
Abstract
Eastern Himalayan foothills are known to have optimal agro-climatic conditions for the production of quality citrus fruits including oranges. Among the citrus-growing regions of eastern Himalayas, Wakro in the far eastern state of Arunachal Pradesh is known for its superior quality oranges, popular as the Wakro orange or the Arunachal orange, which has been included in the Geographical Indication Registry by the Government of India. However, during the last few years, Arunachal orange orchards have been severely affected by aphid infestation associated with rapid decline disease, causing catastrophe for the farmers as well as the state economy. Therefore, in 2015, an intensive survey of severely affected orange orchards was carried out to investigate the etiological factors of decline in citrus production. RNA samples extracted from leaf and aphid specimens collected from Wakro orchards were subjected to Citrus tristeza virus (CTV) detection through 3′-UTR specific RT-PCR. Subsequently, ORF1 and CP genetic regions were amplified and clonal-sequencing was performed. Although BLAST search showed close homology of the present sequences with other virulent genotype VT sequences, the detailed phylogenetic analysis demonstrated affinity and clustering of present sequences with VT sequences belonging to the ‘western’ lineage. This finding is considerably distinct from CTV sequences reported from citrus-growing orchards in India and other neighboring countries. Additionally, low diversity of CP gene sequences, recombination patterns, and presence of sequence segments identical to the present ones in other CTV genotypes was also revealed. Our findings suggest that the ancestral nature of present CTV sequences, corroborate well with the proposed origin of CTV in this part of the globe. We here report our finding of the western lineage of CTV virulent genotype VT, which is distinct from CTV molecular epidemiology in other parts of India, and discuss the implications of these findings.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Citrus fruits include oranges, mandarins, grapefruits, lemons, and limes that are widely grown and marketed throughout the world in large quantities (more than 120 million tonnes annually) (FAO 2017). However, the production of citrus is largely restricted by the Citrus tristeza virus (CTV), which is one of the economically most devastating viral pathogens of citrus. CTV is prevalent in almost all major citrus-growing regions of the world and can infect almost all citrus species and hybrids and has caused a loss of more than 100 million citrus trees during the last century (Lee and Bar-Joseph 2000; Bar-Joseph and Dawson 2008; Moreno et al. 2008; Harper 2013). Long-distance spread of CTV occurs via movement of vegetatively propagating material such as infected grafts (primary propagation), while local spread (secondary propagation) is facilitated by diverse aphid species in a semipersistent manner, of which the brown citrus aphid (BrCA, Toxoptera citricida) and the melon aphid (Aphis gossypii) is reported to be the most efficient vectors (Bar-Joseph et al. 1989; Gottwald et al. 1996; Albiach-Martí et al., 2000; Moreno et al. 2008; Harper 2013). CTV is not known to be spread by seeds (Bar-Joseph and Dawson 2008; Moreno et al. 2008).
CTV belongs to the genus Closterovirus, under family Closteroviridae, characterized by long flexuous rod-like virus particles (~2000 × 12 nm) composed of two types of coat protein (major and minor coat proteins), encapsulating a single-stranded, monopartite, linear, positive-sense genomic RNA of approximately 19.3 kilobases (Martelli et al. 2012; Harper et al. 2013; Biswas et al. 2017). The CTV gRNA consists of at least 12 open reading frames flanked by untranslated regions (UTRs) at both the 5′ and 3′ ends (Harper et al. 2013). The ORF1a segment of the ORF1 encodes a large polyprotein having papain-like protease (P-Pro), Methyltransferase (Mt), and Helicase (Hel) activities, while the ORF1b encodes RNA-dependent RNA polymerase (RdRp), expressed through +1 frameshift mechanism (Albiach-Marti 2012; Martelli et al. 2012; Harper 2013;). ORFs 2–11 encode at least ten distinct proteins, namely p33, p6, HSP70h, p61, CPm, CP, p18, p13, p20, and p23, expressed through subgenomic RNAs (Albiach-Marti 2012; Martelli et al. 2012; Harper 2013).
As per the available ICTV taxonomy report, three distinct strains of CTV have been identified, namely the T30 (mild isolate), T36 (intermediate severity), and VT (severe isolate) (Martelli et al. 2012). However, the availability of complete genome sequences and detailed phylogenetic analyses has led to the identification of at least eight different CTV genotypes distributed throughout the globe, namely the T36, T30, T3, VT, B165, HA16–5, T68, and the resistance breaking RB (Kleynhans and Pietersen 2016). The genotype of infecting CTV is considered an important factor that determines the magnitude of damage (Moreno et al. 2008; Harper 2013).
In India, citrus is the 3rd economically important horticulture crop and is cultivated throughout the tropical and sub-tropical geographical zones, annually producing nearly 10 million tonnes of fruits (ICAR- All India Coordinated Research Project on Fruits 2016-17, 2017; Biswas et al. 2017). Among them, mandarins (Citrus reticulata Blanco) particularly the Nagpur mandarin (Central India), Kinnow mandarin (Northwestern India), Coorg mandarin (Southern India), Darjeeling mandarin, Sikkim mandarin, or Khasi mandarin (Northeastern India, NE India), and the Arunachal Orange/ Wakro Orange/Arunachal Wakro Orange (NE India) are economically important cultivars. Among others, sub-tropical agro-climatic conditions of eastern Himalayan regions (including Darjeeling hills, Sikkim, Arunachal Pradesh) of NE India are known to be optimal for growing quality citrus fruits (Hynniewta et al. 2014; Barbhuiya et al. 2016). Similar to other citrus-growing regions, Indian citriculture is also severely threatened by CTV. CTV has killed an estimated one million citrus trees in India during the last century (Ahlawat 1997). The percentage of disease incidence varies widely (10–90%) throughout India (Ahlawat et al. 1997; Biswas 2008, 2010; Biswas et al. 2017). Some NE Indian orchards in Assam and Meghalaya are reported to have even a 100% incidence of CTV infection (Biswas et al. 2014). Several previous studies have reported prevalence of genotype VT or its recombinants, with an extensive diversity of coat protein (CP) gene in different citrus-growing regions of India (Hilf et al. 2005; Roy et al. 2005; Ghosh et al. 2009; Biswas et al. 2010; Biswas et al. 2012a, 2012b; Sharma et al. 2012).
