
Abstract
Purpose of Review
Over the past years, the ketogenic diet (KD) has been increasingly used as emergency therapy for refractory and super-refractory status epilepticus (RSE/SRSE). The aim of this review was to evaluate the role of the KD in pediatric patients with RSE with a special focus on different types of status epilepticus.
Recent Findings
Over the past decade, several studies have been published on the use of the KD in children with RSE/SRSE. Based on our previous studies on dietary therapy in myoclonic status epilepticus and status epilepticus in epileptic encephalopathies, we consider the KD may work well in particular types of status epilepticus, especially those occurring in the epileptic encephalopathies.
Summary
When patients with RSE/SRSE do not respond to benzodiazepines, IV antiepileptic drugs, and anesthetics, the KD may be an effective and tolerable option to be considered earlier in the treatment algorithm. The development of a consensus may optimize the use of this treatment in patients with epilepsy in critical care. There is a need for randomized controlled trials to confirm what children would be the best candidates for the use of the KD as well as the time of dietary therapy initiation for the treatment of RSE/SRSE.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
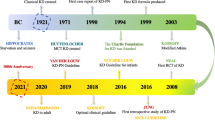
The ketogenic diet (KD) is a high-fat, low-carbohydrate diet, adequate-protein diet that has been used since 1921 for the nonpharmacological treatment of refractory epilepsy [1,2,3]. In spite of a decline in the use of the diet due to the introduction of new antiepileptic drugs, since the 1990s, there has been renewed interest in the KD, as epilepsy becomes drug resistant in approximately one third of children. The KD has been shown to work particularly well in certain epilepsy syndromes, especially the epileptic encephalopathies [4, 5•]. Considerable evidence and consensus guidelines are available for the use of the KD in the treatment of epilepsy [6••]. Over the past years, the diet has been increasingly used as emergency therapy for refractory status epilepticus in children and adults [7,8,9].
Status epilepticus (SE) is commonly defined as a seizure activity of 5 min or more or recurrent seizure activity without a return to baseline between seizures. In the treatment algorithm for SE, the first therapy of choice is benzodiazepines. If the seizures do not respond to therapy within 30 min, treatment with intravenous anti-epileptic drugs, such as phenytoin, phenobarbital, or valproate, is started. If the patient does not respond to this treatment after 2 h, the patient is considered to have refractory SE (RSE). In patients with RSE, intravenous anesthetic agents are indicated [10].
If the RSE returns after the reduction or discontinuation of anesthetics, the patients are diagnosed with super-refractory SE (SRSE). Of all patients with SE admitted to hospital, around 15% develop SRSE [10].
RSE and SRSE are associated with high rates of morbidity and mortality [11]. Additionally, long-term treatment with intravenous anesthetic drugs has been associated with negative outcomes contributing to the high morbidity and mortality rate [12•].
Currently, there is no consensus on how to care for patients with RSE who do not respond to aggressive treatment, as no systematic studies have been conducted, and management mainly depends on expert experience.
Since the first study by Bodenant [13] in an adult patient with partial RSE who had a favorable outcome 7 days after introduction of the KD in association with antiepileptic drugs, the KD is increasingly used for emergency situations in RSE and SRSE.
The aim of this review was to discuss the role of the KD in pediatric patients with RSE with a special focus on different types of status epilepticus.
Different forms of RSE and the KD
SE are categorized into different forms: (1) convulsive status epilepticus associated with generalized tonic-clonic seizures; (2) nonconvulsive status epilepticus (NCSE), a state of ongoing going seizure activity for at least 30 min, with cognitive or behavioral changes, but without convulsive clinical manifestations, diagnosed based on ictal EEG abnormalities and clinical features; and (3) repeated partial seizures manifested as focal motor signs, focal sensory symptoms, or focal impairment of function (e.g., aphasia) not associated with altered awareness (epilepsia partialis continua) [14].
Etiologies may be divided into five categories: immunological disorders, mitochondrial disorders, uncommon infectious diseases, drugs or toxins, and rare genetic diseases [10]. Many cases occur de novo, without a previous history of epilepsy. Such cases have been considered to have new-onset refractory status epilepticus (NORSE), presenting with RSE without a clear acute or active structural, toxic, or metabolic cause [15•]. Febrile infection-related epilepsy syndrome is a subcategory of NORSE that requires a prior febrile infection starting between 2 weeks and 24 h prior to onset of refractory status epilepticus, with or without fever at onset of status epilepticus [16••].
It is important to consider that there are as many types of SE as there are types of epileptic seizures. These types of SE do not occur in an isolated fashion and overlap exists. Different types of SE may occur in the same syndrome.
In our experience, specifically in children, the types of RSE or SRSE that most likely respond to KD therapy are:
- A.
Refractory focal and sometimes generalized status epilepticus, primarily observed in focal epilepsies of different etiologies
- B.
