
Abstract
Duchenne muscular dystrophy is one of the most common inherited genetic diseases and is caused by mutations to the DMD gene that encodes the dystrophin protein. Recent advances in genome editing and gene therapy offer hope for the development of potential therapeutics. Truncated versions of the DMD gene can be delivered to the affected tissues with viral vectors and show promising results in a variety of animal models. Genome editing with the CRISPR/Cas9 system has recently been used to restore dystrophin expression by deleting one or more exons of the DMD gene in patient cells and in a mouse model that led to functional improvement of muscle strength. Exon skipping with oligonucleotides has been successful in several animal models and evaluated in multiple clinical trials. Next-generation oligonucleotide formulations offer significant promise to build on these results. All these approaches to restoring dystrophin expression are encouraging, but many hurdles remain. This review summarizes the current state of these technologies and summarizes considerations for their future development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Duchenne muscular dystrophy
Duchenne muscular dystrophy (DMD) is a progressive wasting disease of skeletal and cardiac muscle (Falzarano et al. 2015). This X-linked disease affects about 1/3500–1/5000 live male U.S. births, making it one of the most common fatal genetic diseases (Parker et al. 2005; Guiraud et al. 2015). Diagnosis usually occurs between 2 and 5 years of age. DMD patients typically lose ambulation in their teenage years and premature fatality often occurs in the third decade of life due to respiratory and cardiac complications (D’Orsogna et al. 1988; Dittrich et al. 2015). DMD occurs when there is a mutation in the DMD gene leading to a complete lack of the essential musculoskeletal protein dystrophin (Hoffman et al. 1987). The dystrophin protein normally links the actin fibers of the cytoskeleton and intracellular contractile apparatus to the extracellular matrix. Mutations in the DMD gene that cause DMD disrupt the translational reading frame or create a premature stop codon. This results in disruption of the connection between the cytoskeleton and extracellular matrix. This bond is crucial to maintain function during contractile stress in skeletal and cardiac muscle; weakened bonds lead to damage that builds up over time and results in overall loss of muscle function (Lapidos et al. 2004). There are a variety of DMD mutations that disrupt the translational reading frame: 6–10 % duplications, 30–35 % point mutations, and 65 % deletions of gene segments (Grimm et al. 1994; Nallamilli et al. 2014). Symptoms are managed primarily through corticosteroids, physical therapy, and consideration of cardiac complications (Wagner et al. 2007; Baxter 2010; Goemans et al. 2013b; Pane et al. 2014; Falzarano et al. 2015; van Westering et al. 2015). Anti-inflammatory steroids slow the disease progression (Ricotti et al. 2013), but they do not ultimately address the cause of DMD. Currently, there is no effective curative treatment for DMD, and thus there is a clear need for a therapy that addresses both cardiac and skeletal muscle deterioration (Ramos and Chamberlain 2015).
The well-defined genetic cause of DMD makes it a possible candidate for gene therapy. The DMD gene is approximately 2.4 Mb in size and is composed of 79 exons encoding a 14 kb cDNA. It is the largest human gene, which likely contributes to the high rate of mutation. Nearly, one-third of all DMD cases arise from spontaneous mutations in the germline (Grimm et al. 1994; Crow 2000). Some inherited dystrophin mutations maintain the reading frame and result in production of an internally truncated, but partially functional, dystrophin protein (Hoffman et al. 1989; England et al. 1990; Helderman-van den Enden et al. 2010). As a result of the significantly less severe phenotype and later onset, these mutations are classified as Becker muscular dystrophy (BMD), rather than DMD (Fig. 1) (Hoffman et al. 1989; Romero et al. 2004; Helderman-van den Enden et al. 2010). Because of the challenges of delivering the large full-length dystrophin cDNA, many therapeutic approaches have focused on shifting the DMD phenotype to be BMD-like by restoring the expression of a gene harboring internal deletions. This can be achieved by editing the DMD gene through genome editing (Maeder and Gersbach 2016) or skipping exons in the pre-mRNA (Kole and Krieg 2015). Alternatively, exogenous dystrophin cDNA transgenes can be delivered, typically by viral vectors (Hollinger and Chamberlain 2015).

Example mutations in the DMD gene that cause BMD or DMD phenotypes and relate exon removal strategies to restore dystrophin expression
Recent technology advances
Many gene therapies are under development for diseases with clear genetic causes, and rapidly developing technologies are creating new approaches to treat these diseases. Classical gene therapy has traditionally focused on delivering exogenous DNA to substitute for the lost endogenous gene expression. This has been successful in many cases, with several programs showing efficacy and safety in clinical trials (Naldini 2015). In contrast to conventional gene therapy, recent advances in genome editing have enabled the correction of the genetic mutations that are the fundamental cause of the disease (Cox et al. 2015; Maeder and Gersbach 2016). These genome editing platforms include zinc finger nucleases (Urnov et al. 2010; Gersbach et al. 2014), TALENs (Gaj et al. 2013; Joung and Sander 2013), meganucleases (Arnould et al. 2011; Silva et al. 2011), and CRISPR/Cas9 (Hsu et al. 2014). These systems facilitate new opportunities for gene therapy by designing enzymes to modify nearly any site in the human genome. Zinc finger nucleases and TALENs consist of programmable DNA-binding proteins fused to the catalytic domain of the FokI endonuclease to enable targeted cleaving of DNA. Cas9 is naturally a nuclease and can also be used for targeted DNA cleavage when directed by a guide RNA (gRNA). The gRNA contains a constant region, to which Cas9 binds, and a variable sequence that is designed to target a complementary genomic sequence. When a DNA break is created by any of these platforms, naturally occurring DNA repair mechanisms are triggered (Fig. 2). Non-homologous end joining is one possible mechanism of repair in which the broken ends are religated. However, this is an error-prone repair process that can result in small insertions or deletions (indels) where the double-strand break was made. These indels can be used to shift or disrupt the reading frame in the targeted gene or disrupt specific sites involved in exon splicing during mRNA processing. Two nucleases can also be introduced to delete the sequence between the two double-strand breaks. Alternatively, another DNA repair mechanism, homology-directed repair, can be used to introduce specific changes at the targeted genomic site by delivering a DNA donor repair template carrying the intended sequence changes along with the nuclease.

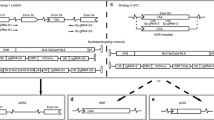
Genome editing strategies to create targeted sequence changes in genomic DNA
Each genome editing platform has its own nuances (Maeder and Gersbach 2016). However, the relative ease of designing and testing gRNAs with the CRISPR/Cas9 method, along with the high frequencies of success with this system, has recently created significant excitement around the potential of rewriting the human genome to treat disease. Nevertheless, all of these genome editing tools have shown success in preclinical models of correction of genetic mutations associated with a plethora of diseases such as sickle cell anemia, X-linked severe combined immunodeficiency, and hemophilia (Urnov et al. 2010; Arnould et al. 2011; Li et al. 2011; Miller et al. 2011; Sebastiano et al. 2011; Zou et al. 2011).