Among the NE Indian states, in Arunachal Pradesh (AP), citrus fruits represent the largest horticulture crop, of which the Arunachal mandarin or the Arunachal Wakro orange or Wakro orange (named after the place of its origin—Wakro, 28.113° N, and 96.812° E, Lohit district) accounts for 90% of total citrus production. Wakro orange is largely organic and has certain unique features that make it superior to other mandarin oranges grown in other parts of India. High vitamin C and total soluble solids (TSS) content, medium acidity sweet-sour taste, loose and easily removable medium-thick peel, good size, better juice content, and attractive orange color of this variety are attributed to characteristic agro-climatic conditions of AP (Geographical Indications Journal 2014). Owing to its distinctive characteristics, Wakro Orange has been registered under the Geographical Indications of Goods (Registration and Protection) Act, 1999 (Geographical Indications Journal 2014) by the Government of India.
Unfortunately, during the last few years, Wakro orange has been witnessing a sharp decline in production, severely affecting the economy of orange growers as well as of the state (Arunachal Govt, 2019; The Assam Tribune, 2019; Business Standard, 2019). A recent survey conducted by the Arunachal Pradesh Horticulture Research and Development Mission (APHRDM) found this “Citrus Decline” situation highly alarming, where farmers were even forced to abandon hundreds of hectares of orchards, destroyed by the rapidly spreading decline disease (Arunachal Govt, 2019). After a survey, APHRDM officials suspected bacterial and/or viral infection, nutrient deficiency, etc. (Arunachal Govt, 2019). Despite the presence of diverse aphid species including T. aurantii and T. citricida (Geographical Indications Journal 2014) and an indication of CTV infection in several quick declining citrus orchards (ICAR-NEH, 2014), molecular epidemiology of CTV in this part of India has not been studied systematically. This dearth of information may be largely attributable to the location of Wakro Orange orchards in tough terrains in some of the remotest parts of India.
Our present study was aimed at detecting, identifying, and characterizing the CTV associated with the rapid decline of Wakro oranges in this region.
Materials and methods
Sample collection
Responding to the request from the district horticultural department, a survey of severely declining orange orchards (Citrus reticulata Blanco, local cultivars of Wakro orange) in the Wakro circle (also known as the ‘orange bowl of the State’) of the Lohit district (Arunachal Pradesh, India) was conducted in March 2015. Four severely affected orchards, showing poor health and symptoms of CTV infection (chlorosis, vein clearing, stem pitting), were surveyed. These orchards were situated at Karhe (27° 54.20′ N 96° 17.38′ E; 365 m ASL), Cibi (27° 44.77′ N 96° 20.24′ E; 345 m ASL), Kangjang (27° 48.15′ N 96° 20.78′ E; 460 m ASL), and Mawai (27° 48.15′ N 96° 20.75′ E; 330 m ASL). Additionally, a state-managed healthy orchard (no symptoms of CTV) situated at Mawai (27° 49.66′ N 96° 19.91′ E; 326 m ASL) was also surveyed. A total of 40 plant samples (8 trees randomly sampled per orchard, 4–5 tender leaves from each tree pooled in a sample tube) and 4 aphid samples (50–60 animals pooled per sample, collected from 4 declining orchards) were collected from the surveyed orchards. The location of the study site and some of the symptoms observed in the affected orchards are shown in Fig. 1.

a Map showing the extreme northeast Indian state of Arunachal Pradesh (in light green and indicated by an arrowhead). Neighboring countries are denoted by alpha-2 country codes: MM, Myanmar; BD, Bangladesh; BT, Bhutan; NP, Nepal; PK, Pakistan. b Map of Arunachal Pradesh showing the Lohit district (in orange color). Panels c to i show fruits from diseased trees, affected orchards, aphid colonies on leaves, and affected leaves with disease symptoms, observed during field survey. Blank base maps were obtained from the d-maps website (https://d-maps.com/index.php?lang=en) and annotated using Microsoft Office PowerPoint 2007 and Microsoft Windows Paint software
Immediately after collection, leaf samples along with petiole were washed thoroughly in distilled water, dabbed, chopped, and immersed in RNAlater (Sigma-Aldrich, St. Louis, USA) filled centrifuge tubes for transportation to the laboratory. In the laboratory, samples preserved in RNAlater were stored in a − 80 °C freezer (New Brunswick Scientific, New Jersey, USA) until further processing.
RNA extraction and detection of CTV RNA
In the laboratory, 20 plant samples (four from each site) and 4 aphid pools were processed independently by two researchers. Approximately 1 g of leaf sample and aphid pools (each composed of 10–15 aphids) was ground in a ball mill (Retsch GmbH, Düsseldorf, Germany). RNA was extracted from the homogenates following TRI reagent protocol, recommended by the manufacturer (Sigma-Aldrich, St. Louis, USA). Finally, RNA pellets were re-suspended in nuclease-free water (Sigma-Aldrich, St. Louis, USA) and stored at −20 °C (short term) or − 80 °C (long term). The integrity of the RNA extracts was checked on denaturing 1% agarose gels and quantity was estimated using a microliter spectrophotometer (Picodrop, Cambridgeshire, UK).
The presence of enzyme inhibitors in the RNA extracts was ruled out by performing quality control RT-PCR with 18S RNA universal primers F-566 (5′-CAGCAGCCG CGGTAATTCC-3′) and R-1200 (5′-CCCGTGTTGAGTCAAATTAAGC-3′), described previously (Hadziavdic et al. 2014). For screening of CTV RNA in the extracts, primers targeting highly conserved 3′-UTR of the CTV genome were used in a nested amplification protocol (Olmos et al. 1999). Briefly, the first round of RT-PCR was done with external primer pairs PEX1 (5′-TAAACAACACACACTCTAAGG-3′) and PEX2 (5′-CATCTGATTGAAGTGGAC-3′), followed by a second round of amplification with internal primer pairs PIN1 (5′-GGTTCACGCATACGTTAAGCCTCACTT-3′) and PIN2 (5′-TATCACTAGACAATAACCGGATGGGTA-3′) (Olmos et al. 1999). An amplicon of ~132 bp indicated the presence of CTV RNA in the corresponding extract. The specificity of the 132 bp amplicons was verified by restriction digestion with Alu I followed by direct sequencing. Alu I restriction digestion pattern of the 132-bp amplicon was predicted using a sequence alignment of 3′-UTR from 70 CTV reference genomes belonging to various genetic types, retrieved from the GenBank. All RT-PCRs were performed in a single-tube format using a commercially available kit containing a combination of enhanced AMV reverse transcriptase (eAMV™-RT) and JumpStart™ DNA polymerase (Sigma-Aldrich, St. Louis, USA). Appropriate control reactions were included during each reaction.