The pure form of myoclonic status epilepticus
- C.
Non-convulsive and/or electrical status epilepticus in the framework of the epileptic encephalopathies
The majority of studies on the use of the KD have been conducted in adults and children with simple or complex RSE, mostly of an immune-mediated etiology. These patients mainly had motor RSE. Cases with focal non-convulsive RSE and generalized RSE have also been published [8, 9, 17••, 18,19,20,21,22,23,24,25,26,27,28,29]. Studies on the use of the KD in children with RSE/SRSE are listed in Table 1.
The second type of status epilepticus that may respond particularly well to the KD is the pure form of myoclonic status epilepticus.
Myoclonic status epilepticus, a term used for different electroclinical presentations with different prognostic and treatment implications, has been described in generalized epilepsy syndromes, neurodegenerative disease, infectious or inflammatory neurologic disease, toxic-metabolic states, and following anoxic brain injury [10].
Our group published two patients with myoclonic RSE. One of them, with myoclonic epilepsy of unknown etiology, had a 75–90% seizure reduction, and the other with progressive encephalopathy associated with myoclonic epilepsy had a 50% seizure reduction. Both patients retained good tolerability for the diet [30].
Myoclonic status epilepticus in non-progressive encephalopathy is associated with a particular electroclinical pattern seen in patients with a genetic etiology, such as Angelman syndrome and diffuse developmental cortical dysplasia [31].
In a second study, six patients with nonprogressive encephalopathy and myoclonic RSE treated with the KD were presented. Seizure reduction was more than 75% in two, more than 50% in three, and only one had a less than 50% decrease in seizures [32•].
In patients with refractory myoclonic status epilepticus, regardless of the type of epileptic syndrome and etiology, the diet should be considered earlier in the course of the treatment.
The third type of SE responding well to the KD is the non-convulsive and/or electrical status epilepticus. This NCSE may be subtle and difficult to recognize on EEG and EEG polygraphy. In children, the majority of epileptic encephalopathies are associated with non-convulsive and electrical status epilepticus, including electrical status epilepticus during sleep (ESES) and hypsarrhythmia. West syndrome and ESES are typical examples of electrical status epilepticus.
The KD is found to work well in ESES or continuous spikes and waves during slow sleep (CSWSS). In a series by our group, 12 patients with ESES were treated with the diet. One became seizure free, one had a more than 75% seizure decrease, and two a more than 50% and two a less than 50% decrease, while two discontinued because of adverse effects and three did not respond to the diet [33]. In a series of 45 patients with drug-resistant epilepsy secondary to malformations of cortical development, the group with a better response to dietary therapy correspond to those with unilateral polymicrogyria associated with ESES [34•].
In children with NCSE, we probably decide to use the diet at the moment when the electro-clinical features are compatible with non-convulsive and electrical status epilepticus.
Dravet syndrome is typically associated with all three types of RSE: The febrile, mainly generalized motor status, myoclonic status epilepticus, and the obtundation state, the latter two of which are the characteristic NCSE of the syndrome. In our series of patients with Dravet syndrome, 10 of the 13 children who remained on the diet had a significant reduction in number of seizures. In children who did not have a considerable decrease in seizures, an improvement in quality of life was observed and the number of antiepileptic drugs could be reduced [35]. Dressler et al. [36] and Tian et al. [37•] demonstrated that KD is effective in the control of SE in patients with Dravet syndrome. In our study, four of five patients with status epilepticus responded well to the diet and did not repeat the event [35]. In two animal models, one for Dravet syndrome and one for generalized epilepsy with febrile seizures plus (GEFS+), the effectiveness of the diet in refractory patients with SCN1A mutations was shown [38].
Lennox-Gastaut syndrome is another epileptic encephalopathy characterized by multiple seizure types, of which in SE tonic and absence seizures are the most typical. In our series of 15 patients who stayed on the diet for at least 18 months, half showed a more than 50% reduction in seizures, and 20% achieved seizure freedom [39].
Patients with migrating focal epilepsy of infancy may develop focal or multifocal RSE with typical electroclinical manifestations for which the KD in our experience is a good option that could be used earlier in the treatment as shown in our series of three patients, of whom two responded well. This particular type of focal status epilepticus may be convulsive or non- convulsive: The convulsive form may include myoclonic jerks [40].
FIRES is a severe epileptic encephalopathy associated with SRSE in patients without epilepsy or a relevant preexisting neurological disorder. Different treatment strategies have been proposed for this condition, but clinical outcomes are poor. Different studies have shown high responder rates in patients with FIRES [21, 26••, 28, 29•, 41]. In FIRES, the types of SRSE are often focal or nonconvulsive with altered consciousness [29•]. In our series of 12 patients with FIRES, two patients were treated with the KD in the acute phase. One had a good and sustained response with a 50–75% seizure reduction and the other had a seizure reduction of less than 50% [41].
