
Abstract
Purpose of review
The purpose of this article is to review (1) the molecular mechanisms and hormonal factors involved in phosphate handling and regulation, (2) how to appropriately interpret serum and urine phosphate measurements in pediatric patients, (3) the pathophysiology of hypophosphatemic and hyperphosphatemic conditions, and (4) current strategies for treatment of hypophosphatemia and hyperphosphatemia in pediatric patients.
Recent findings
For decades, treatment of some hypophosphatemic conditions like X-linked hypophosphatemic rickets (XLH), characterized by a primary increase in fibroblast growth factor 23 (FGF23) activity, consisted of non-specific therapy with phosphate supplementation and calcitriol administration. However, in the past few years, burosumab, a targeted anti-FGF23 antibody, has been developed, representing a promising new medication for the treatment of pediatric XLH patients. The treatment of hyperphosphatemic conditions like chronic kidney disease (CKD) consists of dietary phosphate restriction and enteral phosphate binders; however, the development of new binders and inhibitors of cellular phosphate transporters may offer additional treatment options in the future.
Summary
The evaluation and treatment of disorders of phosphate balance in children is complex, as numerous interrelated mechanisms and hormones are involved in phosphate handling and regulation. Knowledge of the pathophysiology of hypophosphatemic and hyperphosphatemic conditions informs optimal diagnostic and treatment strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phosphate is critically important in many aspects of human physiology, including skeletal development, bone mineralization, membrane composition, nucleotide structure, and cellular signaling [1, 2]. Appropriate handling of phosphate is essential in growing children, as they require a positive phosphate balance to achieve optimal skeletal growth and mineralization, with osteomalacia and rickets-like phenotypes resulting from negative phosphate balances.
Phosphate handling and regulation
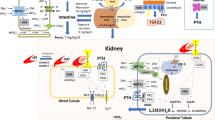
Phosphate homeostasis depends on the regulated partitioning of phosphate among multiple organ systems and fluid compartments, including the gastrointestinal system, kidneys, bone, extracellular fluid and serum, and intracellular fluid and soft tissue (Fig. 1). Phosphate regulation is a complex process involving many different transporters, enzymes, and hormones. Dietary phosphate, the recommended daily allowances of which vary by age (Table 1) [4], is absorbed from the gastrointestinal tract via both transcellular and paracellular mechanisms. Transcellular phosphate absorption is mediated by sodium phosphate cotransporter 2b (NaPi2b), the expression of which is upregulated by 1,25(OH)2 vitamin D3 (1,25D) and downregulated by high luminal phosphate concentrations [5•]. PiT-1 and PiT-2 are other sodium-dependent phosphate cotransporters that may contribute to active transcellular phosphate absorption and may be regulated by changes in dietary phosphate [5•]. Paracellular phosphate absorption is driven by transepithelial phosphate concentration gradients, and paracellular absorption increases as luminal phosphate concentrations increase [5•].

Overview of daily phosphate balance. In order to maintain phosphate balance, the amount of phosphate excreted by the kidneys is equal to the net amount of phosphate absorbed from the intestinal tract. Adapted from Bacchetta and Salusky, Am J Kidney Dis, 2012 [3]
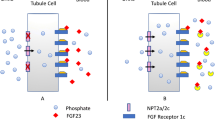
Circulating phosphate is freely filtered by the kidney. Under basal conditions, 80–90% of the phosphate filtered by the glomeruli into the urinary space is reabsorbed by the proximal tubules [6]. Transcellular phosphate absorption in the proximal tubule cells is mediated by apical sodium phosphate cotransporter 2a (NaPi2a), sodium phosphate cotransporter 2c (NaPi2c), and PiT-2 [7]. The main regulators of these proximal tubule transporters are fibroblast growth factor 23 (FGF23), parathyroid hormone (PTH), and phosphate concentrations. FGF23—a predominantly bone-derived hormone, PTH, and high phosphate levels all induce endocytosis and lysosomal degradation of NaPi2a, NaPi2c, and PiT-2, thus lessening proximal tubule phosphate reabsorption and increasing phosphaturia [7]. In addition to being phosphaturic hormones, FGF23 and PTH also affect vitamin D metabolism. FGF23 decreases expression of renal 1α-hydroxylase, the enzyme that converts nutritional 25OH vitamin D3 (25D) to active 1,25D, and increases expression of renal 24-hydroxylase, the enzyme that converts 25D and 1,25D to inactive metabolites, therefore overall decreasing renal 1,25D production [7]. Conversely, PTH increases expression of renal 1α-hydroxylase and decreases expression of renal 24-hydroxylase, thus overall increasing renal 1,25D production [7]. Whereas FGF23 increases phosphaturia and decreases 1,25D-mediated enteral phosphate absorption, PTH increases phosphaturia but enhances 1,25D-mediated enteral phosphate absorption; therefore, the overall phosphate-lowering effects of FGF23 may be more pronounced than those of PTH.
