiFSI. J. Electrochem. Soc. 166, A1860–A1866 (2019). https://doi.org/10.1149/2.0381910jes
Takada, K., Yamada, Y., Yamada, A.: Optimized nonflammable concentrated electrolytes by introducing a low-dielectric diluent. ACS Appl Mater Interfaces 11, 35770–35776 (2019). https://doi.org/10.1021/acsami.9b12709
Song, H.Y., Jung, M.H., Jeong, S.K.: Improving electrochemical performance at graphite negative electrodes in concentrated electrolyte solutions by addition of 1,2-dichloroethane. Appl. Sci. 9, 4647 (2019). https://doi.org/10.3390/app9214647
Miao, R.R., Yang, J., Feng, X.J., et al.: Novel dual-salts electrolyte solution for dendrite-free lithium-metal based rechargeable batteries with high cycle reversibility. J. Power Sources 271, 291–297 (2014). https://doi.org/10.1016/j.jpowsour.2014.08.011
Xiong, S.Z., Xie, K., Diao, Y., et al.: Characterization of the solid electrolyte interphase on lithium anode for preventing the shuttle mechanism in lithium-sulfur batteries. J. Power Sources 246, 840–845 (2014). https://doi.org/10.1016/j.jpowsour.2013.08.041
Duangdangchote, S., Krittayavathananon, A., Phattharasupakun, N., et al.: Insight into the effect of additives widely used in lithium-sulfur batteries. Chem. Commun. 55, 13951–13954 (2019). https://doi.org/10.1039/c9cc06504ka
Hou, T.Z., Xu, W.T., Chen, X., et al.: Lithium bond chemistry in lithium-sulfur batteries. Angew. Chem. Int. Ed. 56, 8178–8182 (2017). https://doi.org/10.1002/anie.201704324
Zhang, X.Q., Chen, X., Hou, L.P., et al.: Regulating anions in the solvation sheath of lithium ions for stable lithium metal batteries. ACS Energy Lett. 4, 411–416 (2019). https://doi.org/10.1021/acsenergylett.8b02376
Lee, S.H., Hwang, J.Y., Ming, J., et al.: Toward the sustainable lithium metal batteries with a new electrolyte solvation chemistry. Adv. Energy Mater. 10, 2000567 (2020). https://doi.org/10.1002/aenm.202000567
Ouyang, Y., Guo, Y.P., Li, D., et al.: Single additive with dual functional-ions for stabilizing lithium anodes. ACS Appl. Mater. Interfaces 11, 11360–11368 (2019). https://doi.org/10.1021/acsami.8b21420
Shimizu, M., Umeki, M., Arai, S.: Suppressing the effect of lithium dendritic growth by the addition of magnesium bis(trifluoromethanesulfonyl)amide. Phys Chem Chem Phys 20, 1127–1133 (2018). https://doi.org/10.1039/c7cp06057b
Zeng, W.D., Cheng, M.M.C., Ng, S.K.Y.: Effects of transition metal cation additives on the passivation of lithium metal anode in Li-S batteries. Electrochim. Acta 319, 511–517 (2019). https://doi.org/10.1016/j.electacta.2019.06.177
Tu, Z.Y., Choudhury, S., Zachman, M.J., et al.: Fast ion transport at solid-solid interfaces in hybrid battery anodes. Nat. Energy 3, 310–316 (2018). https://doi.org/10.1038/s41560-018-0096-1
Liang, X., Pang, Q., Kochetkov, I.R., et al.: A facile surface chemistry route to a stabilized lithium metal anode. Nat. Energy 2, 17119 (2017). https://doi.org/10.1038/nenergy.2017.119
Xu, X.F., Zhou, D., Qin, X.Y., et al.: A room-temperature sodium-sulfur battery with high capacity and stable cycling performance. Nat. Commun. 9, 3870 (2018). https://doi.org/10.1038/s41467-018-06443-3
Pang, Q., Liang, X., Kochetkov, I.R., et al.: Stabilizing lithium plating by a biphasic surface layer formed in situ. Angew. Chem. Int. Ed. 57, 9795–9798 (2018). https://doi.org/10.1002/anie.201805456