Amplification, cloning, and sequencing of CTV major CP and RdRp genetic regions
For amplification of the CP gene sequence, we used previously described PCR primers- HCP1 (5′-ATGGACGACGAAACAAAGAA-3′) and HCP2 (5′-TCAACGTGTGTTGAATTTCC-3′), which amplify complete CP ORF of 672 bps (Huang et al. 2004). The ORF1 encoding complete RdRp ORF (RNA dependent RNA polymerase) was amplified using two pairs of overlapping primers, namely SY17 (5′- TTAAGTTGTATTAACGAGTTTC-3′) and SY18 (5′-AGTAGCTCGAACTTTGAGAC-3′) and SY19 (5′- ATTGGGAACTCTTTGGTTAC-3′) and SY20 (5′- CGCTCGAATTTTATAAGTCC-3′), as described earlier (Vives et al. 2005).
Following PCR, amplicons were purified, ligated into pTZ57R/T vectors using a commercially available kit (Fermentas, Vilnius, Lithuania), and were used to transform XL1-Blue competent bacterial cells. For each amplicon, 5–10 positive clones were randomly selected and cultured and plasmid DNA was purified using a miniprep kit (Sigma-Aldrich, St. Louis, USA). Subsequently, the plasmid DNA was amplified using standard vector-specific universal primers (M13F and M13R). Purified amplicons were sent to a commercial sequencing facility (AgriGenome Labs Pvt. Ltd., Kochi, India) and sequence analyzed on a 3730xl DNA Analyzer (Applied Biosystems, Foster City, USA).
Molecular evolutionary analysis
After trimming vector sequences from sequence data, alignment, manual editing, and assembly (for overlapping amplicons) were done using the Bioedit version 7.2.5 (Hall, 1999). Sequence alignments were subsequently analyzed through the Elimdupes program (https://www.hiv.lanl.gov/content/sequence/elimdupesv2/elimdupes.html) to identify identical or near-identical sequences among clonal-sequences, based on inter-sequence percent nucleotide identity (PNI). From each group of identical or near-identical sequences (PNI ≥ 98), one representative sequence was selected for further analyses and submission to the GenBank. The integrity of protein-coding nucleotide sequences was verified through the ORFfinder program (www.ncbi.nlm.nih.gov/orffinder/). Sequences were then BLAST (BLASTn, Megablast) searched (Altschul et al. 1990) to recognize and retrieve homologous sequences available in the GenBank non-redundant (nr) database. Additionally, well-characterized CTV sequences enlisted in the ICTV species list (Martelli et al. 2012) and reviewed in recent literature (Harper 2013; Biswas et al. 2017) were also retrieved from GenBank for further analyses.
Sequences were aligned using the MUSCLE algorithm (Edgar 2004) followed by molecular evolutionary analyses using MEGA version 6 (Tamura et al. 2013). The best-fitting nucleotide substitution model was determined from the lowest Bayesian Information Criterion (BIC) score. Evolutionary distances were calculated using the maximum composite likelihood method and phylogenetic relatedness was determined employing the neighbor-joining (NJ) algorithm. Ambiguous nucleotides and gap data positions in the alignment were excluded from the analyses. To keep the phylogenetic tree uncluttered, among similar or nearly similar GenBank sequences, identical or near-identical sequences arising from the same site/time/study were kept to a minimum. Consistencies of tree branches were computed by 1000 bootstrap replications of the datasets.
To avert ambiguities in sequence relatedness, which arise from recombination events, we reconstructed Neighbor Network (NeighborNet or NN), using SplitsTree version 4.14.1 (Huson and Bryant 2006), with Kimura-2 parameter distance correction algorithm. Data gaps and parsimony uninformative sites were excluded from the calculations. NN reconstruction algorithms accommodate recombination events and represent sequence relatedness in more accurate topologies (Woolley et al., 2008).
To examine evidence of recombination events in present sequences, aligned datasets were analyzed through different recombination detection algorithms, namely RDP, GENECONV, BOOTSCAN, MAXCHI, CHIMAERA, SISCAN, and 3SEQ, incorporated in the RDP version 4.72 (Martin et al. 2015). Computations were done on default settings with standard Bonferroni correction. Recombination events with P < 0.01 were further verified by analysis of breakpoint plots and phylogenetic trees (unweighted pair group method with arithmetic mean; UPGMA) generated by the program. Recombination events detected by at least two different algorithms were considered true events.
Results
Detection, amplification, cloning, and sequencing of CTV gRNA from citrus plants
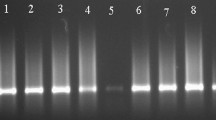
3′-UTR amplicons of the expected size were reproducibly amplifiable in all but one leaf RNA extracts from the affected orchards (Fig. 2a). However, none of the extracts from the healthy orchard yielded 3′-UTR amplicons (Fig. 2a). Additionally, 3′-UTR amplicons were also amplifiable in three of the four aphid pools, collected from declining orchards (Fig. 2a). Specificity of the 3′-UTR amplicons was further confirmed by digestion with restriction endonuclease Alu I, which yielded two fragments of 86 bp and 46 bp, as anticipated from in silico study results (Fig. 2b), followed by direct sequencing. CP and RdRp encoding genetic regions were amplified from the 3′-UTR RT-PCR positive leaf RNA extracts, followed by cloning-sequencing. Nevertheless, despite repeated attempts, we could not amplify these genetic regions in any of the aphid pool RNA extracts.

