
Abstract
The existing conventional pharmacopeial methods generally used by the pharma industry are easy to perform and reasonably priced, but they require a long time and traditional buffers to obtain the results. This technique was discovered to be lacking in ruggedness as a result of the employment of a conventional column and the usage of a dual-buffer mobile phase in the previous study. There are also no methods available for determining the concentration of ceftazidime in a solution using Rapid Resolution Liquid Chromatography. The goal of this research was to develop and evaluate a unique and previously unpublished liquid chromatography technology for the selected drug. The chromatographic elution was achieved through phosphate buffer solution (0.01 mol/L) and acetonitrile (90:10, v/v) and a flow rate of 1.0 mL/min. The retention time was estimated to be 1.7 min with the Agilent Eclipse C8, 100 mm length, 4.6 mm internal diameter, 1.8 μm particle size column with a flow rate of 1.0 mL/min. A photodiode array detector is used to monitor the eluate at 270 nm, and the temperature of the column is maintained at 35 °C. The method validation parameters yielded good results, including range, linearity, precision, accuracy, specificity, and recovery. The calibration curve was linear from 70 to 130 μg/mL, with a correlation coefficient of 0.9994. The inter-day and intra-day precision was less than 1%. The accuracy was studied, and the recovery test indicated a mean absolute of 99.0% and 101.0% for ceftazidime. The degradation products resulting from the stress studies did not interfere with the detection of ceftazidime, and the assay is thus stability-indicating. In the present study, the detection was carried out more rapidly with the Rapid Resolution Liquid Chromatography method than with the traditional high-performance liquid chromatography method, which indicates that this method is both practical and unconventional to be implemented.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Cephalosporins, also known as cephem, are a class of beta-lactam antibiotics derived from the bacteria Acremonium, originally known as "Cephalosporium." They are used to treat a variety of infections. Cephalosporins are a subclass of beta-lactam antibiotics that include cephamycin and other related compounds. The 7-amino cephalosporanic acid, which is formed as a result of the hydrolysis of cephalosporin C generated during fermentation, is a crucial intermediate in the semi-synthesis of a considerable number of cephalosporins (Maiti and Bidinger 1981).
Ceftazidime is a third-generation semisynthetic cephalosporin with high antibacterial activity. It is frequently used to treat common bacterial infections, including those caused by Proteus species and Pseudomonas aeruginosa, as well as those associated with many other bacteria. Additionally, it is usually recognized as the antibiotic of preference for bacterial infections induced by Enterobacter, Klebsiella, Providencia, Proteus, Haemophylus, and Serratia species, among others (Bergman et al. 2021;D'Cunha R et al. 2018; Raj et al. 2013; Yeh et al. 2005; Ye et al. 2008).
These infections include those of the bone and joints, biliary tract, infections in immunocompromised patients (neutropenic patients), cystic fibrosis (infection in respiratory tract), endophthalmitis, peritonitis, meningitis, pneumonia, skin infections (including burns and ulceration), septicaemia, and urinary tract infections (Maiti and Bidinger 1981, Yan et al. 2012; Ye et al. 2008; Andrasi et al. 2007, Cunha R et al. 2018b; Hefnawy et al. 1999; Khan et al. 2011; Li et al. 2005; Patil and Jacob 2012; Shields et al. 2018; Samnidou et al. 2003). The ability to penetrate bacterial cell walls efficiently, its resistance to bacterial enzyme degradation, its high intrinsic activity against bacterial cell targets, its broad spectrum of activity, and its extremely low toxicity are just a few of the many advantages of this compound. Other advantages include its extensive tissue penetration, metabolic stability, and low level of protein binding in the blood (D’Cunha et al. 2018a, 2018b).
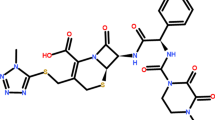
Figure S1 depicts the chemical structure of ceftazidime pentahydrate, which is an antibiotic. Injections of ceftazidime, either as the sodium salt or in solution with arginine, are used to administer the medication. Ceftazidime is widely dispersed in bodily tissues and fluids; it penetrates the placenta and is found in breast milk (Jun Y B et al. 1995).
The absorption of ceftazidime further into the aqueous humor of the eye is relatively good following systemic therapy with the antibiotic (Maiti and Bidinger 1981; Abdellatef et al. 2000; Tyczkowska et al. 1992). On limited occasions, it has been demonstrated that acceptable doses for treatment of eye bacterial infections generated by gram-positive as well as unequivocal gram-negative pathogens can be achieved through systemic inoculation (Jun 1995).