In addition to the recent advances in genome editing, several other approaches for gene therapy for DMD have been extensively evaluated. Oligonucleotide-mediated exon skipping can be utilized to ‘skip’ targeted exons in the pre-mRNA to address point mutations or deleterious frame shifts. This method is currently being widely explored for DMD and two drug candidates have been assessed in clinical trials. More recent advances in this area include improved chemical formulations and expression of these oligonucleotides from viral vectors. In fact, efficient exogenous gene delivery to skeletal and cardiac muscle is possible with viral vectors, but is most effective with the size-restricted adeno-associated virus (AAV). Therefore, the large size of the DMD gene is a challenge for using this method for DMD. There have also been many cell-based approaches evaluated for delivery of the full-length DMD gene through fusion of donor cells to host myofibers by injection of skeletal myoblasts, bone marrow-derived cells, or other stem cells. Some of these strategies have even been tested in clinical trials, but there has been limited success due to poor cell survival and migration from the injection site (Gussoni et al. 1992; Law et al. 1992; Tremblay et al. 1993; Mendell et al. 1995; Jin et al. 2005; Farini et al. 2009; Palmieri et al. 2010; Farini et al. 2012). Furthermore, the systemic nature of DMD and the cardiac and pulmonary complications that lead to premature fatality make cell-based treatments less feasible at this time. Small molecule drugs are also being developed to suppress translation termination caused by nonsense mutations, which generate premature stop codons, and have showed dystrophin restoration in vitro and in vivo (Welch et al. 2007; Gonzalez-Hilarion et al. 2012). Thus, this review summarizes recent developments in gene-based methods that restore dystrophin expression, including gene and cell therapy, genome editing, and exon skipping.
Gene and cell therapy
Several variations of dystrophin cDNA delivery to muscle are under evaluation in ongoing studies. The dystrophin cDNA can be delivered as naked plasmid, as has been assessed in the mdx mouse resulting in stably expressed dystrophin in 1–5 % of myofibers (Zhang et al. 2004). This principle was applied in a clinical trial where 6 out of 9 patients had low but present levels of dystrophin (Romero et al. 2004). However, this plasmid-mediated gene delivery approach can only be applied locally at the injection site and provides only transient dystrophin expression and, therefore, is currently not able to generate therapeutic benefit for DMD patients.
Efficiency of gene transfer and expression levels can be increased using a viral delivery system rather than plasmid DNA. However, the full-length dystrophin cDNA exceeds packaging limits of many viral vectors. The cDNA can be split up into three parts and delivered through co-injection of three viruses, where the expression cassette is reconstituted in vivo via trans-splicing or homologous recombination (Koo et al. 2014; Lostal et al. 2014). Although the efficiency of triple trans-splicing and reconstitution may be low, optimization of the co-injection may be a viable way to express the full-length dystrophin cDNA. A gene therapy utilizing adeno-associated virus (AAV) is particularly compelling as AAV has been shown to have high and persistent levels of in vivo transduction and gene expression in skeletal and cardiac muscle (Wang et al. 2005). Additionally, the virus remains predominantly episomal so it does not pose the same level of risk of non-specific genomic integration and insertional mutagenesis as has been documented for lentivirus (Ehrhardt et al. 2006; Penaud-Budloo et al. 2008). The positive results for both safety and efficacy in many ongoing clinical trials with AAV vectors for diverse conditions, and an approved product in Europe based on intramuscular injection of AAV (Glybera), also support this approach (Naldini 2015).
To develop an approach using a single AAV vector, truncated versions of the dystrophin cDNA have been created termed mini-dystrophin and micro-dystrophin. The DMD gene contains repetitive domains that can be removed to truncate the size of the dystrophin cDNA while retaining significant functionality. Minidystrophins are based on deletion mutations found in very mildly affected BMD patients, whereas microsydstrophins were engineered based on the minimum requirement of the gene for normal dystrophin function (Athanasopoulos et al. 2004). Both have been codon optimized for enhanced expression levels (Kornegay et al. 2010; Athanasopoulos et al. 2011). Early studies in mouse models confirmed that viral delivery of truncated dystrophin cDNAs restores myofiber morphology, histology, and cell membrane integrity (Wang et al. 2000). Follow-up work showed an increase of contractile force in treated muscles (Watchko et al. 2002; Yoshimura et al. 2004; Gregorevic et al. 2008) and protection against eccentric contraction-induced injury (Liu et al. 2005), as well as increased lifespan (Gregorevic et al. 2006; Wang et al. 2009). Further, groups have assessed AAV-mediated expression in mouse cardiac tissue (Yue et al. 2003), and shown improved cardiomyopathy index and ameliorated electrocardiographic abnormalities (Bostick et al. 2008), and protection against dobutamine-stress induced cardiac death (Bostick et al. 2011, 2012). Several studies have also assessed persistence, immunogenicity, and function of microdystrophin expression following AAV delivery to dog models of DMD (Ohshima et al. 2009; Koo et al. 2011; Shin et al. 2013). In particular, delivery of microdystrophin with a modified AAV-9 vector to multiple muscles showed persistent microdystrophin expression and function, improved muscle pathology, and increased muscle force in a dog model of DMD (Shin et al. 2013). These results in a large animal model are promising for the clinical translation of gene therapy for DMD.
In fact, AAV delivery of minidystrophin was assessed in a clinical trial that included intramuscular injection with various doses of AAV expressing minidystrophin. However, there were very few dystrophin-positive fibers in only some patients even though the viral genomes were easily detectable in muscle biopsies (Mendell et al. 2010). It appears that the immune system played a role in the lackluster results; T cells targeting dystrophin epitopes were detected in the blood of many patients, representing a possible immune response to the foreign epitope (Mendell et al. 2010). However, some patients were also found to harbor these T cells prior to gene therapy with the AAV-minidystrophin. There currently lacks a clear explanation for this anti-dystrophin immune response and whether it was aggravated by minidystrophin expression. Moving forward, newer mini- and micro-dystrophin constructs can be engineered to avoid immunogenic neoantigens and patients can be screened for immunity to epitopes in the therapeutic construct. Continued study of possible immune responses and development of immunosuppression regimens will be of utmost concern for all future approaches for dystrophin restoration in DMD patients.
Another strategy for delivery of dystrophin gene sequences to muscle is the administration of cells that engraft into muscle tissues and fuse into muscle fibers. These cells may be allogeneic cells from healthy patients, or autologous cells derived from the DMD patient that have been engineered ex vivo to express dystrophin. In particular, reconstitution of the satellite cell pool, the progenitor cells of skeletal muscle, will be of utmost importance for muscle diseases. Induced pluripotent stem cells from DMD mouse models have been shown to have regenerative potential after correction, and display engraftment after systemic delivery (Filareto et al. 2013). Furthermore, transplantation of myogenic precursors derived from pluripotent cells produces dystrophin-positive myofibers that have improved contractile properties (Darabi et al. 2012). The knowledge gained from studies of satellite cell reconstitution will help inform all gene-based approaches to treating DMD, and targeting these cells is likely important to establishing therapeutic benefit that will last the lifetime of the patient.