a Representative agarose gel showing 132 bp amplicon from CTV 3′-UTR RT-PCR. Lanes N, No Template Control (NTC); L1 to L4 (declining orchards), L5 (healthy orchard) RNA from leaf extracts; A1 to A3, RNA extracts from aphid pools. Marker, M1 100 bp DNA ladder. b Representative agarose gel showing Alu I digested products of 132 bp amplicons. Lanes U, uncut amplicon; L1 to L4, amplicons from leaf extracts (declining orchards); A1 to A3, amplicons from aphid pools. Marker, M2 pUC19 DNA/Msp I digested; M3 50 bp DNA ladder
After cloning-sequencing, a total of 141 CP sequences and 126 RdRp sequences were available for further analyses. On manual inspection of the CP or RdRp sequence alignments, variability at a few specific sites was noticed. Thus, to segregate sequences, CP and RdRp sequences were separately analyzed through the ElimDupes program. Finally, 3 different groups of identical CP gene sequences and 2 different groups of identical RdRp gene sequences were documented. Among these groups, 3 CP gene sequences (S21, S24, and S28) and 2 RdRp gene sequences (S5 and S9), each representing a different group of identical sequences, were selected for further analyses (phylogenetic, recombination) and submission to the GenBank. S21, S24, and S28 represented 69 (49%), 18 (12.7%), and 54 (38.3%) of the total 141 CP clonal-sequences, respectively. S5 and S9 represented 47 (37.3%) and 79 (62.7%) of the total 126 RdRp clonal-sequences, respectively.
Diversity of the sequences and results of BLAST analysis
Among the complete CP gene sequences, S21 and S24 were found to share a PNI of 99% among themselves. While sequence S28 was significantly divergent from S21 and S24, sharing a PNI score of only 93% (S28 vs. S21 and S24, P < 0.01). On the other hand, the two representative RdRp sequences S5 and S9 shared a PNI of 98% among themselves.
In BLASTn search, CP sequences S21 and S24 were found to be highly homologous (99% identity at 100% query coverage) to several sequences isolated from different parts of India as well as other countries, viz. Eastern and NE India (HM573451, GQ392063, GQ475551- GQ475553, LT576375), Western India (GQ475563, GQ475564), Southern India (EU869297), Northern India (JN974902, FJ001829), Uruguay (KU900351), Argentina (EU579381), South Africa (KU883267), Reunion Islands (AY660010), and Malaysia (HQ012413). Conversely, sequence S28 was found to be highly homologous (99% identity at 100% query coverage) to sequences recently isolated from different parts of NE India (GQ475549, KR185332; KR185331, KR185329, KR105771; KR259640, KR259641, KR080487, LN997804) as well as from New Zealand (AY896563).
On the other hand, in BLASTn analysis, RdRp sequences- S5 and S9 were found to be highly similar (97–98% identity at 100% query coverage) to CTV genotype VT sequences from the USA (KC517493, EU937519, KC517494; KC517492; KU361339), Greece (KC262793), China (JQ911664, KU720382, JQ061137), Spain (DQ151548), Italy (KJ790175, KC748392), Japan (AB046398), and Israel (U56902). Interestingly, present sequences were reasonably divergent from two distinct complete genomes CTV sequences isolated from India, namely the Kpg3 from Eastern India (HM573451, 94% identity at 100% query coverage) and the B-165 from southern India (EU076703; 86% identity at 100% query coverage).
Phylogenetic affinities of the CP and RdRp genetic regions
In the CP gene phylogenetic tree, sequences from various parts of India and other countries are segregated into eight distinct clades previously classified as genogroups I through VIII (Tarafdar et al. 2013). Clustering of present sequences S21 and S24 corroborated with BLASTn results, demonstrating their affinity with group VII of CP gene sequences (Fig. 3). Conversely, sequence S28 along with two other sequences (AY896563 from New Zealand and KR259640 from Sikkim, NE India) acquired an intermediate position between group I sequence clade (which comprise sequences mostly isolated from NE India) and group VIII sequence (KC590504 from Tirupati, south India) (Fig. 3). This intermediate position of sequence S28 was more prominent in NN analysis (Fig. 4). Although the clustering pattern of the CP gene sequence in NN analysis was largely analogous to NJ phylogenetic tree, extensive networking within and among various CP gene groups was evident in NN analysis (Fig. 4).

Phylogenetic tree reconstructed for CP (coat protein) gene sequences generated in the present study along with 96 other GenBank sequences, submitted by other researchers. Sequences from the present study (S21, S24, and S28) are marked by black solid squares. CP clades were classified as groups I to VIII following previous literature (Tarafdar et al. 2013). Evolutionary distances were calculated using the maximum composite likelihood (MCL) method and phylogenetic relatedness was recreated using the neighbor-joining (NJ) method. The percentage of bootstrap support for each cluster (>75%) is shown at nodes

Neighbor Net of CP gene sequences reconstructed using the dataset previously used for reconstruction of NJ tree in Fig. 3. The position of the three CP gene sequences generated in the present study (S21, S24, and S28) is indicated by red arrowheads
On the other hand, RdRp sequences corresponding to S5 and S9 phylogenetically clustered with well-characterized CTV genotype VT ‘western’ lineage sequences from different countries, while maintaining considerable distance from the unique VT sequence isolate (Kpg3; HM573451) from another part of NE India (Fig. 5). These results largely substantiated BLASTn results. Notably, NN reconstructed with RdRp sequences were substantially simpler, having scarcer evidence of inter and intragenotype networks (Fig. 6), sharply contrasting the CP gene NN (Fig. 4).