Ye, H., Yin, Y.X., Zhang, S.F., et al.: Synergism of Al-containing solid electrolyte interphase layer and Al-based colloidal particles for stable lithium anode. Nano Energy 36, 411–417 (2017). https://doi.org/10.1016/j.nanoen.2017.04.056
Ding, F., Xu, W., Chen, X.L., et al.: Effects of cesium cations in lithium deposition via self-healing electrostatic shield mechanism. J. Phys. Chem. C 118, 4043–4049 (2014). https://doi.org/10.1021/jp4127754
Zhang, Y.H., Qian, J.F., Xu, W., et al.: Dendrite-free lithium deposition with self-aligned nanorod structure. Nano Lett. 14, 6889–6896 (2014). https://doi.org/10.1021/nl5039117
Pasquale, M.A., Gassa, L.M., Arvia, A.J.: Copper electrodeposition from an acidic plating bath containing accelerating and inhibiting organic additives. Electrochim. Acta 53, 5891–5904 (2008). https://doi.org/10.1016/j.electacta.2008.03.073
Böttcher, T., Duda, B., Kalinovich, N., et al.: Syntheses of novel delocalized cations and fluorinated anions, new fluorinated solvents and additives for lithium ion batteries. Prog. Solid State Chem. 42, 202–217 (2014). https://doi.org/10.1016/j.progsolidstchem.2014.04.013
Liu, S.F., Ji, X., Piao, N., et al.: An inorganic-rich solid electrolyte interphase for advanced lithium-metal batteries in carbonate electrolytes. Angew. Chem. Int. Ed. 60, 3661–3671 (2021). https://doi.org/10.1002/anie.202012005
Guo, J., Wen, Z.Y., Wu, M.F., et al.: Vinylene carbonate-LiNO3: a hybrid additive in carbonic ester electrolytes for SEI modification on Li metal anode. Electrochem. Commun. 51, 59–63 (2015). https://doi.org/10.1016/j.elecom.2014.12.008
Liu, M., Cheng, Z., Qian, K., et al.: Efficient Li-metal plating/stripping in carbonate electrolytes using a LiNO3-gel polymer electrolyte, monitored by operando neutron depth profiling. Chem. Mater. 31, 4564–4574 (2019). https://doi.org/10.1021/acs.chemmater.9b01325
Qi, S.H., He, J., Liu, J.D., et al.: Phosphonium bromides regulating solid electrolyte interphase components and optimizing solvation sheath structure for suppressing lithium dendrite growth. Adv. Funct. Mater. 31, 2009013 (2021). https://doi.org/10.1002/adfm.202009013
Cheng, X.B., Yan, C., Peng, H.J., et al.: Sulfurized solid electrolyte interphases with a rapid Li+ diffusion on dendrite-free Li metal anodes. Energy Storage Mater. 10, 199–205 (2018). https://doi.org/10.1016/j.ensm.2017.03.008
Roh, Y., Kim, Y.-J., Lee, J.H., et al.: Sustainable formation of sulfur-enriched solid electrolyte interface on a Li metal electrode by sulfur chain-containing polymer electrolyte interfacial layers. ACS Appl. Energy Mater. 3, 10070–10079 (2020). https://doi.org/10.1021/acsaem.0c01758
Xu, J.G., Tian, H.K., Qi, J., et al.: Mechanical and electronic stabilization of solid electrolyte interphase with sulfite additive for lithium metal batteries. J. Electrochem. Soc. 166, A3201–A3206 (2019). https://doi.org/10.1149/2.0331914jes
Dai, H.L., Xi, K., Liu, X., et al.: Cationic surfactant-based electrolyte additives for uniform lithium deposition via lithiophobic repulsion mechanisms. J. Am. Chem. Soc. 140, 17515–17521 (2018). https://doi.org/10.1021/jacs.8b08963
Wang, Q., Yang, C.K., Zhang, Y.F., et al.: Surface-based Li+ complex enables uniform lithium deposition for stable lithium metal anodes. ACS Appl. Energy Mater. 2, 4602–4608 (2019). https://doi.org/10.1021/acsaem.9b00929