Genome editing
There are several possible approaches for applying genome editing to the correction of DMD. The majority of DMD-causing mutations are deletions that disturb the translational reading frame (Fig. 1). By removing additional exons around the inherited deletion, the reading frame can be restored. Exon 51 of the human DMD gene has been a primary target for this approach as removal of this exon would address about 13 % of the patient population, which represents the largest population segment that can be addressed by removal of a single exon (Helderman-van den Enden et al. 2010). A large deletion of exons 45–55 has also been tested, as this single approach would capture mutations in a much larger segment of the gene and address 60–65 % of DMD patient mutations (Flanigan et al. 2009; Lu et al. 2011; Aoki et al. 2012). This large deletion has been observed in BMD patients and typically presents as a mild phenotype, suggesting that this region of the protein is dispensable. Targeted deletions of both of these regions have shown dystrophin restoration in patient-derived myoblasts (Ousterout et al. 2015a, b) and human induced pluripotent stem cells (Li et al. 2015; Young et al. 2016). These ex vivo edited cells have been transplanted into the mouse models to show feasibility of dystrophin expression in vivo. Utilization of genome editing tools for targeted exon deletions is a leading approach for applying gene editing for treatment of DMD.
Another approach for DMD treatment using genome editing is to create targeted frameshifts in the gene. Nucleases directed to sites within exons around the deleted region of the gene can create indels that restore the translational reading frame. Meganucleases were used to generate indels that restore the reading frame of a modified DMD gene containing synthetic nuclease target sites, successfully restoring dystrophin expression in myoblasts in vitro and in muscle fibers through plasmid electroporation in vivo (Chapdelaine et al. 2010). This same approach was later extended to targeting DMD gene sequences with TALENs and restoring dystrophin expression in patient-derived cells (Ousterout et al. 2013). However, this strategy is limited by the stochastic nature of indel generation, such that only a fraction of the edited sequences lead to the correct reading frame, and each unique indel will produce novel epitopes of unknown immunogenicity.
Lastly, homologous recombination can be used to restore the reading frame. For example, exons 45 through 52 were successfully inserted into intron 44 in DMD myoblasts (Popplewell et al. 2013), and a nonsense mutation in exon 23 in the mdx mouse model was corrected with homology-directed repair (Long et al. 2014). However, the efficiency of homologous recombination is typically lower than NHEJ, and this repair mechanism is also downregulated in post-mitotic cells such as muscle fibers. As gene editing technologies continue to develop, including the development of methods for reducing NHEJ activity that competes with HDR (Chu et al. 2015), this approach may become more feasible for preclinical development.
Genome editing of the dystrophin gene has been successful in cultured myoblasts, induced pluripotent stem cells, and fibroblasts with zinc finger nucleases, TALENs, meganucleases, and CRISPR/Cas9 systems. Another important approach for restoring dystrophin expression uses phage integrases to insert dystrophin gene sequences from plasmid DNA into genomic target sites either ex vivo in human myoblasts (Quenneville et al. 2004) or in vivo in mdx muscles (Bertoni et al. 2006). This site-specific integration of the transgene leads to sustained expression, in contrast to typical plasmid delivery. However, there are still some concerns regarding efficiency, systemic delivery to large animals, and potential unpredictability of the genomic insertion sites.
Recently, several groups showed efficacy of in vivo gene editing of the DMD gene utilizing NHEJ to create targeted deletions. (Long et al. 2015; Nelson et al. 2015; Tabebordbar et al. 2015; Xu et al. 2015; Iyombe-Engembe et al. 2016). In particular, delivery of the CRISPR/Cas9 system with AAV vectors that are currently in clinical trials for other neuromuscular diseases restored dystrophin expression in skeletal and cardiac muscle in the mdx mouse model (Table 1). Furthermore, both local and systemic delivery of these AAV systems in adult and neonatal mice increased muscle strength. The efficacy of systemic delivery is of particular interest for treatment of all the muscle groups affected by DMD. These studies are the first report of a phenotypic improvement via genome editing in an animal model of muscular dystrophy. These results also demonstrated that even low levels of genome editing and correction are sufficient to produce many dystrophin-positive muscle fibers and a dramatic increase in dystrophin protein expression since each muscle fiber contains hundreds of individual nuclei. Furthermore, Tabebordbar et al. showed that the satellite cells, the stem cells of skeletal muscle, are also edited via AAV-delivered CRISPR/Cas9. This suggests that the level of gene editing and dystrophin restoration will increase as these progenitor cells continue to repopulate the muscle tissues.
Although genome editing is a relatively new approach for DMD correction, similar tools have already moved into the clinic for ex vivo editing of T cells and hematopoietic stem cells and are moving quickly along the clinical pipeline for applications in vivo (Maeder and Gersbach 2016). Recently, FDA approval for a clinical trial of gene editing in the liver to treat hemophilia was announced (Gibney 2016). This first human trial using gene editing tools will help establish safety, paving way for future gene editing therapies.
Exon skipping
Antisense oligonucleotides (AONs) are small single-stranded chemically modified nucleic acids that are designed to target specific gene transcripts. The small size is crucial for delivery, and chemical modifications affect stability, solubility, toxicity, affinity, and degradation resistance. For treatment of DMD, AONs are primarily used to alter pre-mRNA splicing, such that specific exons in the dystrophin mRNA are removed during the splicing process (Touznik et al. 2014; Jirka and Aartsma-Rus 2015). The targeted sequence is spliced out with the flanking introns as the AON essentially hides the exon splice sites from the splicing machinery (Fig. 3). By skipping specific exons, the reading frame of the transcript can be restored. AON-mediated exon skipping has shown tremendous success in preclinical studies in mouse models (Kole and Krieg 2015). Two leading AON chemistries for drug development are 2′-O-methyl phosphorothioate (2OMePs) AONs, and phosphorodiamidate morpholino oligomers (PMOs). The 2OMePs chemistry has a negative charge, whereas PMOs are uncharged at physiological pH (Kole and Krieg 2015).

Strategy of using antisense oligonucleotides (AONs) to ‘skip’ an exon during mRNA processing and restore the dystrophin reading frame
Dystrophin restoration has been achieved in vivo in the mdx mouse model by targeting the splice site of exon 23 using the 2OMePs chemistry (Heemskerk et al. 2010) and the PMO chemistry (Alter et al. 2006). Both AON chemistries have also been evaluated in vitro in myoblasts from a dog model, with the PMO chemistry restoring higher levels of dystrophin than the 2OMePs chemistry (McClorey et al. 2006), as well as in vivo through systemic delivery to dog models of DMD (Yokota et al. 2009). They have also both been used in clinical trials to target exon 51 in the human dystrophin gene (Aartsma-Rus et al. 2014). A primary outcome assessed in DMD clinical trials is the six-minute walk test (6MWT), also called the six-minute walk distance (6MWD), an outcome measure used in a variety of clinical trials (McDonald et al. 2010a, b). The test essentially determines the distance that a patient can walk within six minutes. This test aims to assess the systems involved in walking as a measure of disease progression (Crapo et al. 2002).