In the literature, there are numerous analytical methodologies for the investigation of cephalosporins, notably ceftazidime, which has an empirical formula of C22H22N6O7S2 with a molecular weight of 546.58 g/mol. A number of techniques, such as spectroscopy, HPLC, capillary electrophoresis, fluorimetry, titrimetry, and polarography, are employed in research. However, despite the fact that ceftazidime has been studied and commercialized for its therapeutic effects, there is just a single pharmacopoeial treatise approved for its HPLC determination.
Several methods for determining the amount of ceftazidime in injectable powder have recently been published by Moreno and Salgado, including microbiological assay, HPLC, and spectrophotometry, among other methods (Maiti and Bidinger 1981; D’Cunha et al. 2018b). To this end, the goal of this research was to develop and evaluate a unique and previously unpublished high-performance liquid chromatography technology.
Active substance content in drug substances and drug products that is less than specified can have a negative impact on the patient's safety and, in certain cases, can result in life-threatening situations. The content of the active ingredients listed above may change during the manufacturing process or when storage conditions deteriorate. These drug assays are critical for drug efficacy. According to regulatory guidelines such as ICH, USFDA, MHRA, and others, its content or tests must be confined to specific values. Despite the fact that there are a few ways of determining the composition of ceftazidime for pure API, high-performance liquid chromatography, amperometric detection (Yan et al. 2012), polarography (Bergman et al. 2021; Yeh et al. 2005; Jun 1995; Khan et al. 2011; Li 2005), fluorometry (Ramya Kuber and Sravanthi 2017), and spectrometry (Amer Saleh Mahdi et al. 2019).
To determine ceftazidime in a liquid chromatographic assay, the USP Pharmacopeial method (USP 42) calls for the use of anhydrous dibasic sodium phosphate and monobasic potassium phosphate, acetonitrile as the mobile phase, a LC system with ultraviolet detection, and a Spherisorb ODS column with irregular particle size and unendcapped. This technique was discovered to be lacking in ruggedness as a result of the employment of a conventional column and the usage of a dual-buffer mobile phase in the previous study. There are also no methods available for determining the concentration of ceftazidime in a solution using Rapid Resolution Liquid Chromatography, i.e. RRLC (Kassi et al. 2017).
In order to overcome this issue, we designed a well-defined reverse-phase RRLC technique with photodiode array detection that was both efficient and effective. Aside from that, two methods were used to assess the specificity of the method: the first involved spiking the contaminants into the sample; the second involved executing multiple degradation procedures on the sample. After that, at the Orchid Chemicals and Pharmaceuticals Ltd. Research and Development Center, a proprietary RRLC method for determining the ceftazidime content was developed. To ensure that the established assay method exhibits stability-indicating features, it is tested for linearity, specificity, precision, robustness, accuracy, stability, and ruggedness in analytical solution, among other qualities. ICH and USFDA regulations have been followed in the validation of the technique in order to verify that it consistently meets the standards and quality characteristics that have been established (Guy 2014; Hiremath and Mruthyunjayaswamy 2008; ICH 2006).
2 Materials and Methods
2.1 Chemicals and Reagents
Ceftazidime bulk drug (active pharmaceutical ingredients), pharmaceutical dosage form, and its impurity standards were obtained from Orchid Pharma Limited, Chennai. Analytical grade reagents such as sodium di-hydrogen phosphate, phosphoric acid, hydrochloric acid, sodium hydroxide, and peroxide were purchased from Merck's private limited specialties, Mumbai, India. Also, HPLC grade solvents such as water, methanol, and acetonitrile were also purchased from Rankem Private Limited, Mumbai.
2.2 RRLC Operating Conditions
The Agilent 1200 RRLC system, which included the G1312B Binary Pump SL, the G1379B Micro Vacuum Degasser, the G1367C High Performance HiP Autosampler SL, the G1316B Thermostatted Column Compartment SL, and the G1315C Diode Array Detector SL, was used for method development and validation (DAD). The Waters Empower 2 Software was used to process and assess the chromatograms. The Sartorius microanalytical balance was used for all weighing activities.
The chromatographic column was used for the method development and validation studies on Zorbax Eclipse plus C8 (100 × 4.6 mm, 1.8 μm) which was manufactured by Agilent Technologies. The optimized method was isocratic with various combinations of 0.01 M sodium di-hydrogen phosphate (buffer) and acetonitrile. The buffer was filtered through a 0.22-µm filter paper using a glass vacuum-filtration apparatus. The mobile phase was a mixture of buffer and acetonitrile at a ratio of 90:10 v/v and degassed. The diluent used for the entire study was 0.02 M disodium hydrogen phosphate adjusted to a pH of 7.0 with 10% v/v phosphoric acid, which was filtered through 0.22 µm of porosity membrane filter paper and degassed. The flow rate was set at 1.0 mL/min, the temperature of the column compartment was maintained at 35 °C, and the temperature of the sample was controlled at 5 °C. The wavelength of the detector was controlled in the entire UV region but was also set at a single wavelength of 254 nm and the injection volume was set at 5 μL. The chromatographic run time was set at 4.0 min.