Wang, X.S., Mai, W.C., Guan, X.C., et al.: Recent advances of electroplating additives enabling lithium metal anodes to applicable battery techniques. Energy Environ. Mater. 4, 284–292 (2021). https://doi.org/10.1002/eem2.12109
Guo, J.Q., Chen, Y.P., Xiao, Y.B., et al.: Flame-retardant composite gel polymer electrolyte with a dual acceleration conduction mechanism for lithium ion batteries. Chem. Eng. J. 422, 130526 (2021). https://doi.org/10.1016/j.cej.2021.130526
Moffat, T.P., Wheeler, D., Kim, S.K., et al.: Curvature enhanced adsorbate coverage model for electrodeposition. J. Electrochem. Soc. 153, C127 (2006). https://doi.org/10.1149/1.2165580
Chen, K.H., Wood, K.N., Kazyak, E., et al.: Dead lithium: mass transport effects on voltage, capacity, and failure of lithium metal anodes. J. Mater. Chem. A 5, 11671–11681 (2017). https://doi.org/10.1039/c7ta00371d10.1039/c7ta00371d
Liang, L.Y., Chen, L.Q.: Nonlinear phase field model for electrodeposition in electrochemical systems. Appl. Phys. Lett. 105, 263903 (2014). https://doi.org/10.1063/1.4905341
Mullins, W.W., Sekerka, R.F.: Stability of a planar interface during solidification of a dilute binary alloy. J. Appl. Phys. 35, 444–451 (1964). https://doi.org/10.1063/1.1713333
Mullins, W.W., Sekerka, R.F.: Morphological stability of a particle growing by diffusion or heat flow. J. Appl. Phys. 34, 323–329 (1963). https://doi.org/10.1063/1.1702607
Ahmad, Z., Viswanathan, V.: Stability of electrodeposition at solid-solid interfaces and implications for metal anodes. Phys. Rev. Lett. 119, 056003 (2017). https://doi.org/10.1103/physrevlett.119.056003
Ahmad, Z., Viswanathan, V.: Role of anisotropy in determining stability of electrodeposition at solid-solid interfaces. Phys. Rev. Mater. 1, 055403 (2017). https://doi.org/10.1103/physrevmaterials.1.055403
Tikekar, M.D., Archer, L.A., Koch, D.L.: Stabilizing electrodeposition in elastic solid electrolytes containing immobilized anions. Sci. Adv. 2, e1600320 (2016). https://doi.org/10.1126/sciadv.1600320
Monroe, C., Newman, J.: Dendrite growth in lithium/polymer systems. J. Electrochem. Soc. 150, A1377 (2003). https://doi.org/10.1149/1.1606686
Monroe, C., Newman, J.: The impact of elastic deformation on deposition kinetics at lithium/polymer interfaces. J. Electrochem. Soc. 152, A396 (2005). https://doi.org/10.1149/1.1850854
Monroe, C., Newman, J.: The effect of interfacial deformation on electrodeposition kinetics. J. Electrochem. Soc. 151, A880 (2004). https://doi.org/10.1149/1.1710893
Legrand, N., Knosp, B., Desprez, P., et al.: Physical characterization of the charging process of a Li-ion battery and prediction of Li plating by electrochemical modelling. J. Power Sources 245, 208–216 (2014). https://doi.org/10.1016/j.jpowsour.2013.06.130
Doh, C.H., Han, B.C., Jin, B.S., et al.: Structures and formation energies of LixC6 (x = 1–3) and its homologues for lithium rechargeable batteries. Bull. Korean Chem. Soc. 32, 2045–2050 (2011). https://doi.org/10.5012/bkcs.2011.32.6.2045
Mistry, A., Fear, C., Carter, R., et al.: Electrolyte confinement alters lithium electrodeposition. ACS Energy Lett. 4, 156–162 (2019). https://doi.org/10.1021/acsenergylett.8b02003
