
Abstract
Purpose of Review
Neurostimulation is an electrical therapy for obstructive sleep apnea and is now an approved therapy option for patients whom positive airway pressure (PAP) therapy is not tolerated. This review describes its implementation, efficacy and safety, the available multi-year clinical outcomes for stimulation devices, and future prospects.
Recent Findings
The clinical literature on upper airway neurostimulation was surveyed from July 2014 to December 2021, with a focus on the origins of new therapies, the components of devices, evidences for clinical utility, and adverse events. The basic science literature began as demonstrations of muscle actions leading to neurostimulator prototypes that brought industry interest to clinical therapy. Currently, Inspire® and Nyxoah Genio® are the only two neurostimulators available in the USA. Inspire®, FDA-approved in 2014 as first-in-class therapy, is a hypoglossal nerve (cranial nerve XII) stimulator that is time-coordinated with breathing to prevent upper airway collapse, and its use has the longest experience in the clinic. Given the general narrow inclusion criteria (BMI < 35, ideally < 32), AHI 15–65/h, and a favorable anterior–posterior velopharyngeal collapse pattern on drug-induced sleep endoscopy (DISE), ~ 65% of patients intolerant to PAP therapy achieve clinical success (AHI < 20/h with a reduction of < 50% in AHI) with Inspire® across many centers. In addition to symptomatic relief, adverse events are mild and self-limited after the initial implant surgery, with rarely needed adjustment or replacement of the implantable generator and electrode. The Nyxoah Genio® (2021 FDA approval as a breakthrough device) is strategically different, placing the electrode near the insertion of CNXII bilaterally into the genioglossus muscle and utilizing an external power generator with proprietary programming and activation patterns, Nyxoah Genio® was approved to address concentric collapse, and it is in phase III trials.
Summary
Hypoglossal nerve stimulation is a reasonable second-in-line alternative for selected patients when first-in-line therapeutic options fail. Considering the recent technological advances in micro implantation for smart remote programming and surveillance, the next generation of neurostimulation devices will be more compact, especially when efferent co-activation and/or afferent-efferent patterning seems feasible.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurostimulation therapy represents an innovative clinical approach and therapy for obstructive sleep apnea (OSA). Unlike the traditional OSA therapy, implantable neurostimulation devices involve hardware and software interfaces between the human nervous system and programmable controllers that can address multi-leveled upper airway structures and functional support. Hardware for OSA therapy uses electrical stimulation through electrodes to address instability caused by the neuroanatomic pathogenesis of upper airway apnea, i.e., obstructive sleep apnea and hypopnea. Some devices include feedback capability from input signals (i.e., the pressure generated by inspiratory efforts or inspiratory movement of the chest wall). Three neural interface devices were or are entered into phase I clinical trials for patients suffering from OSA.
OSA is a chronic disease, with a world-wide prevalence across clinical and population estimates of ~ 1 billion people [1]. When correctly diagnosed and optimally managed, there is an individual health benefit with improved sleep continuity, refreshing sleep, decreased waketime drowsiness and fatigue, and reduced blood pressure, with behavioral effects that reduce workplace absenteeism and presenteeism, and inattention related errors or driving crashes. The obstructive sleep apnea hypopnea syndrome—snoring, restless sleep, daytime impairments, and cardiopulmonary morbidity—was first treated with tracheostomy which bypassed the problematic upper airway, and proved the disorder as upper airway obstruction by reversing the illness, but did not address the root cause.
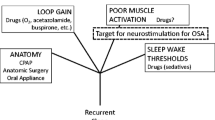
As a neurostimulation target, OSA is attractive because there is a critical role in reduced efferent muscle activation at the onset of sleep-related intermittent closure which can occur in the nasopharynx, oropharynx, and the hypopharynx. Given an upper airway vulnerable to collapse, the reductions in respiratory drive with sleep along with inadequate muscle recruitment during an event and high instability gain in the respiratory control system lead to repetitive bouts of airway compromise. Arousals or reflex activation reopens the airway (Fig. 1). So the onset is a reduction in neuromuscular tone with sleep and a vulnerable airway; if the airway is not vulnerable, a non-obstructive or central apnea or hypopnea will occur due to gain instability during sleep.