2.3 Standard Preparation
Dissolve an accurately weighed quantity of ceftazidime reference standard in the mobile phase to obtain a solution having a known concentration of about 0.1 mg/mL.
2.4 Sample Preparation for a Bulk Drug (API)
Dissolve an accurately weighed quantity of ceftazidime sample (API) in the mobile phase to obtain a solution having a known concentration of about 0.1 mg/mL.
2.4.1 Sample preparation for Pharmaceutical Dosage Form (Ceftazidime for Injection)
Dissolve an accurately weighed quantity of ceftazidime for injection in the mobile phase to obtain a solution having a known concentration of about 0.1 mg/mL of ceftazidime.
2.5 Method Validation
In the current optimized RRLC method, all impurities and degradation products are well resolved from the ceftazidime peak. Validation of the new analytical method was performed in accordance with current ICH criteria. The process of validating an analytical procedure involves establishing, through laboratory research, that the procedure's performance characteristics fulfill the requirements for its intended usage. System suitability, specificity, linearity, accuracy, precision, and accuracy, ruggedness, and robustness were also examined for validation (Bliesner 2006; FDA 2015).
2.5.1 Specificity
Specificity is the ability of the method to measure the analysis response in the presence of its potential impurities. Specificity was also studied by performing forced degradation studies using acid, alkali hydrolysis, peroxide, thermal, photolytic, and humidity degradation studies, and also interference of the degradation products was also investigated.
The specificity of the developed RRLC method for ceftazidime in bulk drug and pharmaceutical dosage was carried out in unspiked and spiked with the presence of its impurities, namely, Impurity-A (2RS,6R,7R)-7-[[(Z)-2-(2-aminothiazol-4-yl)-2-[(1-carboxy1-ethylethoxy) imino]acetyl]amino]-8-oxo-3-[(1-pyridinio)methyl]-5-thia-1-azabicyclo[4.2.0]oct-3-ene-2- carboxylate (∆-2-ceftazidime), Impurity-B ((6R,7R)-7-[[(E)-2-(2-aminothiazol-4-yl)-2-[(1-carboxy1-methylethoxy)imino]acetyl]amino]-8-oxo-3-[(1-pyridinio)methyl]-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate), Impurity-C (6R,7R)-2-carboxy-8-oxo-3-(pyridiniomethyl)-5-thia-1- azabicyclo[4.2.0]oct-2-en-7-aminium dichloride), Impurity D [6R,7R)-7-[[(Z)-2-[[2-(1,1-dimethylethoxy)-1,1-dimethyl-2-oxoethoxy]imino]-2-[2 [(triphenylmethyl)amino]thiazol4-yl]acetyl]amino]-8-oxo-3-(pyridiniomethyl)-5-thia-1- azabicyclo[4.2.0]oct-2-ene-2-carboxylate), Impurity E ((6R,7R)-7-[[(Z)-2-(2-ammoniothiazol-4-yl)-2-[[2-(1,1-dimethylethoxy)-1,1-dimethyl-2-oxoethoxy]imino]acetyl]amino]-8-oxo-3-(pyridiniomethyl)- 5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate chloride, Impurity F (Pyridine), respectively. To prove that the method is specific, all specified impurities of ceftazidime were prepared and injected individually and also spiked in the sample at specification level to check for interferences and peak purity. The difference in the assay value between the spiked and unspiked solutions was calculated.
2.5.2 Calibration Curve (Linearity of RRLC Method)
Linearity is the ability of an analytical procedure to obtain a response that is directly proportional to the concentration (amount) of an analyte in the sample. If the method is linear, the test results are directly or by well-defined mathematical transformation proportional to the concentration of analyte in samples within a given range [26]. The evaluation is performed by plotting a graph with concentrations versus the peak area of ceftazidime. Thus, a series of solutions of ceftazidime were prepared at a concentration ranging from 70 to 130 μg/mL. The calibration curves were plotted over 7 different concentrations in the range of 70–130 μg/mL. Aliquots of standard working solution (0.7, 0.8, 0.9, 1.0, 1.1, 1.2, and 1.3 mL) were transferred to a series of 10-mL volumetric flasks and diluted to the mark with the mobile phase. Under the operating chromatographic conditions mentioned above, aliquots (5 μL) from each solution were injected in duplicate. A graph was plotted with concentration on the X-axis versus area on the Y-axis, and thus, the correlation coefficient was determined.