Phylogenetic tree reconstructed from 1860 bases of 5′ half of CTV genome (corresponding to partial ORF1a and ORF1b coding regions) from two sequences generated in this study (S5 and S9) and 52 others well-characterized complete genomes retrieved from the GenBank. Sequences from the present study are marked by black solid rounds. Evolutionary distances were calculated using the maximum composite likelihood (MCL) method and phylogenetic relatedness was recreated using the neighbor-joining (NJ) method. The percentage of bootstrap support for each cluster (>75%) is shown at the respective node. Genotypes and lineages are mentioned on the right-hand side

Neighbor Net reconstructed from 1860 bases of 5′ half of CTV genome (corresponding to partial ORF1a and ORF1b coding regions) using the dataset previously used for reconstruction of NJ tree in Fig. 5. The position of the two-sequence generated in the present study (S5 and S9) is indicated by red arrowheads
Analyses of recombination
Among the two RdRp sequences generated in this study, sequence S5 had evidence of recombination (detected by BOOTSCAN, MAXCHI, SISCAN, and 3SEQ with a probability of 1.346 × 10−02, 3.033 × 10−06, 2.166 × 10−06, and 1.510 × 10−02, respectively) between the present sequence S9 (minor parent) and a sequence resembling an isolate from China (major parent JQ061137, belonging to the ‘Asian’ lineage of genotype VT) (Fig. 7a). Interestingly, evidence of sequence S9 as the major parent was detected in all the four reference sequences belonging to genotype T68 (EU076703, India; FJ525436, New Zealand; JQ965169, USA; and KC333868, South Africa), while as a minor parent in both the reference sequences belonging to genotype T3 (EU857538, New Zealand and KC525952, USA) included in the present analysis.

Recombination patterns were observed in the sequences S5 (a) and S28 (b), generated in the present study. Each figure is followed by a BOOTSCAN plot showing the breakpoints corresponding to the recombination event. Plots represent % of permuted trees on the y axis
On the other hand, on analyzing the CP gene sequence dataset, the RDP program could not detect any evidence of recombination within sequences S21 and S24. However, isolate S28 was found to have a sequence fragment that had evidence of recombination (detected by SiScan and 3Seq with a probability of 1.243 × 10−10 and 1.336 × 10−02, respectively) between a sequence isolated from Reunion Island (major parent, AY660010, belonging to group VII) and a sequence (isolate AG28) from Tinsukia, NE India (minor parent, KC590498, belonging to group I) (Fig. 7b). Nevertheless, considering the remote origin of sequence AY660010, and its intimate phylogenetic relatedness with sequence S21 (Fig. 3), it is logical to interpret that an immediate ancestor (probably originating in NE India) of these two sequences might have been the major parent.
Discussion
The NE Indian biogeographic zone is an important part of the Indo-Burma biodiversity hotspot region, where the great Himalayan Mountains meet Peninsular India. NE India comes under the Hindustan (Indian) center of origin of a cultivated plant, proposed by Nikolai Vavilov, and is considered to be the native place for numerous tropical and sub-tropical fruits including citrus fruits (Martin et al. 2009; Hummer and Hancock 2015). It is also proposed that CTV has coevolved with its citrus hosts (Bar-Joseph et al. 1989; Martin et al. 2009). CTV is now ubiquitous and is the most devastating pathogen of citrus (Harper et al. 2013). Similar to other citrus-growing regions of the world, CTV is highly prevalent in sub-Himalayan regions of NE India, where it has severely affected and wiped out several orchards resulting in massive economic losses (Biswas et al. 2016; Biswas et al. 2017). Nevertheless, our understanding of CTV epidemiology in NE India is mostly restricted to accessible orchards in the States of Assam, Meghalaya, Nagaland, and Sikkim. To the best of our knowledge, there is no reliable literature on the molecular epidemiology of CTV in the state of Arunachal Pradesh and this study is the first to report CTV molecular epidemiology in one of the oldest and remotest citrus-growing regions of India.
Previous studies have demonstrated an evolutionary asymmetry between the 5′ and 3′ halves of the CTV genome, where the 5′ (including the ORF1) is highly conserved and encodes genotype-specific phylogenetic signals while the 3′ (including the CP) shows relatively high genetic diversity (Albiach-Martí et al., 2000; Hilf and Garnsey, 2000; Hilf et al. 2005; Roy and Brlansky 2010; Harper 2013). Based on these observations, a ~ 400 nucleotide region of ORF1a (corresponding to partial P-Pro domain) has been widely used for determining CTV genotypes (Roy et al. 2005; Biswas et al. 2017). In the present study, we amplified and analyzed a comparatively longer genetic fragment (1860 nts corresponding to partial ORF1a and ORF1b coding regions). We observed that phylogenetic clustering of GenBank reference sequences obtained based on this 1860 nt genetic region was highly concordant with phylogeny derived from complete genome sequences in a previous study (Harper, 2013). Nevertheless, analyses of this genetic region in the present isolates demonstrated the presence of virulent genotype VT, substantiating with previous studies from the Indian subcontinent (Hilf et al. 2005; Roy et al. 2005; Biswas et al. 2010; Sharma et al. 2012; Biswas et al. 2012a; Biswas et al. 2012b; Singh et al. 2013; Tarafdar et al. 2013; Palchoudhury et al. 2017). Interestingly, present isolates clustered with the ‘western’ lineage clade of VT sequences in the phylogeny. Since the Wakro region geo-physically connects the NE Indian part of the Indian subcontinent with China and other parts of Southeast Asia, phylogenetic affiliation of present sequences to either ‘Indian’ or ‘Asian’ lineage was therefore highly anticipated. The Western and the Asian lineages signify evolution and global dispersal of two distinct variants of genotype VT in Western and Asian countries, respectively (Harper 2013), the Indian lineage has been proposed to accommodate the decline inducing recombinant strain ‘Kpg3’ (GenBank accession HM573451), which do not cluster within either of the two lineages in phylogenies (Biswas et al. 2012a; Harper 2013).
Among extant CTV genotypes distributed globally, the highest genetic diversity and widest geographical distribution of VT support its ancient origin. A previous study based on mixed effects model of evolution (MEME) analysis of ORF1a suggested that the western lineage of VT is the most ancestral type known so far, from which all other lineages, including the Asian lineage, have evolved through ‘episodic diversification events’ (Harper et al. 2013). Considering this geographical region as the proposed center of origin of CTV, Western lineage CTV sequences found in the present study appear to represent the original ‘ancestral’ CTV genotype. In such a scenario, the natural spatio-temporal dispersal of this original virus to Southeast Asia and peninsular India might have resulted in the evolution of Asian and Indian lineages, respectively. In contrast, phylogenetic clustering of present sequences with genotype VT sequences from western countries amply supports its relatively recent introduction to western countries. Earlier, it has been proposed that CTV evolved and diverged into major clades in this region followed by their dispersal across Southeast Asia, the Indian subcontinent, and islands of the Indian Ocean long ago, while their dissemination to Europe, Africa, and the Americas took place only recently (~400–500 years ago) (Albiach-Martí et al., 2000; Moreno et al. 2008; Silva et al. 2012).