The use of the KD in RSE/SRSE
There are no clear guidelines for the use of the KD for the treatment of RSE/SRSE. Nevertheless, over the years, experience with the use of the KD in the ICU has been growing [8, 9, 17••, 18••, 19••, 20••, 21••, 22••, 23••, 24••, 25••, 26••, 27••, 28••, 29••].
When using the diet in patients with RSE and SRSE, conditions specific to critically ill patients may complicate the treatment, such as the impossibility of enteral feeding, the concomitant use of steroids, preexisting acidotic states, and a lack of dieticians with expertise in ketogenic formulations [26••].
Contraindications to start the enteral KD in patients with RSE are: Known fatty acid oxidation disorder or pyruvate carboxylase deficiency, unstable metabolic condition, hemodynamic or cardiopulmonary instability, inability to tolerate enteral nutrition, ileus and liver failure, total cholesterol > 300 mg/dL, coagulopathy, concurrent hemodialysis or plasmapheresis, and exposure to propofol within the previous 24 h [17••].
The KD is generally initiated at a low ratio (around 2–3:1 in enteral and 1:1 in parenteral KD administration) and then gradually increased to a 3–4:1 ratio. Initially, the amount of calories administered through the KD is restricted to one third of the estimated energy needs (75% of recommended dietary allowance) and is subsequently increased by one third every 1–3 days to the full estimated energy needs to prevent hypertriglyceridemia or hyperamylasemia induced by a high intake or infusion of lipids [26••, 29•].
It is important to control if dextrose is removed from all fluids and medications, for example antiepileptic drugs and saline, with assistance from the hospital pharmacists [26••, 29].
In the study by Farias-Moeller et al., no difference was found between fasted and non-fasted patients. Patients who respond to the diet can be weaned off anesthetic infusions between 7 and 10 days [26••].
In certain cases, the diet is useful to resolve the SE and improve the general state of the patient to facilitate other treatments. In one of our patients, an infant boy with hemimegalencephaly and SRSE, the diet was started with good response and hemispherectomy could be performed.
Up to now, the youngest patient with SRSE that was safely treated with the KD was a 9-week-old infant in a neonatal ICU [24].
Effectiveness of the KD in RSE/SRSE
In patients with RSE and SRSE, the effectiveness of the KD may be difficult to evaluate due to the simultaneous use of other therapies, such as pharmacologic coma, different AEDs, and immunotherapy [29]. Nevertheless, in different pediatric series evaluating the diet, a success rate of around 75% was found (see Table 1).
When dietary therapy was successful, it worked within 7–10 days [7, 25, 27, 42].
Adverse effects and tolerability of the KD in RSE
Adverse events of KD therapy in RSE include aspiration pneumonia, gastroesophageal reflux, gastrointestinal symptoms, hypertriglyceridemia, metabolic acidosis, hypoglycemia, hyponatremia, kidney stones, elevated liver enzymes, and weight loss [17••, 28]. The adverse effects are usually mild and transient and may be resolved using symptomatic treatment. They rarely lead to the need to discontinue the diet.
In a prospective multicenter study of adult patients with SRSE, metabolic acidosis was found to be the most notable KD-related adverse effect. Monitoring of this condition and aggressive bicarbonate supplementation is important [17••].
Another common complication is hypoglycemia and therefore frequent glycemia monitoring is required. To correct hypoglycemia, an intravenous glucose bolus should be used, as continuous infusion will interrupt ketosis [43].
In critical-care patients, gastrointestinal disorders are common due to long-term treatment with intravenous anesthetic drugs, immobility, decreased bowel movements, and prolonged mechanical ventilation. Prevention and treatment of gastrointestinal disturbances is important as they may complicate KD administration [28].
In the study by Park et al., seven out of nine patients discontinued the diet because of adverse effects, even though the KD was successful in treating the seizures. Therefore, patients should be closely monitored for adverse events and when recognized rapid intervention is necessary to maintain the diet [28].
Timing of diet initiation
Overall, there is heterogeneity regarding timing of implementation of KD, time to ketosis, and clinical outcomes [15]. Currently, the KD is initiated late in the course of the disease at a mean of 24 days [8, 23,24,25,26]. In the study by Farias Moeller et al. a growing trend towards earlier consideration of the KD in the treatment algorithm for RSE/SRSE was observed, from 28 days after SRSE in the first part of the study to an average of 14.8 days in the second half [26••].
Park et al. suggested the KD could be considered 48 h after anesthetic agents are withdrawn or tapered and seizures recur, there are active bowel movements, and there is no evidence of active respiratory tract or systemic infection [28].
Peng et al. [29] found that outcome was better in patients with FIRES when the KD was started within 15 days after SRSE onset. As the KD has shown to work particularly well in certain epileptic syndromes, it may be hypothesized that in certain etiologies, the KD should be started earlier in the course of the RSE/SRSE.