2.5.3 Precision
The precision of an analytical method is the degree of agreement between the individual test results obtained when the method is applied to multiple sampling of a homogenous sample. The precision of an analytical method is usually expressed as the relative standard deviation of a series of measurements (Sabir et al. 2013). Precision measurements were taken in two ways: system precision and precision measurement methods. The system's precision is checked by injecting ceftazidime to ensure that the analytical system is working properly. The retention time and area response of six determinations of the standard solution were measured and calculated for relative standard deviation. The second one, method precision, is analyzed ceftazidime sample six times, and the percentage of RSD is calculated.
The system precision was assessed by measuring the peak response of ceftazidime for 10 replicate injections of the analytical standard solution with a concentration of 100 µg/mL. In the case of method precision, this was achieved by injecting six individual sample preparations of ceftazidime (API) and ceftazidime for injection (dosage) at a concentration of 100 µg/mL of ceftazidime with mobile phase diluent. The intermediate precision was carried out by estimating the response of six individual sample preparations of ceftazidime (API) and ceftazidime for injection (dosage) when injected 6 times inter-day and intra-day. The findings are recorded in terms of the relative standard deviation.
2.5.4 Accuracy
The accuracy of an analytical procedure expresses the closeness of agreement between the value which is accepted either as a conventional true value or as an accepted reference value and the value that was found [ICH Q2 (R1)].
The accuracy of the method was determined by preparing the sample solution at three different levels (70%, 100%, and 130%) of the nominal sample concentration in triplicate and analyzing it as per the proposed method. The percentage recovery of each level was calculated and found to be within the acceptance criteria, which indicates the accuracy of the method.
2.5.5 Robustness
Robustness of an analytical method is the property that indicates insensitivity to changes in known operational parameters (e.g. flow rate, column oven temperature, and mobile phase composition) on the results of the method and hence its suitability for its defined purpose [Sahu, P. K., (2018)]. First is the effect of variation of flow rate, which is evaluated by changing the flow by ± 10% from the specified; the second is the change of column temperature by ± 5 units from the specified; and the last one is the change of organic modifier in mobile phase-B by ± 10% from the specified and checking the change in system suitability parameters.The robustness studies on method precision were performed by making minor changes in chromatographic conditions such as flow rate (± 10%): 1.1 mL and 0.9 mL, the composition of organic modifier in the mobile phase (± 2%) buffer: acetonitrile (880: 120 and 920: 80), the wavelength of the detector (± 5 nm) at 265 nm and 275 nm, and the column oven temperature (± 5 °C) at 40 °C and 30 °C, respectively.
2.5.6 Ruggedness
The ruggedness of the proposed method was determined by analyzing the ceftazidime (API) and ceftazidime for injection solutions by two different analysts using two different columns on different days on different instruments.
2.5.7 System Suitability Test
The system suitability test was conducted to determine the resolution and reproducibility of the system for the analysis to be performed using 6 replicate injections of a reference solution containing 100 (μg/mL) of ceftazidime. The calculated parameters are retention time, peak area, tailing factor (peak symmetry), and potential plate, respectively.
2.5.8 Forced Degradation
Forced degradation studies were performed on ceftazidime to develop a stability-indicating method and to establish the specificity of the proposed method [ICH-Q2 (R1)]. The stress solutions are made with a ceftazidime API and dosage sample that will be utilized throughout the research. The concentration and diluent addition are the same in all of the degradations; the only change is the degradant utilized and the time spent before injecting the sample.
2.5.8.1 Hydrolytic Degradation Under Acidic Conditions
A known quantity of ceftazidime samples is treated with 5 mL of 1.0 N HCl solution. The solution is maintained at room temperature for 4 h, neutralized with 1.0 N sodium hydroxide solution, and diluted to obtain a solution having a known concentration of about 100 (μg/mL) and injected into a chromatograph.
2.5.8.2 Hydrolytic Degradation Under Alkaline Conditions
A known quantity of ceftazidime samples is treated with 5 mL of 0.1 N NaOH solution. The solution is maintained at room temperature for 4 h and diluted to obtain a solution having a known concentration of about 100 (μg/mL) and injected into a chromatograph.
2.5.8.3 Oxidative Degradation
A known quantity of ceftazidime samples is treated with 2 mL of 30% hydrogen peroxide solution. The solution is maintained at room temperature for 4 h, neutralized with 0.1 N HCl solution, and diluted to obtain a solution having a known concentration of about 100 (μg/mL) and injected into a chromatograph.