López, C.M., Vaughey, J.T., Dees, D.W.: Morphological transitions on lithium metal anodes. J. Electrochem. Soc. 156, A726 (2009). https://doi.org/10.1149/1.3158548
Cohen, Y.S., Cohen, Y., Aurbach, D.: Micromorphological studies of lithium electrodes in alkyl carbonate solutions using in situ atomic force microscopy. J. Phys. Chem. B 104, 12282–12291 (2000). https://doi.org/10.1021/jp002526b
Fan, H.L., Gao, C.H., Jiang, H., et al.: A systematical study on the electrodeposition process of metallic lithium. J. Energy Chem. 49, 59–70 (2020). https://doi.org/10.1016/j.jechem.2020.01.013
Liu, H., Cheng, X.B., Xu, R., et al.: Plating/stripping behavior of actual lithium metal anode. Adv. Energy Mater. 9, 1902254 (2019). https://doi.org/10.1002/aenm.201902254
Chen, L., Zhang, H.W., Liang, L.Y., et al.: Modulation of dendritic patterns during electrodeposition: a nonlinear phase-field model. J. Power Sources 300, 376–385 (2015). https://doi.org/10.1016/j.jpowsour.2015.09.055
Pinson, M.B., Bazant, M.Z.: Theory of SEI formation in rechargeable batteries: capacity fade, accelerated aging and lifetime prediction. J. Electrochem. Soc. 160, A243–A250 (2012). https://doi.org/10.1149/2.044302jes
Williamson, M.J., Tromp, R.M., Vereecken, P.M., et al.: Dynamic microscopy of nanoscale cluster growth at the solid-liquid interface. Nat. Mater. 2, 532–536 (2003). https://doi.org/10.1038/nmat944
Liang, Z., Lin, D.C., Zhao, J., et al.: Composite lithium metal anode by melt infusion of lithium into a 3D conducting scaffold with lithiophilic coating. PNAS 113, 2862–2867 (2016). https://doi.org/10.1073/pnas.1518188113
Liu, Y.Y., Lin, D.C., Liang, Z., et al.: Lithium-coated polymeric matrix as a minimum volume-change and dendrite-free lithium metal anode. Nat. Commun. 7, 10992 (2016). https://doi.org/10.1038/ncomms10992
Zhang, Y., Luo, W., Wang, C.W., et al.: High-capacity, low-tortuosity, and channel-guided lithium metal anode. PNAS 114, 3584–3589 (2017). https://doi.org/10.1073/pnas.1618871114
Li, Y.J., Jiao, J.Y., Bi, J.P., et al.: Controlled deposition of Li metal. Nano Energy 32, 241–246 (2017). https://doi.org/10.1016/j.nanoen.2016.12.030
Li, Q., Zhu, S.P., Lu, Y.Y.: 3D porous Cu current collector/Li-metal composite anode for stable lithium-metal batteries. Adv. Funct. Mater. 27, 1606422 (2017). https://doi.org/10.1002/adfm.201606422
Huang, S.B., Zhang, W.F., Ming, H., et al.: Chemical energy release driven lithiophilic layer on 1 m2 commercial brass mesh toward highly stable lithium metal batteries. Nano Lett. 19, 1832–1837 (2019). https://doi.org/10.1021/acs.nanolett.8b04919
Zou, P.C., Wang, Y., Chiang, S.W., et al.: Directing lateral growth of lithium dendrites in micro-compartmented anode arrays for safe lithium metal batteries. Nat. Commun. 9, 464 (2018). https://doi.org/10.1038/s41467-018-02888-8
Yang, G.H., Chen, J.D., Xiao, P.T., et al.: Graphene anchored on Cu foam as a lithiophilic 3D current collector for a stable and dendrite-free lithium metal anode. J. Mater. Chem. A 6, 9899–9905 (2018). https://doi.org/10.1039/c8ta02810a
Wang, Y.Y., Wang, Z.J., Lei, D.N., et al.: Spherical Li deposited inside 3D Cu skeleton as anode with ultrastable performance. ACS Appl. Mater. Interfaces 10, 20244–20249 (2018). https://doi.org/10.1021/acsami.8b04881