Possible mechanisms of action of the KD
Although the underlying mechanisms are still unclear, the KD has shown to have anticonvulsant and neuroprotective as well as anti-inflammatory properties [17••, 44]. The diet mimics the fasting state which leads the body to start using fats instead of carbohydrates as the primary energy source. The most important feature of the KD is the production of ketone bodies by the liver [45].
The KD is potentially involved in multiple pathophysiologic mechanisms, such as neurotransmitter systems, including gamma-aminobutyric acid, glutamate, and adenosine, responsible for neuronal hyperexcitability, ion channel modifications, glucose level changes, and mitochondrial changes [45].
Additionally, it was demonstrated how polyunsaturated fatty acids (PUFAs) through the diet may have anticonvulsant effects in patients with refractory epilepsy due to ion-channel diseases, particularly in those related to voltage-gated sodium and potassium channels. PUFAs are not a typical component of the KD and therefore are considered the result of endogenous production and export into the circulation [46].
Regarding the ion-channel related mutations in the SCN1A gene encoding the α1 subunit of the voltage-gated sodium channel are found in between 70 and 80% of patients with Dravet syndrome and in some families with genetic epilepsy with febrile seizures plus [47]. These findings may explain the good response of these patients to the KD [35]. Other epileptic encephalopathies with myoclonic SE, such as myoclonic status epilepticus in non-progressive epileptic encephalopathies associated with the SCN8A gene, have also been found to respond well to the KD, supporting the ion-channel-related mechanism [32•].
It is currently believed that most probably a combination of different mechanisms finally contributes to the antiseizure effect of the KD [45].
Parental administration of the KD
Most of the patients with RSE and SRSE can be treated with the classic ketogenic formula through a nasogastric tube; however, intravenous administration has also been used.
When enteral KD therapy is impossible due to multiple medical therapies, such as anesthetic agents, or the clinical status of the patient, who may have intestinal dysmotility, malabsorptive status, and/or perfusion abnormalities, parenteral administration of the KD may simplify access to this treatment. Furthermore, children with seizures may develop intolerance to enteral feeding and could benefit from short- or long-term parenteral KD [48].
Two of seven patients with FIRES in the study by Peng et al. [29] who required bowel rest due to upper gastrointestinal hemorrhage or gastroplegia received the KD via parenteral nutrition. They were subsequently successfully converted to the enteral diet.
We recently studied three patients who had been on the KD because of epilepsy of infancy with migrating focal seizures, who were acutely unable to absorb nutrients through the intestinal tract because of appendicitis and intestinal bleeding—that were unrelated to the diet—and required complete bowel rest. In all three children, seizure control was maintained when transitioning from enteral to parenteral nutrition and also when switching back from parenteral to enteral nutrition [49]. Therefore, parenteral KD therapy may also be useful as a temporary bridge towards the enteral KD [48].
There are currently no official guidelines for the use of the parenteral use of the KD. Nevertheless, Dressler et al. have recently developed an algorithm based on the guidelines of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) to standardize calculation of the individual components of parenteral KD therapy. Although in half of the patients in this series ketosis was lower than on the oral KD, seizures remained controlled. [50•].
Discussion
Over the past years, there has been an increasing role for dietary therapy in the treatment of refractory focal as well as generalized status epilepticus.
For adequate decision-making regarding the use of dietary treatment in patients with RSE, not only the etiologies, but also types of epilepsy and epilepsy syndromes as well as electroclinical features should be considered.
Different types of refractory status epilepticus are likely to respond to the KD, such as refractory simple or complex focal and sometimes generalized status epilepticus, the pure form of myoclonic status epilepticus, and non-convulsive or electrical status epilepticus (epileptic encephalopathies).
Up to now, different series of pediatric patients have been reported [18,19,20,21,22,23,24,25,26,27,28,29] (see Table 1) with the youngest patient being 9 weeks of life [24].
In 2014 in our first study on the use of the KD in patients with RSE [39], we reported 10 patients aged 2 to 9 years (mean 5 years) who had simple or complex focal RSE and were placed on the KD. The patients mainly had repetitive and nearly continuous focal status epilepticus refractory to the conventional protocol treatment of SE.
Patients with FIRES seem to be particularly good candidates for KD therapy, and the diet should be considered early in the treatment protocol in these cases [41, 42]. Peng et al. [29] found that those in whom RSE duration was less than 15 days before KD initiation had a better prognosis.
The KD has shown to be a good treatment option for patients with myoclonic-astatic epilepsy [51]. We published two children with refractory myoclonic SE who demonstrated a good response to the KD in terms of reduction of seizure frequency and tolerability [30].