2.5.8.4 Thermal Induced Degradation
For thermal degradation, around 1000 mg of a sample that had been held at 90 °C for 48 h, it was precisely weighed to produce a solution with a known concentration of about 100 (μg/mL), which was injected into a chromatograph.
2.5.8.5 Photodegradation
For photolytic degradation, a precisely weighed quantity of ceftazidime samples was dissolved in diluent to generate a solution with a known concentration of around 100 (μg/mL) and injected after being subjected to 200 W h/m2 of UV light and 1.2 million lux hours of visible light into a photostability chamber.
2.5.8.6 Humidity Degradation
An appropriately weighed quantity of ceftazidime samples was dissolved in diluent and injected after being exposed to 97% RH at room temperature for 48 h for humidity degradation (Somnath D. Bhinge et al. 2016).
3 Results and Discussion
3.1 Method Development
A reverse phase rapid resolution liquid chromatographic method was developed to determine the content of ceftazidime from its impurities. A reverse phase rapid resolution liquid chromatographic method was developed. During initial development, formic acid for acetic pH and triethylamine for the basic pH range were used. A non-polar stationary phase octadecylsilane (C-18) column has been used for method development. Ceftazidime pentahydrate shows the strongest pKa value of about 2.82 and a logP value of about -2.5. It is polar in nature. Since there are no other alternatives, reverse phase (polar) chromatographic analysis is the only option available for determining the content of ceftazidime. Throughout the mobile phase study, volatile buffers and organic solvents such as acetonitrile have been used for the separation of all impurities from ceftazidime. It was observed that there were not any distinct effects observed with peak shapes, resolution, and also that all the impurities were eluted in a higher organic ratio with the mobile phase. After several injections, the peak shape was distorted due to a decrease in column efficiency. Non-volatile salt buffers were used to prevent peak tailing, and the method was optimized using a sodium phosphate buffer mixed with acetonitrile in isocratic mode on the Zorbax Eclipse plus C8 (100 4.6 mm, 1.8) column. The mobile phase was a mixture of buffer and acetonitrile at a ratio of 90:10 v/v. The diluent used for the entire study was 0.02 M disodium hydrogen phosphate adjusted to a pH of 7.0 with 10% v/v phosphoric acid, which gave the optimum solubility. The flow rate was set at 1.0 mL/min, the temperature of the column compartment was maintained at 35 °C, and the temperature of the sample was controlled at 5 °C. The wavelength of the detector was controlled in the entire UV region but was also set at a single wavelength of 254 nm and the injection volume was set at 5 μL. The chromatographic run time was set at 4.0 min. We achieved good peak shapes and reproducibility.
The sequence of injections starts from blank, standard solution (system suitability), sample solution, and spiked sample solutions. Further degradation studies were performed to check the specificity, which is not shown here and is fully studied in the validation part. There was no inference observed from the blank chromatogram, which is shown in Figure S1a when compared with the standard solution (system suitability) and the sample chromatogram. The standard solution chromatogram is shown in Figure S1b. Finally, the sample and the spiked chromatograms are shown in Figures S2a and Sb, and the spiked data show the specificity of the method.
3.2 Method Validation
3.2.1 Specificity
The results of specificity are shown in Table S1, and peak purity plots indicate that the ceftazidime peaks were homogeneous and no co-eluents were present. This confirms the specificity of the method. The moisture content of the ceftazidime (API) and ceftazidime for injection (dosage form) samples was found to be 0.69 and 2.38% w/w, and the values were used for all assay calculations. The specificity of the method was evaluated by injecting the diluents and the sample solution prepared as per the proposed method to check for interference, if any, at the retention time of the ceftazidime peak from the diluents. There was no peak eluting at the retention time of ceftazidime from the diluents.
The specificity of the method was further evaluated by analyzing the sample solution spiked with known related substances of ceftazidime at a 1% level in triplicate. The assay difference between the related substances spiked sample preparation and the unspiked preparation of ceftazidime (API) and ceftazidime for injection (dosage form) was found to be 0.17 and 0.87% w/w. The acceptance criteria for specificity include the assay difference between spiked and unspiked samples, which is not more than 1%. The peak purity plots, which indicate the ceftazidime peaks, are homogeneous and no co-eluents are present. The purity angle should be less than the purity threshold. This confirms the specificity of the method.
3.2.2 Stress Degradation
The results of the degradation of stress are summarized in Table S2. This indicates the effectiveness of the developed method for the separation of degradation products from ceftazidime (API) and ceftazidime for injection (dosage form) for peak injection and thus the specificity of the method.