The 2OMePs-based AON targeting exon 51 was evaluated in several clinical trials and roughly 300 patients (Goemans et al. 2011; Voit et al. 2014). In a placebo-controlled phase 2 study, this drug was delivered subcutaneously twice weekly during the first 3 weeks, then either weekly or intermittently at a dose of 6 mg/kg (Voit et al. 2014). At week 25, the average distance traveled by the weekly treated patients increased compared to placebo controls; however by week 49, there was no statistical difference between the treated and placebo patients (Voit et al. 2014). Data from a phase 1-2a study showed that dystrophin levels in treated patients vary from 1.5- to 8.2-fold above baseline levels as measured by western blot (Goemans et al. 2011). In an open-label extension trial, eight patients had stable 6MWTs for 177 weeks (Jirka and Aartsma-Rus 2015). In a double-blind, placebo-controlled phase 3 study, patients were given 6 mg/kg of drug or placebo weekly for 48 weeks; this study also failed to achieve statistical significance in the 6MWT. However, post hoc analysis suggests that the mixed population of patients 5–16 years old with varying disease severity makes it extremely challenging to find statistical differences (McDonald et al. 2010a; Goemans et al. 2013a; McDonald et al. 2013; Pane et al. 2014; Jirka and Aartsma-Rus 2015). Safety and tolerability risks of the 2OMePs chemistry are also a concern, as the therapy can be associated with injection site reactions, proteinuria, thrombocytopenia, vascular injury, and renal injury. These are likely due to the negatively charged AON interacting with immune cell receptors like toll-like receptors (Kole and Krieg 2015). This can ultimately lead to kidney inflammation, as seen in some patients in the clinical trial (Kole and Krieg 2015).
The PMO AON has been injected intravenously in 31 DMD patients with doses up to 50 mg/kg every week. The study reported dystrophin restoration in 30–60 % of muscle fibers from a biopsy taken at 48 weeks post-treatment (Cirak et al. 2011; Mendell et al. 2013). Six patients showed stable 6MWT results for 120 weeks (Cirak et al. 2011; Mendell et al. 2013). Although there was an overall decline in the 6MWT, the patients performed better than historical controls indicating that the treatment is slowing down the disease progression (Jirka and Aartsma-Rus 2015). In a more recent clinical study at the same dose, treatment with the PMO AON led to a slower rate of decline in ambulation over 3 years as assessed by the 6MWT compared to historical controls (Mendell et al. 2016). Side effects of this drug are minimal in patients, with occasional transient proteinuria, headaches, and procedural pain related to biopsy and catheter placement. There is also a lack of adverse side effects in mice and nonhuman primates even at doses more than ten-fold greater than the clinical dose (Sazani et al. 2010, 2011a, b).
Although treatment for exon 51 skipping is the furthest along in the regulatory process, there are also ongoing trials targeting exons 44, 45, and 53 through exon skipping (Lee and Yokota 2013). Exon skipping is a mutation-specific approach, such that every patient would need to be genotyped and matched to a therapy that will correct their specific reading frame mutations. Currently, there is a focus on skipping single exons as a proof-of-principle for the overall approach. However, this approach may prove challenging for the patients with duplications of one or more exons. In this case, the AONs would target both the original and duplicated copies, which will generally result in an out-of-frame transcript. Thus, more than 1 exon will need to be targeted for these patients. This approach requires a combination of several AONs being delivered as a ‘cocktail’ of drugs to skip larger regions of the transcript. If a cocktail for exons 45–55 were effective, as has been done in mouse studies, this could treat a large cohort of >60 % of all DMD patients (Aoki et al. 2012). An exception is the treatment of mutations that occur in the first few exons, which may be treatable by stimulating translation from an internal ribosomal entry site (IRES) within exon 5, as was done by AON-mediated skipping of exon 2 in a mouse model of exon 2 duplication (Wein et al. 2014).
The impressive preclinical animal data support the concept of therapeutic AON-mediated exon skipping, but the modest clinical trial results suggest that improvements to delivery, pharmacodynamics, and pharmacokinetics are necessary to significantly improve patient outcomes. More recent AON formulations, such as the tricyclo-DNA oligomers, show improved uptake in multiple tissues after systemic administration, including therapeutic benefit in the heart (Goyenvalle et al. 2015). Additionally, the incorporation of cell-penetrating peptides into AONs can similarly assist in tissue penetration, particularly facilitating delivery to the heart (Wu et al. 2008; Yin et al. 2008; McClorey and Wood 2015). Finally, the expression of exon skipping AONs linked to small nuclear RNAs such as U7 enables efficient delivery and prolonged expression of AONs in skeletal and cardiac muscle by AAV vectors (Goyenvalle et al. 2004, 2012). The success of this approach in dog models of DMD is promising for its continued development (Bish et al. 2012; Vulin et al. 2012; Le Guiner et al. 2014).
Conclusions and future directions
There are several promising gene-based strategies under development for restoring dystrophin expression to treat DMD. Exon skipping appears to slow the disease progression in clinical studies and is under consideration for regulatory approval. However, AON-based exon skipping therapies only transiently restore dystrophin expression and, therefore, would require regularly timed injections for the lifetime of the patient (Voit et al. 2014; Mendell et al. 2013). Additionally, thus far exon skipping has only shown a reduced rate of decline in patient function and does not address the need for a curative treatment. New AON formulations or delivery strategies are under development to treat the fatal cardiac complications. Exon skipping drugs have been pioneering in informing the regulatory process for DMD drugs and gene therapies. Continued refinement of this process, including the development of robust clinical endpoints and biomarkers, will dramatically shape the design of future clinical trials.
Gene therapy and genome editing are both restricted by the limitations of delivery with AAV vectors, particularly as the result of the limited packaging size (Gaj et al. 2016). Additionally, the large amounts of viral vector necessary for systemic delivery present economic and feasibility challenges to manufacturing. Thus, continued development of gene delivery technologies will be important to advancing both these fields (Nelson and Gersbach 2016). Another concern is the restriction of the expression of modified DMD genes or gene editing components to specific tissues, which may be aided by new or existing muscle-tropic AAV serotypes (Madigan and Asokan 2016) and muscle-specific promoters (Himeda et al. 2011). The preclinical animal data following mini- and micro-dystrophin delivery are very exciting and have now led to efforts to evaluate its clinical safety and efficacy. A remaining concern is to what extent mini- or micro-dystrophin will address DMD symptoms in humans, particularly the cardiomyopathy, as these truncated proteins presumably do not possess the full wild-type functionality.