Wang, S.H., Yin, Y.X., Zuo, T.T., et al.: Stable Li metal anodes via regulating lithium plating/stripping in vertically aligned microchannels. Adv. Mater. 29, 1703729 (2017). https://doi.org/10.1002/adma.201703729
Li, Z.H., Li, X.L., Zhou, L., et al.: A synergistic strategy for stable lithium metal anodes using 3D fluorine-doped graphene shuttle-implanted porous carbon networks. Nano Energy 49, 179–185 (2018). https://doi.org/10.1016/j.nanoen.2018.04.040
Yu, L., Canfield, N.L., Chen, S.R., et al.: Enhanced stability of lithium metal anode by using a 3D porous nickel substrate. ChemElectroChem 5, 761–769 (2018). https://doi.org/10.1002/celc.201701250
Zhang, R., Wen, S.W., Wang, N., et al.: N-doped graphene modified 3D porous Cu current collector toward microscale homogeneous Li deposition for Li metal anodes. Adv. Energy Mater. 8, 1800914 (2018). https://doi.org/10.1002/aenm.201800914
Shen, L., Shi, P.R., Hao, X.G., et al.: Progress on lithium dendrite suppression strategies from the interior to exterior by hierarchical structure designs. Small 16, 2000699 (2020). https://doi.org/10.1002/smll.202000699
Xue, W.J., Huang, M.J., Li, Y.T., et al.: Ultra-high-voltage Ni-rich layered cathodes in practical Li metal batteries enabled by a sulfonamide-based electrolyte. Nat. Energy 6, 495–505 (2021). https://doi.org/10.1038/s41560-021-00792-y
Shin, D.Y., Ahn, H.J.: Interfacial engineering of a heteroatom-doped graphene layer on patterned aluminum foil for ultrafast lithium storage kinetics. ACS Appl. Mater. Interfaces 12, 19210–19217 (2020). https://doi.org/10.1021/acsami.0c01774
Kim, S.J., Moon, S.H., Kim, M.C., et al.: Micro-patterned 3D Si electrodes fabricated using an imprinting process for high-performance lithium-ion batteries. J. Appl. Electrochem. 48, 1057–1068 (2018). https://doi.org/10.1007/s10800-018-1234-y
Kim, Y.J., Jin, H.S., Lee, D.H., et al.: Guided lithium deposition by surface micro-patterning of lithium-metal electrodes. ChemElectroChem 5, 3169–3175 (2018). https://doi.org/10.1002/celc.201800694
Li, Q., Quan, B.G., Li, W.J., et al.: Electro-plating and stripping behavior on lithium metal electrode with ordered three-dimensional structure. Nano Energy 45, 463–470 (2018). https://doi.org/10.1016/j.nanoen.2018.01.019
Park, J., Kim, D., Jin, D., et al.: Size effects of micro-pattern on lithium metal surface on the electrochemical performance of lithium metal secondary batteries. J. Power Sources 408, 136–142 (2018). https://doi.org/10.1016/j.jpowsour.2018.09.061
Park, J., Jeong, J., Lee, Y., et al.: Micro-patterned lithium metal anodes with suppressed dendrite formation for post lithium-ion batteries. Adv. Mater. Interfaces 3, 1600140 (2016). https://doi.org/10.1002/admi.201600140
Ryou, M.H., Lee, Y.M., Lee, Y., et al.: Mechanical surface modification of lithium metal: towards improved Li metal anode performance by directed Li plating. Adv. Funct. Mater. 25, 834–841 (2015). https://doi.org/10.1002/adfm.201402953
Zhai, P.B., Wei, Y., Xiao, J., et al.: In situ generation of artificial solid-electrolyte interphases on 3D conducting scaffolds for high-performance lithium-metal anodes. Adv. Energy Mater. 10, 1903339 (2020). https://doi.org/10.1002/aenm.201903339