Pathways that appear to interact in the production of recurrent obstructive apneas and hypopneas over time. First is sleep onset and the closure of a vulnerable upper airway segment as respiratory drive falls. To terminate the even, there is the “arousal” response to an apnea/hypopnea that can result in an overshoot and then undershoot of optimal drive. The muscle activation is inadequate to keep the airway open or reopen a closed airway. Neurostimulation addresses the muscle recruitment. The Insert is from a PUBMED search (www.pubmed.gov-Search terms: neurostimulation, OSA, March 2022) and provides an accounting by year of the number of references over time from 1953 to the present in physiology and human therapy. Prior to 2014 when the STAR trial was published, there were applied physiological and preclinical uses of stimulation to upper airway muscles. The uptick in clinical publications since 2014 HNS approval is a result of clinical cohorts and studies
Today, the most common first and well-documented approach to reverse the symptoms, signs, and metabolic consequences of OSA is continuous positive airway pressure (CPAP) [2], which creates a positive intraluminal pressure. This technology improves quality of life through reductions in wake time sleepiness, disturbed sleep, and lower blood pressure, effects which increase with hours of use [3]. Effects on mortality and stroke are less clearly shown, as the timeline to assess this requires years of successful use [4]. However, at 2 years, ~ 60% are unable to use or tolerate CPAP therapy [5, 6]. Surgical management of the anatomy proposes to address anatomic features of airway narrowing. While effective in some and with improving surgical approaches, many procedures directed at soft tissue are not predictably efficacious nor as durable as one would like [7]. For instance, uvulopalatopharyngoplasty (UPPP) will reduce severe AHI by an average of 30–50%, but residual AHI remains in the mild-to-moderate range (< 20/h) after 2 years [8]. Using oral appliances to protrude the mandible is effective in selected patients and, in the short term, improves snoring, sleepiness, and blood pressure, but durability is limited by dental and TMJ side effects and tolerance over time [9, 10].
Neurostimulation now has an established role when CPAP is not “tolerated,” with or without attempts at oral appliance therapy or after anatomic surgery. The concept is not new [11]. From 1953 to the mid-1990s, citations list experimental designs and outcomes mostly (> 90%) in animal models (Fig. 1). One 1993 study demonstrated percutaneous hypoglossal nerve stimulation (HNS) success in humans and keeping the airway open; however, once the airway had closed, HNS did not easily break an apnea [12]. In 2021 there were 85 citations, all except one in humans (Fig. 1). In 2014, one commercial device, Inspire®, met with FDA approval and is now deployed across the USA and in Europe, with over 20,000 units implanted (number courtesy of Inspire Medical LLC). A second technology, Genio®, developed by Nyxoah, followed the successful completion of the BLAST OSA study and received its European CE Mark in 2019. Preliminary data from Nyxoah’s BETTER SLEEP trial resulted in FDA approved in 2021 for emergency use and is in phase III trials. Both produce electrical stimulation to the cranial nerve (CN) XII nerve (which innervates the genioglossus) to activate its motor units to move structures that open the oropharynx and nasopharynx. Nerve stimulation does not produce a positive intraluminal pressure (like CPAP), alter the upper airway (like anatomic surgery), nor protrude the mandible (like an oral appliance) [13]. The pathway for its acute effects is to address deficient muscle activation pathway in recurrent apneas.
The success of neurostimulation includes preventing collapse of the oropharynx and maintaining an open oro- and velo-pharynx and thus keeping the upper airway open enough to permit uninterrupted sleep. Further details are discussed below. The past 7 years after US and European approvals for clinical use have provided a body of literature of published work with the Inspire®, substantive 1-year outcomes with Genio®, and many commentaries on current use, cost-effectiveness, and surgical refinement. This review is not intended to be exhaustive but will provide guidance to its implementation in clinical practice now and the path forward towards improving this line of therapy.
Eligibility
Neurostimulation is considered a choice for those with moderate to severe OSA in whom CPAP is unsuccessful. One might imagine that it could be considered primary therapy when CPAP is impractical (e.g., facial disfigurement) or contraindicated (bullous disease, chronic pneumothorax, or arachnoid leak). Before incorporating it into a practice, there needs to be core knowledge, skill, and experience in the successful deployment of primary treatment options and an ability to compare and contrast the potential success of neurostimulation compared to these other options for the individual patient and articulate this to other physicians and third-party payers [14]. Like any OSA therapy, however, success will depend upon an ability to keep the nasopharynx, oro-pharynx, and/or hypopharynx patent, permitting uninterrupted sleep [15, 16••].