Phosphate measurement
Measurement of phosphate concentrations in the serum and urine can provide information regarding whether the body is appropriately or inappropriately handling and regulating phosphate. Phosphate concentrations may be measured in mg/dl (conventional units) or in mmol/l (SI units); the conversion formula is mg/dl × 0.323 = mmol/l. Of note, 85% of total body phosphate is contained in the bones and teeth, 14% is in the soft tissues, and only 1% is present in the extracellular fluid; therefore, serum phosphate concentrations—a small fraction of overall phosphate—may not always reflect total body phosphate stores [7].
When interpreting serum phosphate concentrations, it is crucial to recognize that normal ranges of serum phosphate vary by age (Table 2) [4]. Importantly, a serum phosphate concentration that is within the normal range for an adult or adolescent would be consistent with hypophosphatemia in an infant or young child. Additionally, serum phosphate concentrations naturally exhibit diurnal variation, nadiring in the late morning (10a–11a), then increasing to a first peak in the late afternoon (4p–5p), then slightly decreasing before rising again to a second, higher peak in the early morning hours (1a–3a), before steadily decreasing again to the late-morning nadir [8,9,10]. During daytime hours, serum phosphate concentrations can vary considerably, with the difference between the late morning nadir and the late afternoon peak being as much as 1 mg/dl [11]. Therefore, when interpreting serum phosphate concentrations, or comparing serum phosphate concentrations measured on different days, the time of day at which the measurements were made must be considered.
Therefore, appropriate interpretation of serum phosphate concentrations requires awareness of the normal age-related ranges and diurnal variation patterns. Additionally, other factors may artifactually affect serum phosphate concentrations. Hemolyzed samples may have some degree of increased phosphate [12, 13]. Spurious hyperphosphatemia may also be observed in samples contaminated with heparin [14] or tissue plasminogen activator [15, 16] (i.e., drawn from a central venous catheter). Although potential post-prandial changes in serum phosphate may be modest [17], measurement of fasting serum phosphate is likely optimal.
To assess kidney phosphate handing, the fractional tubular reabsorption of phosphate (TRP) can be calculated. TRP calculation requires concurrent measurement of serum phosphate, serum creatinine, urine phosphate, and urine creatinine. TRP is calculated as follows:
A normal TRP is considered to be > 0.85 (corresponding to > 85%) [18]; however, TRP can only be interpreted in the context of the serum phosphate concentration. In general, in the setting of hyperphosphatemia, the TRP should be decreased, and in the setting of hypophosphatemia, the TRP should be increased, consistent with appropriate renal compensation. In the setting of hypophosphatemia, an inappropriately low TRP may be consistent with pathologic renal phosphate wasting.
Renal phosphate handling can be further assessed by calculating the ratio of the tubular maximum reabsorption rate of phosphate to the glomerular filtration rate (TmP/GFR). This parameter reflects the maximum renal tubular phosphate reabsorption in mass per unit volume of glomerular filtrate, and corresponds to the theoretical lower limit of serum phosphate below which all filtered phosphate would be reabsorbed [19]. Calculation of TmP/GFR is as follows:
If the TRP is ≤ 0.86, then there is a linear relationship between the serum phosphate concentration and the urinary phosphate excretion rate [19], and TmP/GFR is defined as
If the TRP is > 0.86, then there is a curvilinear relationship between the serum phosphate concentration and the urinary phosphate excretion rate [19], and TmP/GFR is defined as
If serum phosphate is measured in mg/dl, then TmP/GFR is expressed in mg/dl of glomerular filtrate. Some age-related reference ranges for TmP/GFR have been published as follows: neonates (4.4 to 10.6 mg/dl), 3-month-old infants (4.6 to 10.2 mg/dl), 6-month-old infants (3.6 to 8.0 mg/dl), children and adolescents aged 2–15 years (3.6 to 7.6 mg/dl), and adults (2.5 to 4.2 mg/dl) [19]. Similar to TRP, in the setting of hypophosphatemia, a low TmP/GFR is consistent with inappropriate renal phosphate wasting, whereas a normal (or high) TmP/GFR is consistent with appropriate renal phosphate conservation and a non-renal etiology of the hypophosphatemia. In the setting of hyperphosphatemia with normal kidney function, a high TmP/GFR may be consistent with insufficient renal phosphate excretion.
Hypophosphatemic conditions
As shown in Table 3 and detailed below, hypophosphatemia can result from decreased intestinal phosphate absorption, increased renal phosphate excretion, increased FGF23 activity (resulting in both increased phosphaturia and decreased 1,25D production), or shifts of extracellular phosphate into cells. Based on this differential diagnosis, the initial workup of hypophosphatemia includes calculation of the TRP and TmP/GFR, and may include measurement of serum 25D, 1,25D, PTH, and/or calcium. Circulating FGF23 concentrations are not routinely measured in most clinical laboratories; however, it may be ordered on a send-out basis (Mayo Clinical Laboratories). Depending on the FGF23 assay used, plasma, instead of serum, may be required.