Furthermore, many epileptic encephalopathies are associated with non-convulsive or electrical status epilepticus pointing to a role for dietary treatments. The use of dietary treatment should be considered earlier in the management of the patient with this type of electroclinical status epilepticus [5]. An example is Lennox-Gastaut syndrome, one of the epileptic encephalopathies with a good response to the KD. In patients with LGS who develop non-convulsive RSE, dietary therapy should be considered [39].
Recent studies suggest that the diet may be useful in refractory myoclonic status epilepticus as well [30]. The diet was found to be effective in the control of status epilepticus in patients with Dravet syndrome, associated with febrile, mainly generalized motor status, myoclonic SE, and the obtundation state [36, 37•].
In patients with migrating focal epilepsy of infancy, the KD could also be a good option earlier in the management. In this specific epileptic syndrome, the characteristic manifestations lead to frequent hospital admissions due to recurrent SE [40, 49].
Patients with SRE/SRSE in the intensive care unit are exposed to prolonged anesthetic drug therapy, mechanical ventilation, and multiple AEDs, all of which may increase risk of infection or multiorgan dysfunction. The KD is therefore a welcome treatment option in the management of these patients reducing seizures and facilitating weaning from mechanical ventilation. Additionally, the safety profile of the KD favorably compares to the toxicity of anesthetic and antiepileptic drugs these children receive in the ICU setting [23]. Nevertheless, dietary therapy in these patients is a challenge due to the variability of the etiologies and electroclinical features as well as the clinical complications associated with the young age of most children. If possible, the underlying etiology of the RSE/SRSE should be determined and, in the absence of a contraindication, the KD could be considered in earlier in the treatment algorithm.
The KD for RSE/SRSE should be implemented by a multidisciplinary team, consisting of a dieticians, clinical nutritionist, pharmacist, neurologist, and intensivists. Members of the intensive care team should be trained in the diet, taking into account the special features related to critically ill patients, and be willing to stop antiepileptic drugs during the time the diet needs to work (7–10 days).
A tailored approach is necessary for each individual patient. A crucial aspect is to define the adequate time to initiate the diet in patients with RSE/SRSE. Patients who responded to the diet should be followed-up in the outpatient setting. The diet should not be started if it cannot be continued. [42].
The use of diets as emergency treatment is a future challenge in the management of the patient with RSE/SRSE with an important impact of cost-effectiveness. Guidelines are necessary as the implementation of the diet can be complicated by the already complex treatment regimens of critically ill patients. Furthermore, additional research for the use of enteral and parenteral KD therapy is required as most studies published up to now are single center and retrospective, with a possible selection bias.
Conclusions
When patients with RSE or SRSE do not respond to the treatment algorithm of benzodiazepines, IV AEDs, and anesthetics, the KD may be an effective and tolerable option.
In the absence of a contraindication, the KD may be considered earlier in the treatment algorithm.
It is important to be aware that there is a special group of patients with epileptic encephalopathies in whom the non-convulsive RSE is subtle and difficult to recognize on EEG and EEG polygraphy. In these patients, the diet should be considered early in the course of treatment.
Considering the beneficial effect and taking into account the potential of dietary therapy in RSE/SRSE, the development of a consensus to optimize its use in patients with epilepsy in critical care would be important. There is a need for randomized controlled trials to confirm timing of diet initiation and the best candidates for the use of the KD in RSE and SRSE.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Stafstrom CE. Dietary approaches to epilepsy treatment: old and new options on the menu. Epilepsy Curr. 2004;4(6):215–22.
Freeman JM, Vining EP, Pillas DJ, Pyzik PL, Casey JC, Kelly LM. The efficacy of the ketogenic diet-1998: a prospective evaluation of intervention in 150 children. Pediatrics. 1998;102(6):1358–63.
Caraballo R. Dieta Cetogena en el tratamiento de la epilepsia. Buenos Aires: Editorial Journal; 2016.
Nangia S, Caraballo RH, Kang HC, Nordli DR, Scheffer IE. Is the ketogenic diet effective in specific epilepsy syndromes? Epilepsy Res. 2012 Jul;100(3):252–7. https://doi.org/10.1016/j.eplepsyres.2012.01.015.
• Caraballo RH. The use of the ketogenic diet in the treatment of epileptic encephalopathies. JICNA. 2018;18:66 This study provides an overview of the use of the ketogenic diet in epileptic encephalopathies, both those in which the diet has shown to result in a good response, such as West syndrome, Dravet syndrome, or Lennox-Gastaut syndrome, and those that are more rare or only recently identified, such as epilepsy with focal migrating seizures in infancy, febrile infection-related epilepsy syndrome, or myclonic status in non-progressive encephalopathy, about which little is known.
•• Kossoff EH, Zupec-Kania BA, Auvin S, Ballaban-Gil KR, Christina Bergqvist AG, Blackford R, et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open. 2018;3(2):175–92. https://doi.org/10.1002/epi4.12225 This update of the 2009 consensus guideline for the management of children on ketogenic diet therapy includes new concepts and recent research regarding topics of patient selection, counseling, and evaluation, diet choice and attributes, implementation, supplementation, follow-up, side events, and diet discontinuation.