Genome editing is the newest method to show potential efficacy as a therapeutic in mouse models, but there is significant work remaining before clinical trials can be pursued. Dystrophin expression has been restored using genome editing in DMD patient cells, but all in vivo genome editing, thus far, has been in the mdx mouse model. Because genome editing nucleases are specifically targeted to a particular DNA sequence, the reagents for editing the mouse DMD gene demonstrate proof-of-principle for the technology but are not necessarily translatable to human therapy. Furthermore, any genome editing-based therapy will need to undergo extensive characterization for specificity of on-target activity without modifying potential off-target sites (Bolukbasi et al. 2016). This work can be facilitated by recently described unbiased genome-wide assays of nuclease activity (Tsai et al. 2015; Kim et al. 2016) and next-generation high-fidelity nucleases (Kleinstiver et al. 2016; Slaymaker et al. 2016). This analysis will be particularly important given the possibility of sustained expression and activity of genome editing tools from AAV vectors in post-mitotic cells. Similarly, immune response to the nuclease components derived from bacteria, in addition to any potential responses to the AAV viral proteins or the restored dystrophin protein, is a primary concern that needs additional study. Improvements to cell therapy, including cell survival, engraftment, and distribution, may help overcome these concerns of in vivo genome editing (McCullagh and Perlingeiro 2015).
Overall, there is substantial progress in treating DMD by targeting dystrophin from a variety of methods. Each has unique positive and negative attributes, and likely all avenues require further research and optimization. However, for the first time it is plausible that a DMD treatment that addresses the fundamental cause of the disease is on the horizon.
References
Aartsma-Rus A, Ferlini A, Goemans N, Pasmooij AM, Wells DJ, Bushby K, Vroom E, Balabanov P (2014) Translational and regulatory challenges for exon skipping therapies. Hum Gene Ther 25:885–892
Alter J, Lou F, Rabinowitz A, Yin H, Rosenfeld J, Wilton SD, Partridge TA, Lu QL (2006) Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med 12:175–177
Aoki Y, Yokota T, Nagata T, Nakamura A, Tanihata J, Saito T, Duguez SM, Nagaraju K, Hoffman EP, Partridge T et al (2012) Bodywide skipping of exons 45-55 in dystrophic mdx52 mice by systemic antisense delivery. Proc Natl Acad Sci U S A 109:13763–13768
Arnould S, Delenda C, Grizot S, Desseaux C, Paques F, Silva GH, Smith J (2011) The I-CreI meganuclease and its engineered derivatives: applications from cell modification to gene therapy. Protein Eng Design Selection PEDS 24:27–31
Athanasopoulos T, Graham IR, Foster H, Dickson G (2004) Recombinant adeno-associated viral (rAAV) vectors as therapeutic tools for Duchenne muscular dystrophy (DMD). Gene Ther 11(Suppl 1):S109–S121
Athanasopoulos T, Foster H, Foster K, Dickson G (2011) Codon optimization of the microdystrophin gene for Duchene muscular dystrophy gene therapy. Methods Mol Biol 709:21–37
Baxter P (2010) Diagnosis and management of Duchenne muscular dystrophy. Dev Med Child Neurol 52:313
Bertoni C, Jarrahian S, Wheeler TM, Li Y, Olivares EC, Calos MP, Rando TA (2006) Enhancement of plasmid-mediated gene therapy for muscular dystrophy by directed plasmid integration. Proc Natl Acad Sci USA 103:419–424
Bish LT, Sleeper MM, Forbes SC, Wang B, Reynolds C, Singletary GE, Trafny D, Morine KJ, Sanmiguel J, Cecchini S et al (2012) Long-term restoration of cardiac dystrophin expression in golden retriever muscular dystrophy following rAAV6-mediated exon skipping. Mol Ther 20:580–589
Bolukbasi MF, Gupta A, Wolfe SA (2016) Creating and evaluating accurate CRISPR-Cas9 scalpels for genomic surgery. Nat Methods 13:41–50
Bostick B, Yue YP, Lai Y, Long C, Li DJ, Duan DS (2008) Adeno-associated virus serotype-9 microdystrophin gene therapy ameliorates electrocardiographic abnormalities in mdx mice. Hum Gene Ther 19:851–856
Bostick B, Shin JH, Yue YP, Duan DS (2011) AAV-microdystrophin therapy improves cardiac performance in aged female mdx mice. Mol Ther 19:1826–1832
Bostick B, Shin JH, Yue Y, Wasala NB, Lai Y, Duan D (2012) AAV micro-dystrophin gene therapy alleviates stress-induced cardiac death but not myocardial fibrosis in >21-m-old mdx mice, an end-stage model of Duchenne muscular dystrophy cardiomyopathy. J Mol Cell Cardiol 53:217–222
Chapdelaine P, Pichavant C, Rousseau J, Paques F, Tremblay JP (2010) Meganucleases can restore the reading frame of a mutated dystrophin. Gene Ther 17:846–858
Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kuhn R (2015) Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol 33:543–548
Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ et al (2011) Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378:595–605
Cox DB, Platt RJ, Zhang F (2015) Therapeutic genome editing: prospects and challenges. Nat Med 21:121–131
Crapo RO, Casaburi R, Coates AL, Enright PL, MacIntyre NR, McKay RT, Johnson D, Wanger JS, Zeballos RJ, Bittner V et al (2002) ATS statement: guidelines for the six-minute walk test. Am J Resp Crit Care 166:111–117
Crow JF (2000) The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet 1:40–47
Darabi R, Arpke RW, Irion S, Dimos JT, Grskovic M, Kyba M, Perlingeiro RCR (2012) Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 10:610–619
Dittrich S, Tuerk M, Haaker G, Greim V, Buchholz A, Burkhardt B, Fujak A, Trollmann R, Schmid A, Schroeder R (2015) Cardiomyopathy in duchenne muscular dystrophy: current value of clinical, electrophysiological and imaging findings in children and teenagers. Klin Padiatr 227:225–231
D’Orsogna L, O’Shea JP, Miller G (1988) Cardiomyopathy of Duchenne muscular dystrophy. Pediatr Cardiol 9:205–213
Ehrhardt A, Engler JA, Xu H, Cherry AM, Kay MA (2006) Molecular analysis of chromosomal rearrangements in mammalian cells after phiC31-mediated integration. Hum Gene Ther 17:1077–1094
England SB, Nicholson LVB, Johnson MA, Forrest SM, Love DR, Zubrzyckagaarn EE, Bulman DE, Harris JB, Davies KE (1990) Very mild muscular-dystrophy associated with the deletion of 46-percent of dystrophin. Nature 343:180–182
Falzarano MS, Scotton C, Passarelli C, Ferlini A (2015) Duchenne muscular dystrophy: from diagnosis to therapy. Molecules 20:18168–18184
Farini A, Razini P, Erratico S, Torrente Y, Meregalli M (2009) Cell based therapy for Duchenne muscular dystrophy. J Cell Physiol 221:526–534
Farini A, Villa C, Manescu A, Fiori F, Giuliani A, Razini P, Sitzia C, Del Fraro G, Belicchi M, Meregalli M et al (2012) Novel insight into stem cell trafficking in dystrophic muscles. Int J Nanomed 7:3059–3067