Deng, W., Zhou, X.F., Fang, Q.L., et al.: Microscale lithium metal stored inside cellular graphene scaffold toward advanced metallic lithium anodes. Adv. Energy Mater. 8, 1703152 (2018). https://doi.org/10.1002/aenm.201703152
Yan, K., Lu, Z.D., Lee, H.W., et al.: Selective deposition and stable encapsulation of lithium through heterogeneous seeded growth. Nat. Energy 1, 16010 (2016). https://doi.org/10.1038/nenergy.2016.10
Ahn, J., Park, J., Kim, J.Y., et al.: Insights into lithium surface: stable cycling by controlled 10 μm deep surface relief, reinterpreting the natural surface defect on lithium metal anode. ACS Appl. Energy Mater. 2, 5656–5664 (2019). https://doi.org/10.1021/acsaem.9b00805
Shen, X., Zhang, R., Chen, X., et al.: The failure of solid electrolyte interphase on Li metal anode: structural uniformity or mechanical strength? Adv. Energy Mater. 10, 1903645 (2020). https://doi.org/10.1002/aenm.202070045
Kushima, A., So, K.P., Su, C., et al.: Liquid cell transmission electron microscopy observation of lithium metal growth and dissolution: root growth, dead lithium and lithium flotsams. Nano Energy 32, 271–279 (2017). https://doi.org/10.1016/j.nanoen.2016.12.001
He, Y., Ren, X.D., Xu, Y.B., et al.: Origin of lithium whisker formation and growth under stress. Nat. Nanotechnol. 14, 1042–1047 (2019). https://doi.org/10.1038/s41565-019-0558-z
Ishikawa, K., Ito, Y., Harada, S., et al.: Crystal orientation dependence of precipitate structure of electrodeposited Li metal on Cu current collectors. Cryst. Growth Des. 17, 2379–2385 (2017). https://doi.org/10.1021/acs.cgd.6b01710
White, E.R., Singer, S.B., Augustyn, V., et al.: In situ transmission electron microscopy of lead dendrites and lead ions in aqueous solution. ACS Nano 6, 6308–6317 (2012). https://doi.org/10.1021/nn3017469
Yu, S.H., Huang, X., Brock, J.D., et al.: Regulating key variables and visualizing lithium dendrite growth: an operando X-ray study. J. Am. Chem. Soc. 141, 8441–8449 (2019). https://doi.org/10.1021/jacs.8b13297
Hsieh, Y.C., Leißing, M., Nowak, S., et al.: Quantification of dead lithium via in situ nuclear magnetic resonance spectroscopy. Cell Rep. Phys. Sci. 1, 100139 (2020). https://doi.org/10.1016/j.xcrp.2020.100139
Magnussen, O.M., Hotlos, J., Nichols, R.J., et al.: Atomic structure of Cu adlayers on Au(100) and Au(111) electrodes observed by in situ scanning tunneling microscopy. Phys. Rev. Lett. 64, 2929–2932 (1990). https://doi.org/10.1103/physrevlett.64.2929
Manne, S., Hansma, P.K., Massie, J., et al.: Atomic-resolution electrochemistry with the atomic force microscope: copper deposition on gold. Science 251, 183–186 (1991). https://doi.org/10.1126/science.251.4990.183
Bard, A.J., Fan, F.R.: Introductory lecture. Studies of the liquid/solid interface by scanning tunnelling microscopy and scanning electrochemical microscopy. Faraday Disc. 94, 1–22 (1992). https://doi.org/10.1039/fd9929400001
Schmidt, W.U., Alkire, R.C., Gewirth, A.A.: Mechanic study of copper deposition onto gold surfaces by scaling and spectral analysis of in situ atomic force microscopic images. J. Electrochem. Soc. 143, 3122–3132 (1996). https://doi.org/10.1149/1.1837174
Schneeweiss, M.A., Kolb, D.M.: The initial stages of copper deposition on bare and chemically modified gold electrodes. Phys. Stat. Sol. (a) 173, 51–71 (1999). https://doi.org/10.1002/(SICI)1521-396X(199905)173:1%3c51::AID-PSSA51%3e3.0.CO;2-O