Cervenka MC, Hartman AL, Venkatesan A, Geocadin RG, Kossoff EH. The ketogenic diet for medically and surgically refractory status epilepticus in the neurocritical care unit. Neurocrit Care. 2011 Dec;15(3):519–24. https://doi.org/10.1007/s12028-011-9546-3.
Thakur KT, Probasco JC, Hocker SE, Roehl K, Henry B, Kossoff EH, et al. Ketogenic diet for adults in super-refractory status epilepticus. Neurology. 2014;82(8):665–70. https://doi.org/10.1212/WNL.0000000000000151.
Wusthoff CJ, Kranick SM, Morley JF, Christina Bergqvist AG. The ketogenic diet in treatment of two adults with prolonged nonconvulsive status epilepticus. Epilepsia. 2010 Jun;51(6):1083–5. https://doi.org/10.1111/j.1528-1167.2009.02388.
Shorvon S, Ferlisi M. The treatment of super-refractory status epilepticus: a critical review of available therapies and a clinical treatment protocol. Brain. 2011;134:2802–18.
Sahin M, Menache CC, Holmes GL, Riviello JJ. Outcome of severe refractory status epilepticus in children. Epilepsia. 2001;42(11):1461–7.
• Sculier C, Gaínza-Lein M, Sánchez Fernández I, Loddenkemper T. Long-term outcomes of status epilepticus: a critical assessment. Epilepsia. 2018;59(Suppl Suppl 2):155–69 The findings of this study on the long-term outcomes after a status epilepticus episode in pediatric and adult patients showed that etiology is the main determinant of outcome, that the effect of age or SE duration is often difficult to distinguish from the underlying cause, and that effect of the treatment on long-term outcome after SE is still unknown.
Bodenant M, Moreau C, Sejourné C, Auvin S, Delval A, Cuisset JM, et al. Interest of the ketogenic diet in a refractory status epilepticus in adults. Rev Neurol (Paris). 2008;164(2):194–9. https://doi.org/10.1016/j.neurol.2007.08.009.
Glauser T, Shinnar S, Gloss D, Alldredge B, Arya R, Bainbridge J, et al. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr. 2016;16(1):48–61. https://doi.org/10.5698/1535-7597-16.1.48.
• Vasquez A, Farias-Moeller R, Tatum W. Pediatric refractory and super-refractory status epilepticus. Seizure. 2019;68:62–71. https://doi.org/10.1016/j.seizure.2018.05.012 This review summarizes the available evidence related to pediatric refractory status epilepticus and super-refractory status epilepticus focusing on epidemiology, etiologies, therapeutic approaches, and clinical outcomes.
•• Hirsch LJ, Gaspard N, van Baalen A, Nabbout R, Demeret S, Loddenkemper T, et al. Proposed consensus definitions for new-onset refractory status epilepticus (NORSE), febrile infection-related epilepsy syndrome (FIRES), and related conditions. Epilepsia. 2018;59(4):739–44 This consensus document proposes definitions for New-Onset Refractory Status Epilepticus (NORSE), Febrile Infection-Related Epilepsy Syndrome (FIRES), and related conditions, as well as for Infantile Hemiconvulsion-Hemiplegia and Epilepsy syndrome (IHHE) and for prolonged, refractory and super-refractory status epilepticus to enable improved communication for investigators, physicians, families, patients, and other caregivers.
•• Cervenka MC, Hocker S, Koenig M, Bar B, Henry-Barron B, Kossoff EH, et al. Phase I/II multicenter ketogenic diet study for adult superrefractory status epilepticus. Neurology. 2017;88(10):938–43 This prospective multicenter study of adults patients with super-refractory status epilepticus treated with a ketogenic diet treatment algorithm provides Class IV evidence that in adults with super-refractory status epilepticus, ketogenic therapy is effective in inducing ketosis
Kumada T, Miyajima T, Kimura N, Saito K, Shimomura H, Oda N, et al. Modified Atkins diet for the treatment of nonconvulsive status epilepticus in children. J Child Neurol. 2010;25(4):485–9. https://doi.org/10.1177/0883073809347597.
Villeneuve N, Pinton F, Bahi-Buisson N, Dulac O, Chiron C, Nabbout R. The ketogenic diet improves recently worsened focal epilepsy. Dev Med Child Neurol. 2009 Apr;51(4):276–81. https://doi.org/10.1111/j.1469-8749.2008.03216.x.
Nabbout R, Mazzuca M, Hubert P, Peudennier S, Allaire C, Flurin V, et al. Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES). Epilepsia. 2010;51(10):2033–7. https://doi.org/10.1111/j.1528-1167.2010.02703.x.