Decreased intestinal phosphate absorption
As 1,25D can regulate intestinal phosphate absorption [5•], conditions that decrease the concentration or activity of 1,25D may result in impaired enteral phosphate absorption and hypophosphatemia. The substrate for 1,25D production is 25D, so deficiency of nutritional 25D can result in reduced 1,25D concentrations. As suggested by the American Academy of Pediatrics, nutritional 25D deficiency is defined as a serum 25D concentration of less than 20 ng/ml [20]. The serum concentration of 25D that defines a state of sufficiency remains controversial. The differential diagnosis of 25D deficiency includes limited sunlight exposure, limited 25D intake (e.g., exclusively breastfed infants in the absence of vitamin supplementation), conditions resulting in gastrointestinal malabsorption of 25D (e.g., cystic fibrosis, Crohn’s disease, celiac disease), and increased catabolism of 25D (e.g., chronic use of antiepileptic drugs) [20].
Rare genetic conditions, called vitamin D-dependent rickets, can present with decreased 1,25D activity in the setting of sufficient 25D substrate. Vitamin D-dependent rickets, type 1 (1α-hydroxylase deficiency) is characterized by impaired 1α-hydroxylase conversion of 25D to 1,25D, resulting in normal 25D levels, low 1,25D concentrations, and high PTH (induced by decreased 1,25D activity and/or hypocalcemia) [21]. Vitamin D-dependent rickets, type 2 (vitamin D receptor mutation) is characterized by sufficient 25D to 1,25D conversion but impaired 1,25D binding to the vitamin D receptor, resulting in normal 25D levels, high 1,25D concentrations, and high PTH (induced by decreased 1,25D activity and/or hypocalcemia) [21]. Interestingly, many children with vitamin D-dependent rickets, type 2, have some degree of alopecia (suggesting a role for the vitamin D receptor in the regulation of hair growth) [21].
Lastly, the chronic use of medications that bind phosphate in the intestinal lumen can decrease the amount of phosphate available for enteral absorption. Notably, antacids that contain aluminum, calcium, or magnesium can bind enteral phosphate, and the chronic use of such medications, especially in the setting of a low phosphate diet, may contribute to lower serum phosphate concentrations [22].
Increased renal phosphate excretion
At baseline, the vast majority (80–90%) of phosphate filtered by the glomeruli into the urinary space is reabsorbed by proximal tubule cells [6], mediated by luminal phosphate transporters NaPi2a, NaPi2c, and PiT-2 [7]. Therefore, abnormalities in renal proximal tubule cells or the specific phosphate transporters may result in excessive phosphaturia and hypophosphatemia.
Generalized proximal tubular dysfunction, referred to as Fanconi syndrome, may result in excessive phosphaturia. Etiologies of Fanconi syndrome include congenital diseases, such as cystinosis, tyrosinemia, galactosemia, Dent disease, Wilson disease, and Lowe syndrome, and acquired causes, such as administration of ifosfamide, cisplatin, or aminoglycosides [23]. As Fanconi syndrome is a generalized proximal tubulopathy, the excessive phosphaturia may also be accompanied by decreased bicarbonate reabsorption (proximal (type 2) renal tubular acidosis), increased glucosuria, and/or low molecular weight (tubular) proteinuria.
Rare loss of function mutations in the specific proximal tubule cell phosphate transporters can result in renal phosphate wasting and hypophosphatemia. Mutations in the gene encoding NaPi2c lead to a disease entity known as hereditary hypophosphatemic rickets with hypercalciuria (HHRH). In HHRH, hypophosphatemia stimulates 1α-hydroxylase activity, appropriately increasing 1,25D production [24]. Elevated 1,25D levels increase intestinal calcium absorption and decrease PTH-mediated calcium reabsorption in the renal distal tubules, resulting in hypercalciuria, which increases the risk of kidney stones and nephrocalcinosis [24]. Mutations in the gene encoding NaPi2a have been less frequently described. There are a few case reports describing hypophosphatemic patients with mutations in the genes encoding NaPi2a and the sodium hydrogen exchanger regulatory factor 1 (NHERF1) [6]; NHERF1 is involved in the transcription and proper intracellular trafficking of NaPi2a [6].
As PTH downregulates proximal tubule cell phosphate transporters, hyperparathyroidism can result in decreased phosphate reabsorption and excessive phosphaturia. PTH also increases 1,25D production, promoting enteral phosphate absorption. If PTH-induced increases in 1,25D-mediated enteral phosphate absorption do not fully compensate for PTH-induced phosphaturia, then hypophosphatemia may result.
Increased fibroblast growth factor 23 (FGF23) activity
First characterized in 2000 [25], FGF23 is a predominantly bone-derived hormone that both downregulates proximal tubule cell phosphate transporters, inducing phosphaturia, and decreases 1,25D production, reducing enteral phosphate absorption. Therefore, in the setting of normal kidney function, excessive FGF23 activity can result in hypophosphatemia. Regulation of FGF23 production is complex, involving several different enzymes. Inactivating mutations in some of these enzymes can result in bone overproduction of FGF23, leading to early-onset hypophosphatemic rickets. Similarly, activating mutations of FGF23 itself can also cause early-onset hypophosphatemic rickets.