3.2.3 Linearity
The calibration result for ceftazidime is shown in Table 1. As the area count is plotted against the concentration, it reveals that the method was linear over the specified range, as seen in Fig. 1. The value of the correlation coefficient (R2) was estimated to be 0.9994 from the calibration plot. The Y-equation for ceftazidime concentrations was found to be y = 11,136 x -134,493.

Calibration plot of ceftazidime
3.2.4 Precision
System Precision.
The results of system precision are shown in Table S3. The system precision was found to be 0.14% RSD. The acceptance criterion for system precision of % RSD is not more than 1%, which means that the precision is below the limits.
Method Precision.
The findings of method precision are shown in Table 2. The acceptance criterion for method precision is that the percentage RSD of ceftazidime (API) and ceftazidime for injection (dosage) assay from six-sample preparation is not more than 1%. The % RSD for the assay value of six different sample preparations for ceftazidime (API) and ceftazidime for injection (dosage) was observed to be 0.10 and 0.44%, indicating that the method is precise. The representative chromatograms of blank solution, standard solution, and sample solution are shown in Fig. 1.
Intermediate Precision.
The findings of intermediate precision are shown in Table 3. The acceptance criterion for method precision is that the percentage RSD of ceftazidime (API) and ceftazidime for injection (dosage) assays from six-sample preparation is not more than 1.0%. The percentage RSD for the assay values of six different sample preparations for ceftazidime (API) and ceftazidime for injection (dosage) was observed to be 0.22 and 0.62%, indicating that the method is precise.
3.2.5 Accuracy
The accuracy of the method was determined by preparing the sample solution at three different levels (70, 100, and 130% of the nominal sample concentration) in triplicate and analyzing it as per the proposed method. The percentage recovery of each level was calculated and found to be within the acceptance criteria (99.89–100.69), which indicates the accuracy of the method. The results of accuracy are shown in Table 4 (a & b).
3.2.6 Ruggedness
The ruggedness of the proposed method was calculated using the method and intermediate precision. The mean, standard deviation, and percentage RSD of the assay values are shown in Table S4. Within the acceptance criterion, the standard deviation of ruggedness was found to be 0.18 and 0.38%. The percentage RSD of ruggedness was found to be 0.18 and 0.51, which were found to be well below the acceptance criteria limit of 1%. The observation shows that this method can be used in different laboratories without any significant variation.
3.2.7 Robustness
At these various conditions, the assays of ceftazidime (API) and ceftazidime for injection (dosage) samples were performed in triplicate as per the proposed method. The system suitability criteria were evaluated, and the obtained assay values were compared with the data of method precision. The data given in Tables S5 and S6 indicate that the method is robust under these varied conditions. The robustness of the method was evaluated by deliberately varying the chromatographic conditions such as flow rate (10%), the organic modifier in the mobile phase by 2% absolute, the wavelength of detection by 5 nm, and the column oven temperature at (5 °C). At these varied conditions, the assay of ceftazidime (API) and ceftazidime for injection (dosage) samples was performed in triplicate as per the proposed method, and the robustness criteria were evaluated, and the assay values so obtained were compared with the data of the method precision. The acceptance criterion for robustness is that the overall percentage of RSD should not be more than 1%. All the robustness parameters show that the overall percentage RSD was below 1%, which means that the method is robust.
3.2.8 System Suitability
System suitability must be assessed in order to confirm the chromatographic system's suitability and reproducibility for analysis. System suitability was determined from six replicate injections of the standard solution containing 100 μg/mL of ceftazidime prior to the sample analysis (API). The acceptance conditions were less than 2% relative standard deviation (RSD) for peak areas, the USP tailing factor was less than 2.0, and the USP plate count was more than 5000 for ceftazidime peaks from the standard solution. The critical parameters tested that met the acceptance criteria are shown in Table S7.
3.2.9 Solution Stability
A study to establish the stability of the ceftazidime test solution at 5 °C bench top was conducted on an hourly basis. The area of ceftazidime in the test solution was estimated at regular intervals against the initial test solution area each time.
The cumulative percentage RSD values of ceftazidime (API) and ceftazidime for injection (dosage) were found to be 0.42 and 1.19, respectively, which indicates that the sample solution is stable for at least 24 h at 5 °C. The results of accuracy are shown in Table 5.
4 Conclusion
The method was validated as per the ICH guidelines and acceptable results were obtained. According to the findings of the extensive validation study, the approach is for linearity, specificity, precision, ruggedness, accuracy, solution stability, robustness, and system suitability, and stability-indicating. Using the validation settings, it was possible to produce findings that were acceptable in terms of the acceptance criteria. In this study, it was discovered that the approach was suitable for use in ordinary laboratory analysis and that it had a high degree of accuracy and precision. Based on the results of the extensive validation research that was done, it was determined that the method is stability-indicating and that it may be used throughout the shelf life of the drug product. Many companies investing in fast LC instruments such as the RRLC, UPLC, and UFLC strive hard to develop methods that are faster and cost-effective in terms of money, time, and resources. This method has advantages in terms of minimum analysis time, increased sample throughput and productivity, reproducibility and robustness, high efficiency and resolution, less solvent consumption, development of a fast LC method and rapid validation.