Vaccarezza M, Silva W, Maxit C, Agosta G. Super-refractory status epilepticus: treatment with ketogenic diet in pediatrics. article in Spanish. Rev Neurol. 2012;55(1):20–5.
Caraballo RH, Flesler S, Armeno M, Fortini S, Agustinho A, Mestre G, et al. Ketogenic diet in pediatric patients with refractory focal status epilepticus. Epilepsy Res. 2014;108(10):1912–6. https://doi.org/10.1016/j.eplepsyres.2014.09.033.
O’Connor SE, Ream MA, Richardson C, Mikati MA, Trescher WH, Byler DL, et al. The ketogenic diet for the treatment of pediatric status epilepticus. Pediatr Neurol. 2014 Jan;50(1):101–3. https://doi.org/10.1016/j.pediatrneurol.2013.07.020.
Cobo NH, Sankar R, Murata KK, Sewak SL, Kezele MA, Matsumoto JH. The ketogenic diet as broad-spectrum treatment for super-refractory pediatric status epilepticus: challenges in implementation in the pediatric and neonatal intensive care units. J Child Neurol. 2015;30:259–66.
Appavu B, Vanatta L, Condie J, Kerrigan JF, Jarrar R. Ketogenic diet treatment for pediatric super-refractory status epilepticus. Seizure. 2016;41:62–5. https://doi.org/10.1016/j.seizure.2016.07.006.
•• Farias-Moeller R, Bartolini L, Pasupuleti A, Brittany Cines RD, Kao A, Carpenter JL. A practical approach to ketogenic diet in the pediatric intensive care unit for super-refractory status epilepticus. Neurocrit Care. 2017;26(2):267–72. https://doi.org/10.1007/s12028-016-0312-4 This study describes the experience with a practical approach to initiation of the ketogenic diet for children with super-refractory status epilepticus in the pediatric intensive care unit through which the patients could be successfully weaned off continuous anesthetic infusions.
Arya R, Peariso K, Gainza-Lein M, Harvey J, Bergin A, Brenton JN, et al. Efficacy and safety of ketogenic diet for treatment of pediatric convulsive refractory status epilepticus. Epilepsy Res. 2018;144:1–6 This study provides the results of ketogenic diet therapy in patients with refractory status epilepticus treated with the ketogenic diet at institutions participating in the pediatric Status Epilepticus Research Group suggesting efficacy and safety of the treatment.
Park EG, Lee J, Lee J. The ketogenic diet for super-refractory status epilepticus patients in intensive care units. Brain and Development. 2019;41(5):420–7. https://doi.org/10.1016/j.braindev.2018.12.007.
• Peng P, Peng J, Yin F, Deng X, Chen C, He F, et al. Ketogenic diet as a treatment for super-refractory status epilepticus in febrile infection-related epilepsy syndrome. Front Neurol. 2019;10:423. https://doi.org/10.3389/fneur.2019.00423 The findings of this study show that in children with febrile infection-related epilepsy syndrome, a fatal epileptic encephalopathy associated with super-refractory status epilepticus, the ketogenic diet should be tried earlier in the course of the disease and that the intravenous route of administration may be an alternative for patients who cannot receive the diet enterally.
Caraballo RH, Valenzuela GR, Armeno M, Fortini S, Mestre G, Cresta A. The ketogenic diet in two paediatric patients with refractory myoclonic status epilepticus. Epileptic Disord. 2015 Dec;17(4):491–5. https://doi.org/10.1684/epd.2015.0781.
Caraballo RH, Cersósimo RO, Espeche A, Arroyo HA, Fejerman N. Myoclonic status in nonprogressive encephalopathies: study of 29 cases. Epilepsia. 2007 Jan;48(1):107–13.
• Caraballo R, Darra F, Reyes G, Armeno M, Cresta A, Mestre G, et al. The ketogenic diet in patients with myoclonic status in non-progressive encephalopathy. Seizure. 2017;51:1–5. https://doi.org/10.1016/j.seizure.2017.07.002 The findings of this study suggest that the ketogenic diet is a promising therapy for patients with myoclonic status in non-progressive encephalopathy as those who responded well to the diet cognitive performance and quality of life also improved.
Reyes G, Flesler S, Armeno M, Fortini S, Ariela A, Cresta A, et al. Ketogenic diet in patients with epileptic encephalopathy with electrical status epilepticus during slow sleep. Epilepsy Res. 2015;113:126–31.
• Pasca L, Caraballo RH, De Giorgis V, Reyes JG, Macasaet JA, Masnada S, et al. Ketogenic diet use in children with intractable epilepsy secondary to malformations of cortical development: a two-centre experience. Seizure. 2018;57:34–7. https://doi.org/10.1016/j.seizure.2018.03.005 The findings of this study of pediatric patients with refractory epilepsy due to malformation of cortical development with ESES suggest that the ketogenic diet should be considered these patients when surgery is not a viable option.