X-linked hypophosphatemic (XLH) rickets is the most common inherited form of rickets, and is caused by inactivating mutations in the gene encoding PHEX (phosphate regulating gene with homologies to endopeptidases on the X chromosome) [6]. PHEX deficiency results in upregulation of FGF23 expression and increased circulating FGF23 concentrations. Increased FGF23 activity causes renal phosphate wasting and inappropriately low/normal 1,25D levels. Autosomal dominant hypophosphatemic rickets (ADHR) is a rare disorder characterized by mutations in the gene encoding FGF23 itself that render the FGF23 protein more resistant to proteolytic cleavage [6]. These activating mutations result in increased FGF23 activity, excessive phosphaturia, and inappropriately low/normal 1,25D levels. Autosomal recessive hypophosphatemic rickets (ARHR), type 1 and type 2, are rare disorders characterized by mutations in the genes encoding dentin matrix protein 1 (DMP1) or ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), respectively [6]. Similar to XLH, these mutations can result in increased bone FGF23 production.
Other conditions can also result in increased FGF23 activity. Tumor-induced osteomalacia (TIO) is an acquired, paraneoplastic syndrome of renal phosphate wasting [1]. Most of the tumors associated with TIO are phosphaturic mesenchymal tumors, mixed connective tissue type (PMTMCT), occurring in soft tissue and bone, and ectopically expressing and secreting FGF23 [1]. These tumors are often difficult to detect as they are typically small, slow-growing, and found in a wide variety of anatomic locations [1]. Fibrous dysplasia/McCune-Albright syndrome is a rare, complex condition caused by gain-of-function mutations in the gene encoding the stimulatory alpha subunit of the G protein complex (GNAS) [26]. In this disorder, dysplastic bone is the source of FGF23 overproduction, with more severe bone disease associated with higher degrees of FGF23 overproduction [26]. Lastly, similar to the stabilizing FGF23 mutations of ADHR, certain intravenous iron preparations, including ferric carboxymaltose [27], iron polymaltose [28], and saccharated iron oxide [29], inhibit FGF23 proteolytic cleavage, resulting in increased FGF23 activity and hypophosphatemia [30].
Shifts of extracellular phosphate into cells
Besides decreased intestinal phosphate absorption, increased renal phosphate excretion, and/or increased FGF23 activity, shifts of extracellular phosphate into cells can lower serum phosphate concentrations. Increased insulin levels promote the transport of phosphate into skeletal muscle and liver [22]. Specifically, in refeeding syndrome, the insulin surge can precipitate acute hypophosphatemia [31]. Hungry bone syndrome is a condition characterized by marked uptake of calcium and phosphate into bones post-parathyroidectomy in hyperparathyroid patients [32]. Acute respiratory alkalosis and excessive cellular phosphate consumption by tumor cells can also decrease serum phosphate levels.
Hypophosphatemia treatments
Treatment of hypophosphatemia depends on the underlying cause, as reversal of the underlying etiology may improve serum phosphate concentrations. For instance, treatment of 25D deficiency-induced hypophosphatemia with cholecalciferol or ergocalciferol supplementation can increase phosphate levels. However, in other hypophosphatemic conditions, such as those characterized by renal phosphate wasting, phosphate supplementation and/or administration of calcitriol (exogenous active 1,25D) may be required to increase serum phosphate concentrations. Although treatment with phosphate supplementation and calcitriol can be effective in alleviating hypophosphatemia, medication-associated complications can occur. Specifically, calcitriol can increase serum calcium concentrations, resulting in hypercalcemia and hypercalciuria. Increased urinary calcium can complex with urinary phosphate, engendering renal tubular calcifications and nephrocalcinosis. Additionally, phosphate supplements can complex with calcium, precipitating intermittent hypocalcemia that stimulates PTH release despite concurrent calcitriol-induced PTH suppression. This secondary hyperparathyroidism can worsen phosphaturia. Therefore, when treating with phosphate supplementation and/or calcitriol, in addition to assessing the effects on serum and urine phosphate levels, close monitoring of serum calcium concentrations, the urine calcium/creatinine ratio, and serum PTH is warranted.
For the past several decades, treatment of conditions like XLH has consisted of conventional therapy of phosphate supplementation and calcitriol. Although conventional therapy may improve serum phosphate concentrations, these treatments can have adverse effects, may require administration of oral phosphate supplementation up to five times daily, and may not fully prevent the lower limb deformities and/or suboptimal linear growth associated with XLH. However, in the past few years, a promising new treatment for XLH has emerged—burosumab, a neutralizing antibody to FGF23. As XLH is characterized by a primary increase in FGF23 activity, directly targeting FGF23 should ameliorate XLH-associated symptoms and morbidity. Burosumab is injected subcutaneously every 2 weeks.