References
Abdel-Hamid ME (1998) FSQ spectrophotometric and HPLC analysis of some cephalosporins in the presence of their alkali-induced degradation products. Il Farmaco 53(2):132–138. https://doi.org/10.1016/S0014-827X(97)00021-9
Abdellatef HE, Shalaby AA, Elsaid HM, Ayad MM (2000) Colorimetric and titrimetric methods for determination of some cephalosporins in their pure and dosage forms. Sci Pharm 68(3):263–273. https://doi.org/10.3797/scipharm.aut-00-24
Andrasi M, Buglyo P, Zekany L, Gaspar A (2007) A comparative study of capillary zone electrophoresis and pH-potentiometry for determination of dissociation constants. J Pharm Biomed Anal 44(5):1040–1047. https://doi.org/10.1016/j.jpba.2007.04.024
Bergman J, Harvill L, Hawkins S, Sladky K, Cox S (2021) Determination of ceftazidime in plasma by RP-HPLC and ultraviolet detection. Biomed Chromatogr 35(7)
Bliesner DM (2006) Validating chromatographic methods. John Wiley. 88–92. ISBN: 978–0–471–74147–3
Bhinge SD, Malipatil SM (2016) Development and validation of stability indicating method for simultaneous estimation of cefixime and dicloxacillin using RP-HPLC method. J Taibah Univ Sci Elsevier
D’Cunha R, Bach T, Young BA, Li P, Nalbant D, Zhang J, Winokur P, An G (2018a) Quantification of cefepime, meropenem, piperacillin, and tazobactam in human plasma using a sensitive and robust liquid chromatography-tandem mass spectrometry method, part 1: Assay development and validation. Antimicrob Agents Chemotherapy, 62(9). Doi: https://doi.org/10.1128/AAC.00859-18
D’Cunha R, Bach T, Young BA, Li P, Nalbant D, Zhang J, Winokur P, An G (2018b) Quantification of cefepime, meropenem, piperacillin, and tazobactam in human plasma using a sensitive and robust liquid chromatography-tandem mass spectrometry method, part 1: Assay development and validation. Antimicrob Agents Chemother 62(9):1781–1787. https://doi.org/10.1128/AAC.00859-18
FDA (2015). Guidance for Industry Analytical Procedures and Methods Validation for Drugs and Biologics.
Guy RC (2014) International Conference on harmonisation. Encyclopedia of toxicology: Third Edition, 2(November 1994), 1070–1072. Doi: https://doi.org/10.1016/B978-0-12-386454-3.00861-7
Hefnawy M, El-Shabrawy Y, Belal F (1999) Spectrofluorometric determination of alpha-aminocephalosporins in biological fluids and pharmaceutical preparations. J Pharm Biomed Anal 21(4):703–707. https://doi.org/10.1016/S0731-7085(99)00208-3
Hiremath B, Mruthyunjayaswamy BHM (2008) Development and validation of spectrophotometric methods for determination of ceftazidime in pharmaceutical dosage forms. Acta Pharm 58(3):275–285. https://doi.org/10.2478/V10007-008-0017-0
ICH Q2 (R1) International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (2005) ICH Q2 (R1) Validation of Analytical Procedures: Text and Methodology
ICH. (2006). Q3A(R2) impurities in new drug substances , Q3A(R2) impurities in new drug products
Jun YB (1995) High performance liquid chromatographic analysis of the concentration of ceftazidime in ear lobe serum. CNKI Engineering 15:435–437
Kassi S, Karlovets EV, Tashkun SA, Perevalov VI, Campargue A (2017) Analysis and theoretical modeling of the 18O enriched carbon dioxide spectrum by CRDS near 1.35 μm: (I) 16O12C18O, 16O12C17O, 12C16O2 and 13C16O2. J Quant Spectrosc Radiat Transfer 187:414–425. https://doi.org/10.1016/J.JQSRT.2016.09.002
Khan A, Iqbal Z, Khan MI, Javed K, Khan A, Ahmad L, Shah Y, Nasir F (2011) Simultaneous determination of cefdinir and cefixime in human plasma by RP-HPLC/UV detection method: Method development, optimization, validation, and its application to a pharmacokinetic study. J Chromatogr B Anal Technol Biomed Life Sci 879(24):2423–2429. https://doi.org/10.1016/j.jchromb.2011.06.040
Li H (2005) RP-HPLC determination of two components and related substances in ceftazidime and sulbactam sodium for injection. Chin J Antibiotics, 30(10).