Additionally, owing to the stability of CTV genomes (evolutionary rates being the slowest among RNA viruses), the similarity of genome sequences in distant geographical regions have been suggested to indicate the origin and dispersal of CTV (Albiach-Martí et al., 2000; Moreno et al. 2008; Silva et al. 2012). Interestingly, one of the ORF1 sequences (S5) in this study was found to be recombinant, involving a major parent similar to the Asian lineage genotype VT sequence (JQ061137, China) and present isolate S9 sequence as a minor parent. This finding of an Asian lineage sequence as a major recombinant partner may either be suggestive of remnants of the ‘Asian’ lineage evolved in this geographical region or might be a relatively recent introduction, which cannot be concluded from the present results. Notably, in recombination analysis, the present S9 like sequence was found to be involved in all the genotype T68 and genotype T3 reference sequences included in this study.
On the other hand, previous studies on CP gene sequences have revealed the circulation of diverse phylogroups in different citrus-growing regions, which do not necessarily correlate with the genotype determined by analysis of ORF1 sequences (Rubio et al. 2001; Martin et al. 2009; Biswas et al. 2012b). Such inconsistency between different genetic regions of the CTV genome has been attributed to frequent natural homologous and non-homologous recombination events or gene flow among distinct genetic types occurring particularly in the 3′ half of the CTV genome (Rubio et al. 2001; Martin et al. 2009; Sharma et al. 2012; Biswas et al. 2012a, 2012b; Palchoudhury et al. 2015). Our findings of NN analyses of ORF1 and CP genetic regions showing a sharply discrete pattern of reticulate evolution strongly support these previous observations.
Nevertheless, analyzing CP gene variability, previous studies have reported eight globally circulating CP genetic variants (genogroups I to VIII), of which seven (I, III, IV, V, VI, VII, and VIII) are present throughout the Indian subcontinent while five (I, III, V, VI, and VII) are present in different parts of NE India (Tarafdar et al. 2013; Palchoudhury et al. 2015; Biswas et al. 2017). In contrast, CP gene sequence diversity observed in present samples was limited to only two genogroups (I and VII), which were related to sequences from other parts of sub-Himalayan NE India (Darjeeling, Kalimpong, and Sikkim). We also detected a recombinant CP sequence involving a group VII sequence (resembling present sequence S21) accepting a large fragment from a group I sequence (similar to isolate AG28, GenBank KC590498) reported from Tinsukia, Assam (Biswas et al. 2016; Biswas et al. 2017). Similar recombination events, particularly involving isolate AG28 like CP and ORF1a sequences, have recently been reported from other parts of NE India too (Gautom et al. 2020). However, we could not identify any genogroup VI CP gene sequence, which represents a clade of phylogenetically related sequences exclusively from different parts of NE India. Taken together, limited genetic diversity, evidence of recombination with sequences only from nearby geographical regions, and remnants of the present sequences in other genotypes indicate the antiquity of present CTV sequences from Wakro and provides credible evidence to support prevailing hypotheses on the origin of CTV.
Apart from the CTV genotype, transmitting vector aphid species can also significantly influence the spatio-temporal dynamics and magnitude of infection. Although BrCA (T. citricida) is the most frequent and efficient vectors associated with CTV transmission in India (Ahlawat 1997; Biswas 2008), other aphid vectors (T. aurantii, A. gossypii, Myzus persicae, Dactynotus jaceae, A. craccivora) have also been recognized to transmit CTV (Verma et al. 1965). During the present survey, we could detect T. aurantii in high densities in all the declining orchards (situated at an altitude of 330–426 m). These findings are different from a previous study from Darjeeling, where T. aurantii was dominant in the citrus orchards at high altitudes (>500 m), whereas T. citricida was predominant in the orchards of lower altitude (Ghosh et al. 2015). T. aurantii has earlier been shown to be highly adaptable to wider host range and higher altitudes, particularly in the sub-tropical hills of NE India (Roychoudhuri, 1980; Agarwala and Bhattacharya 1994). Remarkably, we could reproducibly amplify CTV specific 132 bp 3′-UTR genetic region in aphid pools, we could not amplify other genetic fragments (CP or the ORF1) from the aphid pools to implicate their role in transmission.
In conclusion, the present study reveals the molecular epidemiology of CTV from a place that comes under a geographical region, believed to be the origin of the citrus fruits. Due to its remoteness, this region has remained largely unreached and thus maintains pristine conditions in terms of biodiversity. Our results suggesting the circulation of an unanticipated genetic type of CTV with less interaction with CTV sequences only from neighboring geographical areas indicate the ancestral nature of present CTV sequences. However, we acknowledge that a complete genome sequence would be required for a better understanding of the evolution and diversification of CTV.
Data availability statement
Sequence data generated during the current study are available as nucleotide sequences in the NCBI GenBank under accession numbers - KY882459 through KY882476.