Since 2018, four studies have been published presenting clinical data on the efficacy and safety of burosumab in children with XLH [33•]. In one case series and two open-label phase 2 trials of burosumab treatment in pediatric XLH patients (as young as 1-year-old), burosumab increased renal tubular phosphate reabsorption and serum phosphate concentrations, improved linear growth and physical functioning, and decreased bone pain and rickets severity [33•]. In 2019, Imel et al. published a randomized, active-controlled, open-label, phase 3 trial of burosumab vs. conventional therapy (oral phosphate and calcitriol) in 61 children with XLH aged 1–12 years, with a primary endpoint of change in rickets severity at week 40 [34•]. Compared with patients randomized to remain on conventional therapy, patients randomized to switch from conventional therapy to burosumab had significantly greater improvement in rickets severity, and also had greater increases in TmP/GFR, serum phosphate, serum 1,25D, and linear growth [34•]. Based on the clinical trial data presented to date, a consensus statement was published in 2019, recommending the consideration of burosumab in children at least 1-year-old with XLH, and in adolescent XLH patients with overt bone disease refractory to conventional therapy, complications related to conventional therapy, and/or inability to adhere to conventional therapy [35]. Although the pediatric burosumab studies did not observe excessive FGF23 suppression resulting in hyperphosphatemia [33•], close monitoring of serum phosphate concentrations is warranted to inform appropriate dosing adjustments.
Hyperphosphatemic conditions
As shown in Table 4 and detailed below, hyperphosphatemia can result from acute phosphate loading, decreased renal phosphate excretion, decreased FGF23 activity (resulting in both decreased phosphaturia and increased 1,25D production), or shifts of intracellular phosphate out of cells.
Acute phosphate loading
Administration of an acute exogenous phosphate load over several hours can overwhelm the renal capacity for phosphate excretion, increasing serum phosphate concentrations. Such exogenous sources of phosphate include oral sodium phosphate laxatives, sodium phosphate-containing enemas, and intravenous phosphate. Given the potential toxicity associated with these agents [36, 37], care must be taken when prescribing phosphate-containing laxatives and enemas [38]. Specifically, the administration of phosphate-containing enemas is contraindicated in patients with known or suspected gastrointestinal obstruction, and in children younger than 2 years of age [38]. Also, appropriate dosing is paramount, as overdose and excessively frequent dosing can both result in critical toxicity [38].
Decreased renal phosphate excretion
Kidney disease can impair renal phosphate excretion, resulting in hyperphosphatemia. Acute or chronic decreases in glomerular filtration rate can decrease the filtration and urinary excretion of phosphate. However, at least initially, normophosphatemia can be maintained as increased secretion of the phosphaturic hormones FGF23 and PTH will decrease proximal tubular phosphate reabsorption, thus increasing per nephron phosphate excretion. Yet, when the glomerular filtration rate falls below 20 to 25 mL/min, renal phosphate reabsorption is thought to be maximally suppressed. At this point, urinary phosphate excretion may no longer be able to completely offset phosphate intake, resulting in hyperphosphatemia.
As PTH downregulates proximal tubule cell phosphate transporters, hypoparathyroidism can result in increased phosphate reabsorption and insufficient phosphaturia. Hypoparathyroidism can also result in decreased 1,25D production; however, if decreased 1,25D-mediated enteral phosphate absorption does not fully offset the increased renal phosphate reabsorption, then hyperphosphatemia may result.
Decreased fibroblast growth factor 23 (FGF23) activity
Decreased FGF23 activity results in decreased phosphaturia and increased 1,25D production, promoting hyperphosphatemia. Hyperphosphatemic familial tumoral calcinosis (HFTC) is a rare, heterogenous condition characterized by decreased FGF23 activity, hyperphosphatemia, and resultant ectopic soft tissue and vascular calcifications. In HFTC, FGF23 deficiency or resistance results from (1) inactivating mutations in the gene encoding FGF23, (2) inactivating mutations in the gene encoding N-acetylgalactosaminyltransferase 3 (GALNT3), an enzyme that protects FGF23 from proteolytic cleavage, or (3) inactivating mutations in the gene encoding α-Klotho, a critical co-receptor for FGF23 signaling [39, 40].
Shifts of intracellular phosphate out of cells
Shifts of intracellular phosphate out of cells can increase serum phosphate concentrations. In patients with large tumor burdens characterized by rapid cell turnover (lymphomas and some leukemias), release of intracellular phosphate into the circulation (tumor lysis syndrome) may occur with the initiation of cytotoxic therapy, or may occur spontaneously [41]. In rhabdomyolysis, intracellular phosphate is released from damaged cells. Acidosis can also promote a transcellular shift of phosphate out of cells.
Hyperphosphatemia treatments
Treatment of hyperphosphatemia should address the underlying etiology. In pediatric patients with chronic kidney disease (CKD), it is recommended that elevated serum phosphate levels be lowered toward the normal range; doing so often requires concurrent dietary phosphate restriction and enteral phosphate binders [42]. In pediatric CKD patients with hyperphosphatemia, it is suggested that dietary phosphate intake be reduced to 80% of the age-specific daily adequate intake (AI) for infants, or 80% of the recommended dietary allowances (RDA) for older children [42]. However, adherence to a phosphate-restricted diet is difficult and, as protein sources contain high amounts of phosphate, may limit adequate protein intake.