Caraballo RH, Cersósimo RO, Sakr D, Cresta A, Escobal N, Fejerman N. Ketogenic diet in patients with Dravet syndrome. Epilepsia. 2005;46(9):1539–44.
Dressler A, Trimmel-Schwahofer P, Reithofer E, Mühlebner A, Gröppel G, Reiter-Fink E, et al. Efficacy and tolerability of the ketogenic diet in Dravet syndrome—comparison with various standard antiepileptic drug regimen. Epilepsy Res. 2015;109:81–9. https://doi.org/10.1016/j.eplepsyres.2014.10.014.
• Tian X, Chen J, Zhang J, Yang X, Taoyun J, Zhang Y, et al. The efficacy of ketogenic diet in 60 Chinese patients with Dravet syndrome. Front Neurol. 2019;10:625. https://doi.org/10.3389/fneur.2019.00625 This large study on ketogenic diet treatment in patients with Dravet syndrome indicates that the diet has many advantages for these patients, as it works rapidly, is effective in more than half of the children, and has tolerable adverse reactions.
Dutton SB, Sawyer NT, Kalume F, Jumbo-Lucioni P, Borges K, Catterall WA, et al. Protective effect of the ketogenic diet in Scn1a mutant mice. Epilepsia. 2011 Nov;52(11):2050–6. https://doi.org/10.1111/j.1528-1167.2011.03211.x.
Caraballo RH, Fortini S, Flesler S, Armeno M, Ariela A, Cresta A, et al. Ketogenic diet in patients with Lennox-Gastaut syndrome. Seizure. 2014 Oct;23(9):751–5. https://doi.org/10.1016/j.seizure.2014.06.005.
Caraballo R, Noli D, Cachia P. Epilepsy of infancy with migrating focal seizures: three patients treated with the ketogenic diet. Epileptic Disord. 2015 Jun;17(2):194–7.
Caraballo RH, Reyes G, Avaria MFL, Buompadre MC, Gonzalez M, Fortini S, et al. Febrile infection-related epilepsy syndrome: a study of 12 patients. Seizure. 2013;22:553–9. https://doi.org/10.1016/j.seizure.2013.04.005.
Kossoff E, Nabbout R. Use of dietary therapy for status epilepticus. J Child Neurol. 2013;28(8):1049–51.
Gomes D, Pimentel J, Bentes C, Aguiar de Sousa D, Antunes AP, Alvarez A, et al. Consensus protocol for the treatment of super-refractory status epilepticus. Acta Med Port. 2018;31(10):598–605. https://doi.org/10.20344/amp.9679.
Bough KJ, Rho JM. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia. 2007;48:43–58.
Rho JM. How does the ketogenic diet induce anti-seizure effects? Neurosci Lett. 2017;637:4–10. https://doi.org/10.1016/j.neulet.2015.07.034.
Rogawski MA, Löscher W, Rho JM. Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harb Perspect Med. 2016;6(5):a022780. https://doi.org/10.1101/cshperspect.a022780.
Marini C, Scheffer IE, Nabbout R, Suls A, De Jonghe P, Zara F, et al. The genetics of Dravet syndrome. Epilepsia. 2011;52(Suppl 2):24–9. https://doi.org/10.1111/j.1528-1167.2011.02997.x.
Chiusolo F, Diamanti A, Bianchi R, Fusco L, Elia M, Capriati T, et al. From intravenous to enteral ketogenic diet in PICU: a potential treatment strategy for refractory status epilepticus. Eur J Paediatr Neurol. 2016;20(6):843–7. https://doi.org/10.1016/j.ejpn.2016.08.004.
Armeno M, Verini A, Araujo MB, Reyes G, Caraballo RH. Ketogenic parenteral nutrition in three pediatric patients with epilepsy with migrating focal seizures. Epileptic disorders. In press.
•• Dressler A, Haiden N, Trimmel-Schwahofer P, Benninger F, Samueli S, Gröppel G, et al. Ketogenic parenteral nutrition in 17 pediatric patients with epilepsy. Epilepsia Open. 2017;3(1):30–9 This study provides an algorithm for ketogenic parenteral nutrition based on the guidelines of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) for accurate component calculating. Although in half of the patients in this series ketosis was lower than on the oral ketogenic diet, seizures remained controlled.
Caraballo RH, Cersósimo RO, Sakr D, Cresta A, Escobal N, Fejerman N. Ketogenic diet in patients with myoclonic-astatic epilepsy. Epileptic Disord. 2006;8(2):151–5.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Roberto Caraballo declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pediatrics in South America
Rights and permissions
About this article

Cite this article
Caraballo, R. Ketogenic Diet for Refractory Status Epilepticus in Children. Curr Treat Options Peds 5, 417–430 (2019). https://doi.org/10.1007/s40746-019-00185-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40746-019-00185-0