Filareto A, Parker S, Darabi R, Borges L, Iacovino M, Schaaf T, Mayerhofer T, Chamberlain JS, Ervasti JM, McIvor RS et al (2013) An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells. Nat Commun 4:1549
Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, Sampson JB, Mendell JR, Wall C, King WM et al (2009) Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat 30:1657–1666
Gaj T, Gersbach CA, Barbas CF 3rd (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31:397–405
Gaj T, Epstein BE, Schaffer DV (2016) Genome engineering using adeno-associated virus: basic and clinical research applications. Mol Ther 24:458–464
Gersbach CA, Gaj T, Barbas CF 3rd (2014) Synthetic zinc finger proteins: the advent of targeted gene regulation and genome modification technologies. Acc Chem Res 47:2309–2318
Gibney E (2016) The science to look out for in 2016. Nature 529:14–15
Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, Holling T, Janson AA, Platenburg GJ, Sipkens JA et al (2011) Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med 364:1513–1522
Goemans N, Klingels K, van den Hauwe M, Boons S, Verstraete L, Peeters C, Feys H, Buyse G (2013a) Six-minute walk test: reference values and prediction equation in healthy boys aged 5 to 12 years. PLoS ONE 8:e84120
Goemans N, van den Hauwe M, Wilson R, van Impe A, Klingels K, Buyse G (2013b) Ambulatory capacity and disease progression as measured by the 6-minute-walk-distance in Duchenne muscular dystrophy subjects on daily corticosteroids. Neuromuscular disorders: NMD 23:618–623
Gonzalez-Hilarion S, Beghyn T, Jia J, Debreuck N, Berte G, Mamchaoui K, Mouly V, Gruenert DC, Deprez B, Lejeune F (2012) Rescue of nonsense mutations by amlexanox in human cells. Orphanet journal of rare diseases 7:58
Goyenvalle A, Vulin A, Fougerousse F, Leturcq F, Kaplan JC, Garcia L, Danos O (2004) Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science 306:1796–1799
Goyenvalle A, Babbs A, Wright J, Wilkins V, Powell D, Garcia L, Davies KE (2012) Rescue of severely affected dystrophin/utrophin-deficient mice through scAAV-U7snRNA-mediated exon skipping. Hum Mol Genet 21:2559–2571
Goyenvalle A, Griffith G, Babbs A, El Andaloussi S, Ezzat K, Avril A, Dugovic B, Chaussenot R, Ferry A, Voit T et al (2015) Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat Med 21:270–275
Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, Finn E, Adams ME, Froehner SC, Murry CE et al (2006) rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med 12:787–789
Gregorevic P, Blankinship MJ, Allen JM, Chamberlain JS (2008) Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol Ther J Am Soc Gene Ther 16:657–664
Grimm T, Meng G, Liechti-Gallati S, Bettecken T, Muller CR, Muller B (1994) On the origin of deletions and point mutations in Duchenne muscular dystrophy: most deletions arise in oogenesis and most point mutations result from events in spermatogenesis. J Med Genet 31:183–186
Guiraud S, Aartsma-Rus A, Vieira NM, Davies KE, van Ommen GJB, Kunkel LM (2015) The Pathogenesis and Therapy of Muscular Dystrophies. Annu Rev Genom Hum G 16:281
Gussoni E, Pavlath GK, Lanctot AM, Sharma KR, Miller RG, Steinman L, Blau HM (1992) Normal dystrophin transcripts detected in duchenne muscular-dystrophy patients after myoblast transplantation. Nature 356:435–438
Heemskerk H, de Winter C, van Kuik P, Heuvelmans N, Sabatelli P, Rimessi P, Braghetta P, van Ommen GJ, de Kimpe S, Ferlini A et al (2010) Preclinical PK and PD studies on 2′-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol Ther J Am Soc Gene Ther 18:1210–1217
Helderman-van den Enden AT, Straathof CS, Aartsma-Rus A, den Dunnen JT, Verbist BM, Bakker E, Verschuuren JJ, Ginjaar HB (2010) Becker muscular dystrophy patients with deletions around exon 51; a promising outlook for exon skipping therapy in Duchenne patients. Neuromuscular disorders: NMD 20:251–254
Himeda CL, Chen X, Hauschka SD (2011) Design and testing of regulatory cassettes for optimal activity in skeletal and cardiac muscles. Methods Mol Biol 709:3–19
Hoffman EP, Brown RH Jr, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51:919–928
Hoffman EP, Kunkel LM, Angelini C, Clarke A, Johnson M, Harris JB (1989) Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology 39:1011–1017
Hollinger K, Chamberlain JS (2015) Viral vector-mediated gene therapies. Curr Opin Neurol 28:522–527
Hsu PD, Lander ES, Zhang F (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278
Iyombe-Engembe JP, Ouellet DL, Barbeau X, Rousseau J, Chapdelaine P, Lague P, Tremblay JP (2016) efficient restoration of the dystrophin gene reading frame and protein structure in DMD myoblasts using the CinDel method. Mol Ther Nucleic Acids 5:e283
Jin K, Sun Y, Xie L, Mao XO, Childs J, Peel A, Logvinova A, Banwait S, Greenberg DA (2005) Comparison of ischemia-directed migration of neural precursor cells after intrastriatal, intraventricular, or intravenous transplantation in the rat. Neurobiol Dis 18:366–374
Jirka S, Aartsma-Rus A (2015) An update on RNA-targeting therapies for neuromuscular disorders. Curr Opin Neurol 28:515–521
Joung JK, Sander JD (2013) TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14:49–55
Kim D, Kim S, Kim S, Park J, Kim JS (2016) Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res 26:406–415
Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK (2016) High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529:490–495
Kole R, Krieg AM (2015) Exon skipping therapy for Duchenne muscular dystrophy. Adv Drug Deliv Rev 87:104–107
Koo T, Okada T, Athanasopoulos T, Foster H, Takeda S, Dickson G (2011) Long-term functional adeno-associated virus-microdystrophin expression in the dystrophic CXMDj dog. J Gene Med 13:497–506
Koo T, Popplewell L, Athanasopoulos T, Dickson G (2014) Triple trans-splicing adeno-associated virus vectors capable of transferring the coding sequence for full-length dystrophin protein into dystrophic mice. Hum Gene Ther 25:98–108
Kornegay JN, Li J, Bogan JR, Bogan DJ, Chen C, Zheng H, Wang B, Qiao C, Howard JF Jr, Xiao X (2010) Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Mol Ther J Am Soc Gene Ther 18:1501–1508
Lapidos KA, Kakkar R, McNally EM (2004) The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res 94:1023–1031
Law PK, Goodwin TG, Fang Q, Duggirala V, Larkin C, Florendo JA, Kirby DS, Deering MB, Li HJ, Chen M et al (1992) Feasibility, safety, and efficacy of myoblast transfer therapy on Duchenne muscular dystrophy boys. Cell Transplant 1:235–244
Le Guiner C, Montus M, Servais L, Cherel Y, Francois V, Thibaud JL, Wary C, Matot B, Larcher T, Guigand L et al (2014) Forelimb treatment in a large cohort of dystrophic dogs supports delivery of a recombinant AAV for exon skipping in Duchenne patients. Mol Ther 22:1923–1935
Lee JJ, Yokota T (2013) Antisense therapy in neurology. J Personalized Med 3:144–176
Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS, Malani N, Anguela XM, Sharma R, Ivanciu L et al (2011) In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 475:217–221
Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T, Tanaka M, Amano N, Watanabe A, Sakurai H et al (2015) Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem cell reports 4:143–154
Liu MJ, Yue YP, Harper SQ, Grange RW, Chamberlain JS, Duan DS (2005) Adeno-associated virus-mediated microdystrophin expression protects young mdx muscle from contraction-induced injury. Mol Ther 11:245–256
Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN (2014) Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 345:1184–1188
Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN (2015) Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. doi:10.1126/science.aad5725
Lostal W, Kodippili K, Yue Y, Duan D (2014) Full-length dystrophin reconstitution with adeno-associated viral vectors. Hum Gene Ther 25:552–562
Lu QL, Yokota T, Takeda S, Garcia L, Muntoni F, Partridge T (2011) The status of exon skipping as a therapeutic approach to duchenne muscular dystrophy. Mol Ther 19:9–15
Madigan VJ, Asokan A (2016) Engineering AAV receptor footprints for gene therapy. Curr Opin Virol 18:89–96
Maeder ML, Gersbach CA (2016) Genome editing technologies for gene and cell therapy. Mol Ther. doi:10.1038/mt.2016.10
McClorey G, Wood MJ (2015) An overview of the clinical application of antisense oligonucleotides for RNA-targeting therapies. Curr Opin Pharmacol 24:52–58
McClorey G, Moulton HM, Iversen PL, Fletcher S, Wilton SD (2006) Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther 13:1373–1381
McCullagh KJ, Perlingeiro RC (2015) Coaxing stem cells for skeletal muscle repair. Adv Drug Deliv Rev 84:198–207
McDonald CM, Henricson EK, Abresch RT, Florence JM, Eagle M, Gappmaier E, Glanzman AM, Group PG-DS, Spiegel R, Barth J et al (2013) The 6-minute walk test and other endpoints in Duchenne muscular dystrophy: longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve 48:343–356
McDonald C, Henricson E, Abresch R, Han J, Nicorici A, Goude E, Elfring G, Reha A, Hirawat S, Miller L (2010a) The 6-Min walk test in duchenne muscular dystrophy: longitudinal observations. Neurology 74:A219–A219
McDonald CM, Henricson EK, Han JJ, Abresch RT, Nicorici A, Elfring GL, Atkinson L, Reha A, Hirawat S, Miller LL (2010b) The 6-Minute Walk Test as a New Outcome Measure in Duchenne Muscular Dystrophy. Muscle Nerve 41:500–510
Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, Shao J, Kaye EM, Mercuri E, Eteplirsen Study G, Telethon Foundation DMDIN (2016) Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Annals of neurology 79:257–271
Mendell JR, Kissel JT, Amato AA, King W, Signore L, Prior TW, Sahenk Z, Besson S, Mcandrew PE, Rice R et al (1995) Myoblast Transfer in the Treatment of Duchennes Muscular-Dystrophy. New Engl J Med 333:832–838
Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, Bowles D, Gray S, Li C, Galloway G et al (2010) Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med 363:1429–1437
Mendell JR, Rodino-Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP, Alfano L, Gomez AM, Lewis S, Kota J et al (2013) Eteplirsen for the treatment of Duchenne muscular dystrophy. Annals of neurology 74:637–647
Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ et al (2011) A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29:143–148
Naldini L (2015) Gene therapy returns to centre stage. Nature 526:351–360
Nallamilli BR, Ankala A, Hegde M. 2014. Molecular diagnosis of duchenne muscular dystrophy. Current protocols in human genetics/editorial board, Jonathan L Haines et al 83:9 25 21–29 25:29
Nelson CE, Gersbach CA (2016) Engineering delivery vehicles for genome editing. Annu Rev Chem Biomol Eng 7:637–662
Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Rivera RM, Madhavan S, Pan X, Ran FA, Yan WX et al (2015) In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. doi:10.1126/science.aad5143
Ohshima S, Shin JH, Yuasa K, Nishiyama A, Kira J, Okada T, Takeda S (2009) Transduction efficiency and immune response associated with the administration of AAV8 vector into dog skeletal muscle. Mol Ther 17:73–80
Ousterout DG, Perez-Pinera P, Thakore PI, Kabadi AM, Brown MT, Qin X, Fedrigo O, Mouly V, Tremblay JP, Gersbach CA (2013) Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol Ther 21:1718–1726
Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, Gersbach CA (2015a) Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun 6:6244
Ousterout DG, Kabadi AM, Thakore PI, Perez-Pinera P, Brown MT, Majoros WH, Reddy TE, Gersbach CA (2015b) Correction of dystrophin expression in cells from Duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol Ther J Am Soc Gene The 23:523–532
Palmieri B, Tremblay JP, Daniele L (2010) Past, present and future of myoblast transplantation in the treatment of Duchenne muscular dystrophy. Pediatr Transplant 14:813–819
Pane M, Mazzone ES, Sivo S, Sormani MP, Messina S, D’Amico A, Carlesi A, Vita G, Fanelli L, Berardinelli A et al (2014) Long term natural history data in ambulant boys with Duchenne muscular dystrophy: 36-month changes. PLoS One 9:e108205
Parker AE, Robb SA, Chambers J, Davidson AC, Evans K, O’Dowd J, Williams AJ, Howard RS (2005) Analysis of an adult Duchenne muscular dystrophy population. QJM Monthly J Assoc Physicians 98:729–736
Penaud-Budloo M, Le Guiner C, Nowrouzi A, Toromanoff A, Cherel Y, Chenuaud P, Schmidt M, von Kalle C, Rolling F, Moullier P et al (2008) Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle. J Virol 82:7875–7885
Popplewell L, Koo T, Leclerc X, Duclert A, Mamchaoui K, Gouble A, Mouly V, Voit T, Paques F, Cedrone F et al (2013) Gene correction of a duchenne muscular dystrophy mutation by meganuclease-enhanced exon knock-in. Hum Gene Ther 24:692–701
Quenneville SP, Chapdelaine P, Rousseau J, Beaulieu J, Caron NJ, Skuk D, Mills P, Olivares EC, Calos MP, Tremblay JP (2004) Nucleofection of muscle-derived stem cells and myoblasts with phiC31 integrase: stable expression of a full-length-dystrophin fusion gene by human myoblasts. Mol Ther J Am Soc Gene Ther 10:679–687
Ramos J, Chamberlain JS (2015) Gene therapy for duchenne muscular dystrophy. Expert Opinion Orphan Drugs 3:1255–1266
Ricotti V, Ridout DA, Scott E, Quinlivan R, Robb SA, Manzur AY, Muntoni F, NorthStar Clinical N (2013) Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry 84:698–705
Romero NB, Braun S, Benveniste O, Leturcq F, Hogrel JY, Morris GE, Barois A, Eymard B, Payan C, Ortega V et al (2004) Phase I study of dystrophin Duchenne/Becker plasmid-based gene therapy in muscular dystrophy. Hum Gene Ther 15:1065–1076
Sazani P, Weller DL, Shrewsbury SB (2010) Safety pharmacology and genotoxicity evaluation of AVI-4658. Int J Toxicol 29:143–156