References
Agarwala BK, Bhattacharya S (1994) Anholocycly in tropical aphids: population trends and influence of temperature on development reproduction and survival of three aphid species (Homoptera: Aphidoidea). Phytophaga 6:17–27
Ahlawat YS (1997) Viruses greening bacterium and viroids associated with citrus (Citrus species) decline in India. Indian Journal of Agricultural Science 67:51–57
Albiach-Marti MR (2012) Molecular virology and pathogenicity of Citrus tristeza virus. In: Garcia M (Ed.) Viral genomes -molecular structure diversity gene expression mechanisms and host-virus interactions. Available online at https://www.intechopen.com/books/current-issues-in-molecular-virology-viral-genetics-and-biotechnological-applications/the-complex-genetics-of-citrus-tristeza-virus. Accessed on September 30, 2019
Albiach-Martí MR, Mawassi M, Gowda S, Satyanarayana T, Hilf ME, Shanker S, Almira EC, Vives MC, López C, Guerri J, Flores R, Moreno P, Garnsey SM, Dawson WO (2000) Sequences of citrus tristeza virus separated in time and space are essentially identical. Journal of Virology 74:6856–6865
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. Journal of Molecular Biology 215:403–410
Arunachal Govt portal; Citrus decline is alarming. Published on State Portal of Arunachal Pradesh Available online at http://arunachalpradesh.nic.in/csp_ap_portal/pdf/Documents/citrus-decline-alarming-arunachal.pdf Accessed on September 30, 2019
Barbhuiya AR, Khan ML, Dayanandan S (2016) Genetic structure and diversity of natural and domesticated populations of Citrus medica L in the Eastern Himalayan region of Northeast India. Ecology and Evolution 6:3898–3911
Bar-Joseph M, Dawson WO (2008) Citrus tristeza virus. In: Mahy BWJ, Van Regenmortel MHV (eds) Encyclopaedia virology, 3rd edn. Academic Press, Massachusetts, pp 520–525
Bar-Joseph M, Marcus R, Lee RF (1989) The continuous challenge of Citrus tristeza virus control. Annual Review of Phytopathology 27:291–316
Biswas KK (2008) Molecular diagnosis of Citrus tristeza virus in mandarin (Citrus reticulata) orchards of hills of West Bengal. Indian Journal of Virology 19:26–31
Biswas KK (2010) Molecular characterization of Citrus tristeza virus isolates from the Northeastern Himalayan region of India. Archives of Virology 155:959–963
Biswas KK, Tarafdar A, Diwedi S, Lee RF (2012a) Distribution genetic diversity and recombination analysis of Citrus tristeza virus of India. Virus Genes 45:139–148
Biswas KK, Tarafdar A, Sharma SK (2012b) Complete genome of mandarin decline Citrus tristeza virus of Northeastern Himalayan hill region of India: comparative analyses determine recombinant. Archives of Virology 157:579–583
Biswas KK, Tarafdar A, Sharma SK, Singh JK, Dwivedi S, Biswas K, Jayakumar BK (2014) Current status of Citrus tristeza virus incidence and its spatial distribution in citrus growing geographical zones of India. Indian Journal of Agricultural Science 84:8–13
Biswas KK, Pal Choudhuri S, Godara S (2016) Decline of mandarin orange caused by Citrus tristeza virus in Northeast India: conventional and biotechnological management approaches. Journal of Agricultural Engineering and Food Technology 3:236–241
Biswas KK, Palchoudhury S, Ghosh DK (2017) Closterovirus in India: distribution genomics and genetic diversity of Citrus Tristeza. In: Mandal B, Rao GP, Baranwal VK, Jain RK (eds) A century of plant virology in India. Springer Nature, Switzerland, pp 201–216
Business Standard; Orange growth hit in Arunachal Pradesh, January 11 2015 Available online at https://www.business-standard.com/article/pti-stories/orange-growth-hit-in-arunachal-pradesh-115011100455_1.html Accessed on September 30, 2019
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32:1792–1797
FAO (2017) Citrus fruit fresh and processed statistical bulletin 2016. Available at http://www.fao.org/economic/est/est-commodities/citrus-fruit/en/ Accessed on September 27, 2019
Gautom T, Hazarika DJ, Goswami G, Barooah M, Kalita MC, Boro RC (2020) Exploring the genetic diversity and recombination analysis of Citrus tristeza virus isolates prevalent in Northeast India. Indian Phytopathology 73:145–153
Geographical Indications Journal (2014) (volume 61) Geographical Indications Registry, Government of India November 21 2014. Available online at http://www.ipindia.nic.in/journal-gi.htm Accessed on September 30, 2019
Ghosh DK, Aghave B, Roy A, Ahlawat YS (2009) Molecular cloning sequencing and phylogenetic analysis of coat protein gene of a biologically distinct CTV isolate occurring in Central India. Journal of Plant Biochemistry and Biotechnology 18:105–108
Ghosh A, Ghosh A, Lepcha R, Majumdar K, Baranwal BK (2015) Identification and distribution of aphid vectors spreading Citrus tristeza virus in Darjeeling hills and Dooars of India. Journal of Asia Pacific Entomology 18:601–605
Gottwald TR, Garnsey SM, Cambra M, Moreno P, Irey M, Borbon J (1996) Differential Effects of Toxoptera citricida vs Aphis gossypii on the temporal increase and Spatial Patterns of Spread of Citrus Tristeza. Available at https://escholarship.org/uc/item/92v4x1wd. Accessed on September 27, 2019
Hadziavdic K, Lekang K, Lanzen A, Jonassen I, Thompson EM, Troedsson C (2014) Characterization of the 18S rRNA gene for designing universal eukaryote specific primers. PLoS One 9:e87624
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symposium Series 41:95–98
Harper SJ (2013) Citrus tristeza virus: evolution of complex and varied genotypic groups. Frontiers in Microbiology 4:93
Hilf ME, Garnsey SM (2000) Characterization and classification of Citrus tristeza virus isolates by amplification of multiple molecular markers In: Proceedings of 14th conference of the International Organization of Citrus Virologists (IOCV) 18–27
Hilf ME, Mavrodieva VA, Garnsey SM (2005) Genetic marker analysis of a global collection of isolates of Citrus tristeza virus: characterization and distribution of CTV genotypes and association with symptoms. Phytopathology 95:909–917
Huang Z, Rundell PA, Guan X, Powell CA (2004) Detection and isolate differentiation of Citrus tristeza virus in infected trees based on reverse transcription-polymerase chain reaction. Plant Disease 88:625–629
Hummer KE, Hancock JF (2015) Vavilovian centers of plant diversity: implications and impacts. Hortscience 50:780–783
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution 23:254–267