Therefore, most pediatric CKD patients with hyperphosphatemia also require enteral phosphate-binding agents. Enteral binders taken with meals complex with phosphate in the intestinal tract, thus limiting phosphate absorption by inhibiting passive paracellular diffusion [42]. Several phosphate binders are available for clinical use, and the choice of which binder to use should take into account adverse effect profiles, comorbidities, and concomitant medications. Calcium-based phosphate binders, such as calcium carbonate and calcium acetate, are effective in lowering serum phosphate concentrations, but may contribute to hypercalcemia and extraskeletal calcification. In children with CKD, it is suggested that the total dose of elemental calcium provided by calcium-based phosphate binders and by dietary calcium not exceed twice the age-specific AI or RDA for calcium [42].
Non-calcium-based enteral phosphate binders are also available. One of the most commonly used non-calcium-based binders is sevelamer, a phosphate-binding resin that is formulated as sevelamer hydrochloride and sevelamer carbonate. Other non-calcium-based phosphate binders include lanthanum, aluminum, and iron salts. Although effective in lowering serum phosphate concentrations, lanthanum- and aluminum-based binders are not recommended for use in children, as lanthanum may pathologically accumulate in the bone and liver, and aluminum retention may cause neurotoxicity and impaired bone mineralization [42]. Recently, iron-based phosphate binders, such as ferric citrate, have been shown to be effective in both controlling serum phosphate levels and improving iron status [43], but their safety and efficacy in pediatric CKD patients have not been systemically evaluated [42, 44].
Whereas enteral phosphate binders limit paracellular phosphate absorption, other medications are in development that inhibit active transcellular NaPi2b-mediated intestinal phosphate absorption [45]. Nicotinamide, the amide form of vitamin B3, inhibits NaPi2b expression and has been shown to be effective in lowering serum phosphate concentrations in adult CKD patients [45]. However, the safety and efficacy of nicotinamide use in pediatric CKD patients have not been systemically evaluated [45]. Interestingly, a recent pre-clinical study assessed the effects of a NaPi2a inhibitor in mice with normal and impaired kidney function, observing dose-dependent increases in urinary phosphate excretion and decreases in serum phosphate concentrations [46].
Summary
The evaluation and treatment of disorders of phosphate balance in children is complex, requiring a nuanced understanding of the interrelated mechanisms and hormones involved in phosphate handling and regulation. Hypophosphatemia may result from decreased intestinal phosphate absorption, increased renal phosphate excretion, increased FGF23 activity, and/or shifts of extracellular phosphate into cells. Hyperphosphatemia may result from acute phosphate loading, decreased renal phosphate excretion, decreased FGF23 activity, and/or shifts of intracellular phosphate out of cells. Whereas treatment for some hypophosphatemic conditions like XLH historically relied on phosphate supplementation and calcitriol administration, the advent of burosumab, an anti-FGF23 antibody, represents a promising new therapeutic direction. The treatment of hyperphosphatemic conditions like CKD consists of dietary phosphate restriction and enteral phosphate binders; however, the development of new binders and sodium phosphate cotransporter inhibitors may offer additional treatment options in the future.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance
Ruppe MD. Jan de Beur SM. Disorders of phosphate homeostasis. In: Bilezikian JP, editor. Primer on the metabolic bone diseases and disorders of mineral metabolism. 9th ed. American Society for Bone and Mineral Research: Hoboken, NJ; 2019. p. 674–83.
Goretti Penido M, Alon US. Phosphate homeostasis and its role in bone health. Pediatr Nephrol. 2012;27(11):2039–48. https://doi.org/10.1007/s00467-012-2175-z.
Bacchetta J, Salusky IB. Evaluation of hypophosphatemia: lessons from patients with genetic disorders. Am J Kidney Dis. 2012;59(1):152–9. https://doi.org/10.1053/j.ajkd.2011.08.035.
K/DOQI clinical practice guidelines for nutrition in children with chronic kidney disease. Am J Kidney Dis. 2009;53:S1–123.
• Hernando N, Wagner CA. Mechanisms and regulation of intestinal phosphate absorption. Compr Physiol. 2018;8(3):1065–90. https://doi.org/10.1002/cphy.c170024. This review provides a comprehensive summary of the mechanisms underlying phosphate transport and regulation.
Gattineni J, Baum M. Genetic disorders of phosphate regulation. Pediatr Nephrol. 2012;27(9):1477–87. https://doi.org/10.1007/s00467-012-2103-2.
Hernando N, Gagnon KB, Lederer ED. Phosphate transport in epithelial and nonepithelial tissue. Physiol Rev. 2020. https://doi.org/10.1152/physrev.00008.2019.
Jubiz W, Canterbury JM, Reiss E, Tyler FH. Circadian rhythm in serum parathyroid hormone concentration in human subjects: correlation with serum calcium, phosphate, albumin, and growth hormone levels. J Clin Invest. 1972;51(8):2040–6. https://doi.org/10.1172/jci107010.