Maiti RK, Bidinger FR (1981) Martindale-The-Complete-Drug-Reference_-36th-Edition. J Chem Inf Model 53(9):1689–1699
Patil PN, Jacob S (2012) HPLC analysis of cephalosporins and study of different analytical parameters. Int J Pharm Sci 3(1):1–14
Raj K (2013) Sustainable urban habitats and urban water supply: accounting for unaccounted for water in Bangalore City, India. Current Urban Stud 01(04):156–165. https://doi.org/10.4236/cus.2013.14017
Ramya Kuber B, Sravanthi PSK (2017) Analytical method development and validation of Ceftazidime pentahydrate and Tazobactam sodium by RP-HPLC method in bulk and dosage forms. Res J Pharm, Biol Chem Sci 8(3):2447–2457
Sabir AM, Moloy M, Bhasin PS (2013) HPLC method development and validation: a review. Int Res J Pharm 4(4):39–46
Sahu PK, Ramisetti NR, Cecchi T, Swain S, Patro CS, Panda J (2018) An overview of experimental designs in HPLC method development and validation. J Pharm Biomed Anal 147:590–611
Samanidou VF, Hapeshi EA, Papadoyannis IN (2003) Rapid and sensitive high-performance liquid chromatographic determination of four cephalosporin antibiotics in pharmaceuticals and body fluids. J Chromatogr B Anal Technol Biomed Life Sci 788(1):147–158. https://doi.org/10.1016/S1570-0232(02)01040-1
Shields RK, Clancy CJ, Pasculle AW, Press EG, Haidar G, Hao B, Chen L, Kreiswirth BN, Nguyen MH (2018) Verification of ceftazidime-avibactam and ceftolozane-tazobactam susceptibility testing methods against carbapenem-resistant enterobacteriaceae and pseudomonas aeruginosa. J Clin Microbiol, 56(2) Doi: . https://doi.org/10.1128/JCM.01093-17
Tyczkowska KL, Seay SS, Stoskopf MK, Aucoin DP (1992) Determination of ceftazidime in dolphin serum by liquid chromatography with ultraviolet-visible detection and confirmation by thermospray liquid chromatography-mass spectrometry. J Chromatogr B Biomed Sci Appl 576(2):305–313. https://doi.org/10.1016/0378-4347(92)80204-4
Yan D, Li J, Zhang Z, Zhu H (2012) Determination of cephazolin, ceftazidime, and ceftriaxone distribution in nucleus pulposus. Arch Orthop Trauma Surg 132(7):969–973. https://doi.org/10.1007/s00402-012-1514-7
Ye G, Cai X, Wang B, Zhou Z, Yu X, Wang W, Zhang J, Wang Y, Dong J, Jiang Y (2008) Simultaneous determination of vancomycin and ceftazidime in cerebrospinal fluid in craniotomy patients by high-performance liquid chromatography. J Pharm Biomed Anal 48(3):860–865. https://doi.org/10.1016/j.jpba.2008.06.012
Yeh H-H, Yang Y-H, Chou Y-W, Ko J-Y, Chou C-A, Chen S-H (2005) Determination of ceftazidime in plasma and cerebrospinal fluid by micellar electrokinetic chromatography with direct sample injection. Electrophoresis 26(4–5):927–934. https://doi.org/10.1002/ELPS.200410185xxx
Funding
The author, Dr. K. Venkatachalam, is extremely thankful for the financial help for the DST-SERB project of the Department of Science and Technology, India (Ref. No EEQ/2016/000559, Date: 06.02.2017).
Author information
Authors and Affiliations
Contributions
KSB helped in Conceptualization, Methodology, Writing—Original draft preparation, Writing—Reviewing and Editing. SPS contributed to Investigation, Reviewing and Editing. GD Investigation. KV helped in Writing review, Visualization, Supervision and Project administration.
Corresponding author
Ethics declarations
Conflicts of interest
Author declare that they have no conflict of interest.
Consent of the publisher
The author further declares that the article will not be published elsewhere in the same form, in any language, without the written consent of the publisher.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Barnabas, K.S., Suvaitha, S.P., Dhinagaran, G. et al. A Novel Method Development and Validation of Ceftazidime in Bulk Drug and Pharmaceutical Dosage Forms Using RRLC Method. Iran J Sci Technol Trans Sci 46, 1187–1196 (2022). https://doi.org/10.1007/s40995-022-01333-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40995-022-01333-2