Sazani P, Ness KP, Weller DL, Poage D, Nelson K, Shrewsbury AS (2011a) Chemical and mechanistic toxicology evaluation of exon skipping phosphorodiamidate morpholino oligomers in mdx mice. Int J Toxicol 30:322–333
Sazani P, Ness KP, Weller DL, Poage DW, Palyada K, Shrewsbury SB (2011b) Repeat-dose toxicology evaluation in cynomolgus monkeys of AVI-4658, a phosphorodiamidate morpholino oligomer (PMO) drug for the treatment of duchenne muscular dystrophy. Int J Toxicol 30:313–321
Sebastiano V, Maeder ML, Angstman JF, Haddad B, Khayter C, Yeo DT, Goodwin MJ, Hawkins JS, Ramirez CL, Batista LF et al (2011) In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 29:1717–1726
Shin JH, Pan X, Hakim CH, Yang HT, Yue Y, Zhang K, Terjung RL, Duan D (2013) Microdystrophin ameliorates muscular dystrophy in the canine model of duchenne muscular dystrophy. Mol Ther 21:750–757
Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, Paques F (2011) Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther 11:11–27
Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F (2016) Rationally engineered Cas9 nucleases with improved specificity. Science 351:84–88
Tabebordbar M, Zhu K, Cheng JK, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA et al (2015) In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. doi:10.1126/science.aad5177
Touznik A, Lee JJ, Yokota T (2014) New developments in exon skipping and splice modulation therapies for neuromuscular diseases. Expert Opin Biol Therapy 14:809–819
Tremblay JP, Malouin F, Roy R, Huard J, Bouchard JP, Satoh A, Richards CL (1993) Results of a triple blind clinical study of myoblast transplantations without immunosuppressive treatment in young boys with Duchenne muscular dystrophy. Cell Transplant 2:99–112
Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP et al (2015) GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33:187–197
Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD (2010) Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11:636–646
van Westering TL, Betts CA, Wood MJ (2015) Current understanding of molecular pathology and treatment of cardiomyopathy in duchenne muscular dystrophy. Molecules 20:8823–8855
Voit T, Topaloglu H, Straub V, Muntoni F, Deconinck N, Campion G, De Kimpe SJ, Eagle M, Guglieri M, Hood S et al (2014) Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol 13:987–996
Vulin A, Barthelemy I, Goyenvalle A, Thibaud JL, Beley C, Griffith G, Benchaouir R, le Hir M, Unterfinger Y, Lorain S et al (2012) Muscle function recovery in golden retriever muscular dystrophy after AAV1-U7 exon skipping. Mol Ther 20:2120–2133
Wagner KR, Lechtzin N, Judge DP (2007) Current treatment of adult Duchenne muscular dystrophy. Biochim Biophys Acta 1772:229–237
Wang B, Li J, Xiao X (2000) Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci USA 97:13714–13719
Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, Chen C, Li J, Xiao X (2005) Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol 23:321–328
Wang B, Li J, Fu FH, Xiao X (2009) Systemic human minidystrophin gene transfer improves functions and life span of dystrophin and dystrophin/utrophin-deficient mice. J Orthop Res 27:421–426
Watchko J, O’Day T, Wang B, Zhou LQ, Tang Y, Li J, Xiao X (2002) Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum Gene Ther 13:1451–1460
Wein N, Vulin A, Falzarano MS, Szigyarto CA, Maiti B, Findlay A, Heller KN, Uhlen M, Bakthavachalu B, Messina S et al (2014) Translation from a DMD exon 5 IRES results in a functional dystrophin isoform that attenuates dystrophinopathy in humans and mice. Nat Med 20:992–1000
Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S et al (2007) PTC124 targets genetic disorders caused by nonsense mutations. Nature 447:87–91
Wu B, Moulton HM, Iversen PL, Jiang J, Li J, Spurney CF, Sali A, Guerron AD, Nagaraju K, Doran T et al (2008) Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc Natl Acad Sci USA 105:14814–14819
Xu L, Park KH, Zhao L, Xu J, El Refaey M, Gao Y, Zhu H, Ma J, Han R (2015) CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol Ther. doi:10.1038/mt.2015.192
Yin H, Moulton HM, Seow Y, Boyd C, Boutilier J, Iverson P, Wood MJ (2008) Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum Mol Genet 17:3909–3918
Yokota T, Lu QL, Partridge T, Kobayashi M, Nakamura A, Takeda S, Hoffman E (2009) Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol 65:667–676
Yoshimura M, Sakamoto M, Ikemoto M, Mochizuki Y, Yuasa K, Miyagoe-Suzuki Y, Takeda S (2004) AAV vector-mediated microdystrophin expression in a relatively small percentage of mdx myofibers improved the mdx phenotype. Mol Ther J Am Soc Gene Ther 10:821–828
Young CS, Hicks MR, Ermolova NV, Nakano H, Jan M, Younesi S, Karumbayaram S, Kumagai-Cresse C, Wang D, Zack JA et al (2016) A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell. doi:10.1016/j.stem.2016.01.021
Yue YP, Li ZB, Harper SQ, Davisson RL, Chamberlain JS, Duan DS (2003) Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation 108:1626–1632
Zhang G, Ludtke JJ, Thioudellet C, Kleinpeter P, Antoniou M, Herweijer H, Braun S, Wolff JA (2004) Intraarterial delivery of naked plasmid DNA expressing full-length mouse dystrophin in the mdx mouse model of duchenne muscular dystrophy. Hum Gene Ther 15:770–782
Zou J, Mali P, Huang X, Dowey SN, Cheng L (2011) Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood 118:4599–4608
Acknowledgments
This work has been supported by the Muscular Dystrophy Association (MDA277360), a Duke-Coulter Translational Partnership Grant, a Duke/UNC-Chapel Hill CTSA Consortium Collaborative Translational Research Award, a Hartwell Foundation Individual Biomedical Research Award, a March of Dimes Foundation Basil O’Connor Starter Scholar Award, National Institutes of Health (NIH) grant R01AR069085, an NIH Director’s New Innovator Award (DP2-OD008586), and the Office of the Assistant Secretary of Defense for Health Affairs, through the Duchenne Muscular Dystrophy Research Program under Award No. W81XWH-15-1-0469. Opinions, interpretations, conclusions and recommendations are those of the author and are not necessarily endorsed by the NIH or Department of Defense. J.R.H. is supported by a National Science Foundation Graduate Research Fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
CA.G. and J.R.H. have filed patent applications related to genome editing for Duchenne muscular dystrophy. C.A.G. is an advisor to Editas Medicine, a company engaged in development of therapeutic genome editing.
Rights and permissions
About this article

Cite this article
Robinson-Hamm, J.N., Gersbach, C.A. Gene therapies that restore dystrophin expression for the treatment of Duchenne muscular dystrophy. Hum Genet 135, 1029–1040 (2016). https://doi.org/10.1007/s00439-016-1725-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-016-1725-z