Portale AA, Halloran BP, Morris RC Jr. Dietary intake of phosphorus modulates the circadian rhythm in serum concentration of phosphorus. Implications for the renal production of 1,25-dihydroxyvitamin D. J Clin Invest. 1987;80(4):1147–54. https://doi.org/10.1172/jci113172.
Isakova T, Xie H, Barchi-Chung A, Smith K, Sowden N, Epstein M, et al. Daily variability in mineral metabolites in CKD and effects of dietary calcium and calcitriol. Clin J Am Soc Nephrol. 2012;7(5):820–8. https://doi.org/10.2215/cjn.11721111.
Becker GJ, Walker RG, Hewitson TD, Pedagogos E. Phosphate levels--time for a rethink? Nephrol Dial Transplant. 2009;24(8):2321–4. https://doi.org/10.1093/ndt/gfp220.
Lippi G, Salvagno GL, Montagnana M, Brocco G, Guidi GC. Influence of hemolysis on routine clinical chemistry testing. Clin Chem Lab Med. 2006;44(3):311–6. https://doi.org/10.1515/cclm.2006.054.
Perović A, Dolčić M. Influence of hemolysis on clinical chemistry parameters determined with Beckman Coulter tests - detection of clinically significant interference. Scand J Clin Lab Invest. 2019;79(3):154–9. https://doi.org/10.1080/00365513.2019.1576099.
Ball CL, Tobler K, Ross BC, Connors MR, Lyon ME. Spurious hyperphosphatemia due to sample contamination with heparinized saline from an indwelling catheter. Clin Chem Lab Med. 2004;42(1):107–8. https://doi.org/10.1515/cclm.2004.021.
Cachat F, Bardy D, Durussel C, Di Paolo E. Spurious hyperphosphatemia in a patient with alteplase-locked central venous catheter. Pediatr Nephrol. 2006;21(2):301–2. https://doi.org/10.1007/s00467-005-2088-1.
Schiller B, Virk B, Blair M, Wong A, Moran J. Spurious hyperphosphatemia in patients on hemodialysis with catheters. Am J Kidney Dis. 2008;52(3):617–20. https://doi.org/10.1053/j.ajkd.2008.03.033.
Isakova T, Gutierrez O, Shah A, Castaldo L, Holmes J, Lee H, et al. Postprandial mineral metabolism and secondary hyperparathyroidism in early CKD. J Am Soc Nephrol. 2008;19(3):615–23. https://doi.org/10.1681/asn.2007060673.
Slatopolsky E, Bricker NS. The role of phosphorus restriction in the prevention of secondary hyperparathyroidism in chronic renal disease. Kidney Int. 1973;4(2):141–5. https://doi.org/10.1038/ki.1973.92.
Payne RB. Renal tubular reabsorption of phosphate (TmP/GFR): indications and interpretation. Ann Clin Biochem. 1998;35(Pt 2):201–6. https://doi.org/10.1177/000456329803500203.
Diab L, Krebs NF. Vitamin excess and deficiency. Pediatr Rev. 2018;39(4):161–79. https://doi.org/10.1542/pir.2016-0068.
Malloy PJ, Feldman D. Genetic disorders and defects in vitamin d action. Endocrinol Metab Clin North Am. 2010;39(2):333–46, table of contents. https://doi.org/10.1016/j.ecl.2010.02.004.
Liamis G, Milionis HJ, Elisaf M. Medication-induced hypophosphatemia: a review. Qjm. 2010;103(7):449–59. https://doi.org/10.1093/qjmed/hcq039.
Foreman JW. Fanconi syndrome. Pediatr Clin N Am. 2019;66(1):159–67. https://doi.org/10.1016/j.pcl.2018.09.002.
Bergwitz C, Miyamoto KI. Hereditary hypophosphatemic rickets with hypercalciuria: pathophysiology, clinical presentation, diagnosis and therapy. Pflugers Arch. 2019;471(1):149–63. https://doi.org/10.1007/s00424-018-2184-2.
Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26(3):345–8. https://doi.org/10.1038/81664.
Boyce AM, Collins MT. Fibrous dysplasia/McCune-Albright syndrome: a rare, mosaic disease of Gα s activation. Endocr Rev. 2020;41(2):345–70. https://doi.org/10.1210/endrev/bnz011.
Wolf M, Chertow GM, Macdougall IC, Kaper R, Krop J, Strauss W. Randomized trial of intravenous iron-induced hypophosphatemia. JCI Insight. 2018;3(23). https://doi.org/10.1172/jci.insight.124486.
Schouten BJ, Hunt PJ, Livesey JH, Frampton CM, Soule SG. FGF23 elevation and hypophosphatemia after intravenous iron polymaltose: a prospective study. J Clin Endocrinol Metab. 2009;94(7):2332–7. https://doi.org/10.1210/jc.2008-2396.
Yamamoto S, Okada Y, Mori H, Fukumoto S, Tanaka Y. Fibroblast growth factor 23-related osteomalacia caused by the prolonged administration of saccharated ferric oxide. Intern Med. 2012;51(17):2375–8. https://doi.org/10.2169/internalmedicine.51.7450.