Hynniewta M, Malik SK, Rao SR (2014) Genetic diversity and phylogenetic analysis of Citrus (L) from north-east India as revealed by meiosis and molecular analysis of internal transcribed spacer region of rDNA. Meta Gene 2:237–251
ICAR- All India Coordinated Research Project on Fruits 2016-17 (2017). Available online at https://icar.org.in/dare-icar-annual-reports. Accessed on September 27, 2019
ICAR-NEH Annual Report 2014–15. ICAR Research Complex for NEH Region Umroi Road Umiam Meghalaya www.icarneh.ernet.in
Kleynhans J, Pietersen G (2016) Comparison of multiple viral population characterization methods on a candidate cross-protection Citrus tristeza virus (CTV) source. Journal of Virological Methods 237:92–100
Lee RF, Bar-Joseph M (2000) Tristeza. In: Timmer LW, Garnsey SM, Graham JH (eds) Compendium of citrus diseases, 2nd edn. APS Press, Minnesota, pp 61–63
Martelli GP, Agranovsky AA, Bar-Joseph M, Boscia D, Candresse T, Coutts RHA, Dolja VV, Hu JS, Jelkmann W, Karasev AV, Martin RR, Minafra A, Namba S, Vetten HJ (2012) Family Closteroviridae. In: Andrew MQK, Adams MJ, Carstens EB, Leftkowitc EJ (eds) Ninth Report of the International Committee on Taxonomy of Viruses. Academic Press, Massachusetts, pp 987–1001
Martin S, Sambade A, Rubio L, Vives MC, Moya P, Guerri P, Elena SF, Moreno P (2009) Contribution of recombination and selection to molecular evolution of Citrus tristeza virus. Journal of General Virology 90:1527–1538
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evolution 1:vev003
Moreno P, Ambrós S, Albiach-Martí MR, Guerri J, Peña L (2008) Citrus tristeza virus: a pathogen that changed the course of the citrus industry. Molecular Plant Pathology 9:251–268
Olmos A, Cambra M, Esteban O, Gorris MT, Terrada E (1999) New device and method for capture reverse transcription and nested PCR in a single closed-tube. Nucleic Acids Research 27:1564–1565
Palchoudhury S, Sharma SK, Biswas MK, Biswas KK (2015) Diversified Citrus tristeza virus causing decline disease in Khasi mandarin in Manipur State of Northeast India. Journal of Mycology and Plant Pathology 45:317–323
Palchoudhury S, Ghimiray S, Biswas MK, Biswas KK (2017) Citrus tristeza virus variants and their distribution in mandarin orchards in Northeastern Himalayan Hill region of India. International Journal of Current Microbiology and Applied Science 6:1680–1690
Roy A, Brlansky RH (2010) Genome analysis of an orange stem pitting citrus tristeza virus isolate reveals a novel recombinant genotype. Virus Research 151:118–130
Roy A, Manjunath KL, Brlansky RH (2005) Assessment of sequence diversity in the 5′-terminal regions of Citrus tristeza virus from India. Virus Research 113:132–142
Roychoudhuri DN (1980) Aphids of North-east India and Bhutan. Zoological Society of India. pp 76–78
Rubio L, Ayllón MA, Kong P, Fernández A, Polek M, Guerri J, Moreno P, Falk BW (2001) Genetic variation of Citrus tristeza virus isolates from California and Spain: evidence for mixed infections and recombination. Journal of Virology 75:8054–8062
Sharma SK, Tarafdar A, Khatun D, Kumari S, Biswas KK (2012) Intra farm diversity and evidence of genetic recombination of Citrus tristeza virus isolates in Delhi region of India. Journal of Plant Biochemistry and Biotechnology 21:38–43
Silva G, Marques N, Nolasco G (2012) The evolutionary rate of citrus tristeza virus ranks among the rates of the slowest RNA viruses. Journal of General Virology 93:419–429
Singh JK, Tarafdar A, Sharma SK, Biswas KK (2013) Evidence of recombinant Citrus tristeza virus isolate occurring in acid lime cv pant lemon orchard in Uttarakhand Terai Region of Northern Himalaya in India. Indian Journal of Virology 24:35–41
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA 6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution 30:2725–2729
Tarafdar A, Godara S, Dwivedi S, Jayakumar BK, Biswas KK (2013) Characterization of Citrus tristeza virus and determination of genetic variability in North-east and South India. Indian Phytopathology 66:302–307
The Assam Tribune; Citrus decline hits orange bowl of Arunachal, Guwahati Monday January 19 2015 Available online at https://assamtribune.com/scripts/mdetails.asp/?id=jan1915/oth052 Accessed on September 30, 2019
Verma PM, Rao DG, Capoor SP (1965) Transmission of tristeza virus by Aphis craccivora Koch and Dactynotus jaceae (L). Indian Journal of Entomology 27:67–71
Vives MC, Rubio L, Sambade A, Mirkov TE, Moreno P, Guerri J (2005) Evidence of multiple recombination events between two RNA sequence variants within a Citrus tristeza virus isolate. Virology 331:232–237
Woolley SM, Posada D, Crandall KA (2008) A comparison of phylogenetic network methods using computer simulation. PLoS One 3:e1913
Acknowledgements
We acknowledge the excellent contribution of Late Mr. Raghvendra Budhauliya in laboratory experiments. We gratefully thank Dr. BM Mishra, Ex-Deputy Commissioner (Lohit District), Government of Arunachal Pradesh, officials of the Horticulture department (AP), and the orange growers of Wakro for their support and cooperation during the field survey. We acknowledge Dr. YangChen D Bhutia for her help with language editing.
Funding
This research was performed by institutional intramural grants from the Defence Research and Development Organization (DRDO), Ministry of Defence, Government of India.
Author information
Authors and Affiliations
Contributions
This study was conceptualized by Sibnarayan Datta and Vijay Veer. Field surveys, specimen collection, and identification of specimens were performed by Sibnarayan Datta, Reji Gopalakrishnan, Vanlalhmuaka, Mukesh K Meghvansi, Mohan G Vairale, and Safior Rahman. Laboratory experiments were performed by Sibnarayan Datta and Bidisha Das. Data was formally analyzed, prepared for visualizations, and curated by Sibnarayan Datta. Vijay Veer and Sanjai K Dwivedi worked towards fund acquisition, project administration, and overall supervision of the study. The original draft was written by Sibnarayan Datta and all the co-authors reviewed, edited, and approved the manuscript before submission. Vijay Veer and Sanjai K Dwivedi critically reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Datta, S., Das, B., Gopalakrishnan, R. et al. Detection of ‘ancestral’ western lineage of Citrus tristeza virus virulent genotype in declining Arunachal Wakro orange. Trop. plant pathol. 46, 493–505 (2021). https://doi.org/10.1007/s40858-021-00438-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40858-021-00438-0