Hölzle, M.H., Zwing, V., Kolb, D.M.: The influence of steps on the deposition of Cu onto Au(111). Electrochim. Acta 40, 1237–1247 (1995). https://doi.org/10.1016/0013-4686(95)00055-J
Gunawardena, G., Hills, G., Montenegro, I., et al.: Electrochemical nucleation: Part I. General considerations. J. Electroanal. Chem. Interfacial Electrochem. 138, 225–239 (1982). https://doi.org/10.1016/0022-0728(82)85080-8
Vereecken, P.M., Strubbe, K., Gomes, W.P.: An improved procedure for the processing of chronoamperometric data: application to the electrodeposition of Cu upon (100) n-GaAs. J. Electroanal. Chem. 433, 19–31 (1997). https://doi.org/10.1016/S0022-0728(97)00195-2
Radisic, A., Long, J.G., Hoffmann, P.M., et al.: Nucleation and growth of copper on TiN from pyrophosphate solution. J. Electrochem. Soc. 148, C41 (2001). https://doi.org/10.1149/1.1344539
Kamrani Moghaddam, L., Ramezani Paschepari, S., Zaimy, M.A., et al.: The inhibition of epidermal growth factor receptor signaling by hexagonal selenium nanoparticles modified by SiRNA. Cancer Gene Ther. 23, 321–325 (2016). https://doi.org/10.1038/cgt.2016.38
Koch, S.L., Morgan, B.J., Passerini, S., et al.: Density functional theory screening of gas-treatment strategies for stabilization of high energy-density lithium metal anodes. J. Power Sources 296, 150–161 (2015). https://doi.org/10.1016/j.jpowsour.2015.07.027
Ghassemi, H., Au, M., Chen, N., et al.: Real-time observation of lithium fibers growth inside a nanoscale lithium-ion battery. Appl. Phys. Lett. 99, 123113 (2011). https://doi.org/10.1063/1.3643035
Liu, X.H., Zhong, L., Zhang, L.Q., et al.: Lithium fiber growth on the anode in a nanowire lithium ion battery during charging. Appl. Phys. Lett. 98, 183107 (2011). https://doi.org/10.1063/1.3585655
Kelton, K.F.: Crystal nucleation in liquids and glasses. Solid State Phys. 45, 75–177 (1991). https://doi.org/10.1016/S0081-1947(08)60144-7
Li, Y.Q., Zhang, L.Y., Liu, S.F., et al.: Original growth mechanism for ultra-stable dendrite-free potassium metal electrode. Nano Energy 62, 367–375 (2019). https://doi.org/10.1016/j.nanoen.2019.05.020
Mehdi, B.L., Stevens, A., Qian, J.F., et al.: The impact of Li grain size on coulombic efficiency in Li batteries. Sci. Rep. 6, 34267 (2016). https://doi.org/10.1038/srep34267
Yan, K., Wang, J.Y., Zhao, S.Q., et al.: Temperature-dependent nucleation and growth of dendrite-free lithium metal anodes. Angew. Chem. Int. Ed. 58, 11364–11368 (2019). https://doi.org/10.1002/anie.201905251
Acknowledgements
This work was supported primarily by the National Key Research and Development Program of China (2020YFA0710303), National Natural Science Foundation of China (No. 22109025), Natural Science Foundation of Fujian Province, China (2021J05121). B.R. Li and Y. Chao contributed equally to this work.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
There are no conflicts to declare.
Rights and permissions
About this article

Cite this article
Li, B., Chao, Y., Li, M. et al. A Review of Solid Electrolyte Interphase (SEI) and Dendrite Formation in Lithium Batteries. Electrochem. Energy Rev. 6, 7 (2023). https://doi.org/10.1007/s41918-022-00147-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41918-022-00147-5