Zoller H, Schaefer B, Glodny B. Iron-induced hypophosphatemia: an emerging complication. Curr Opin Nephrol Hypertens. 2017;26(4):266–75. https://doi.org/10.1097/mnh.0000000000000329.
Marinella MA. Refeeding syndrome and hypophosphatemia. J Intensive Care Med. 2005;20(3):155–9. https://doi.org/10.1177/0885066605275326.
Jain N, Reilly RF. Hungry bone syndrome. Curr Opin Nephrol Hypertens. 2017;26(4):250–5. https://doi.org/10.1097/mnh.0000000000000327.
• Schindeler A, Biggin A, Munns CF. Clinical evidence for the benefits of Burosumab therapy for X-linked hypophosphatemia (XLH) and other conditions in adults and children. Front Endocrinol (Lausanne). 2020;11:338. https://doi.org/10.3389/fendo.2020.00338. This review summarizes the clinical trial data to date regarding burosumab treatment in adults and children with XLH.
• Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O, et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet. 2019;393(10189):2416–27. https://doi.org/10.1016/s0140-6736(19)30654-3. This study is a randomized, active-controlled, open-label, phase 3 trial of burosumab vs. conventional therapy (oral phosphate and calcitriol) in 61 children with XLH aged 1-12 years, with a primary endpoint of change in rickets severity.
Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019;15(7):435–55. https://doi.org/10.1038/s41581-019-0152-5.
Ladenhauf HN, Stundner O, Spreitzhofer F, Deluggi S. Severe hyperphosphatemia after administration of sodium-phosphate containing laxatives in children: case series and systematic review of literature. Pediatr Surg Int. 2012;28(8):805–14. https://doi.org/10.1007/s00383-012-3124-4.
Becknell B, Smoyer WE, O'Brien NF. Hemodialysis for near-fatal sodium phosphate toxicity in a child receiving sodium phosphate enemas. Pediatr Emerg Care. 2014;30(11):814–7. https://doi.org/10.1097/pec.0000000000000267.
McNamara S, Galt DJ. Regarding near-fatal sodium phosphate toxicity in a child receiving sodium phosphate enemas. Pediatr Emerg Care. 2015;31(8):e7. https://doi.org/10.1097/pec.0000000000000523.
Farrow EG, Imel EA, White KE. Miscellaneous non-inflammatory musculoskeletal conditions. Hyperphosphatemic familial tumoral calcinosis (FGF23, GALNT3 and αKlotho). Best Pract Res Clin Rheumatol. 2011;25(5):735–47. https://doi.org/10.1016/j.berh.2011.10.020.
Boyce AM, Lee AE, Roszko KL, Gafni RI. Hyperphosphatemic tumoral calcinosis: pathogenesis, clinical presentation, and challenges in management. Front Endocrinol (Lausanne). 2020;11:293. https://doi.org/10.3389/fendo.2020.00293.
Cheung WL, Hon KL, Fung CM, Leung AK. Tumor lysis syndrome in childhood malignancies. Drugs Context. 2020;9:1–14. https://doi.org/10.7573/dic.2019-8-2.
Hanudel MR, Moe SM, Salusky IB. Pathophysiology and treatment of chronic kidney disease–mineral and bone disorder. In: Bilezikian JP, editor. Primer on the metabolic bone diseases and disorders of mineral metabolism. 9th ed. Hoboken, NJ: American Society for Bone and Mineral Research; 2019. p. 695–704.
Nakanishi T, Hasuike Y, Nanami M, Yahiro M, Kuragano T. Novel iron-containing phosphate binders and anemia treatment in CKD: oral iron intake revisited. Nephrol Dial Transplant. 2016;31(10):1588–94. https://doi.org/10.1093/ndt/gfv268.
Hanudel MR, Laster M, Ramos G, Gales B, Salusky IB. Clinical experience with the use of ferric citrate as a phosphate binder in pediatric dialysis patients. Pediatr Nephrol. 2018;33(11):2137–42. https://doi.org/10.1007/s00467-018-3999-y.
Fouque D, Vervloet M, Ketteler M. Targeting gastrointestinal transport proteins to control hyperphosphatemia in chronic kidney disease. Drugs. 2018;78(12):1171–86. https://doi.org/10.1007/s40265-018-0950-2.
Thomas L, Xue J, Murali SK, Fenton RA, Dominguez Rieg JA, Rieg T. Pharmacological Npt2a inhibition causes phosphaturia and reduces plasma phosphate in mice with normal and reduced kidney function. J Am Soc Nephrol. 2019;30(11):2128–39. https://doi.org/10.1681/asn.2018121250.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pediatric Nephrology
Rights and permissions
About this article

Cite this article
Hanudel, M.R. Evaluation and Treatment of Disorders of Phosphate Balance. Curr Treat Options Peds 6, 227–240 (2020). https://doi.org/10.1007/s40746-020-00208-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40746-020-00208-1