Ideally, neurostimulation is offered at centers with cooperation among both surgical and medical sleep specialists. Patient selection is critical. In some places, a skilled surgeon also has credentials in sleep medicine, which is useful as OSA does not often present as an isolated sleep disorder. In some centers, 20% of patients present with other sleep disorders (poor hygiene, insomnia, restless legs, narcolepsy, etc.) which will enter into a decision as to predict HNS symptom reduction, surgical success, and adherence. HNS therapy at present is offered to those who are CPAP intolerant before or after anatomic surgery [17]. Assessments before implant include recognition of medical, psychiatric, neurologic, and sleep co-morbidities, and critical assessments of patient expectations. The technology for CPAP and oral appliance therapies has steadily improved as well as the approaches to address adherence through motivational training. An “adequate” attempt, in good faith, preferably with documented adherence monitoring, is ideal to document the first criteria—an inability to use CPAP. Some centers will include in the insurance pre-authorization a personal statement by the patient as to the personal reasons that led to considering HNS and their expectation of outcome. OSA patients referred back to sleep medicine for assessments of HNS will need to restart the process, and often a fresh start with CPAP or oral appliance is successful. Teamwork is as important after the implant, since perhaps as many as 25% are still not adequately treated. In these, combinatorial therapy can achieve therapeutic success not only for residual OSA [18, 19], but also co-morbid sleep disorders (insomnia with low arousal thresholds, poor sleep hygiene, REM behavior disorder, etc.) after implantation. In a few patients, the experience with the HNS is disappointing and in others systemic infection may lead to a consideration of explanting the device.
Drug-Induced Sedation Endoscopy
A procedure called drug-induced sedation (or sleep) endoscopy (DISE) is indicated for assessments of non-PAP therapies like oral appliance and anatomic surgery [20], and almost always is deployed for HNS [21]. There are a number of collections in which comparisons of DISE to awake endoscopy with Mueller maneuvers or imaging attest to the increased information gained by this procedure. In approximately a third of the time, features seen by the internal examination of the airway inform the surgeon about other issues to consider or address [22]. In the development of the Inspire® technology, it was the observation that the manner of closure of the velopharynx under moderate sedation had an impact on outcome. HNS treatment was successful in 81% of patients with an anterior–posterior pattern of velopharyngeal collapse on DISE, while treatment success was achieved in none of the 5 patients with complete concentric collapse [23]. This pattern of anterior–posterior collapse (similar to a garage door closing) being favorable, while a more concentric appearing collapse (similar to a camera shutter), is in the FDA indications for Inspire® [24]. It is a pattern also favorable for an oral appliance [25]. The Nyxoah device has FDA emergency approval to address OSA with concentric collapse. Higher BMIs and higher AHIs have been identified as parameters associated with a concentric collapse of the velum on DISE, but the correlation is modest at best [26]. The most recent analysis finds virtually no difference in outcomes if the BMI upper limit is 35 vs. 32 [27].
Patients undergoing DISE may have reductions in airway area at multiple regions under deep sedation; more often the collapse in the retropalatal region is most common and the hypopharyngeal region is least common [28]. The pattern of anterior–posterior obstruction at the level of the velum cannot be predicted on the basis of other tests [29]. A scoring system was developed to evaluate each of the vulnerable regions, the velum, oropharynx, tongue, and epiglottis of the upper airway—the VOTE classification. A number of intrinsic (airway mucosal folding, palatal orientation, tissue pressure, etc.) and extrinsic (airway stiffness, wall fat, lung volume, etc.) factors can determine internal shape and collapsibility.
A DISE is performed in medically stable patients, under moderate sedation, and with post-procedure monitoring. On the other hand, the DISE procedure in those who are CPAP intolerant can be used to identify other anatomic problems such as lingual tonsil, prolapsing epiglottis, or lack of evidence to uncover evidence for (in)effectiveness of an oral appliance. The risk benefit in this population for a DISE appears favorable.
Technology Interfaces with the Anatomy
The stimulators that have been deployed are listed in Table 1. All are intended to stimulate a muscle which is declining in activation before an apnea [30]. The cranial nerve 12 (CNXII) is the hypoglossal nerve and has a role in the motor control of swallowing, talking, exercise, vomiting, hiccups, and facial expressions among other things. These are all coordinated through voluntary and involuntary networks. This brainstem CNXII nucleus has predominantly efferent, but some afferent inputs are integrated into brainstem respiratory networks that coordinate these different actions [31, 32]. The nerve exits from the ventrolateral side of the medulla oblongata with motor and sensory roots, as well as contributing to the ansa cervicalis. Lateral to the hypoglossal muscle, there is a division into the lateral and medial main branches [33, 34] where phenotypic complexity exists. The optimal outcome for a proximal nerve placement depends on the positioning on the proximal nerve for functional inclusion of protractors and exclusion of fibers that could retract the tongue mass. Relevant to Inspire®, there is crossover of the cranial nerve XII across the midline to the back of the tongue, explaining an appreciable symmetrical protrusion at lower stimulation amplitudes [35]. In the Genio® bilateral hypoglossal nerve approach, the positioning of the electrodes is close to its insertion into the genioglossus bilaterally of the distal portion of the nerve [36] with both intrinsic and retrusor fiber activations observed. In the Inspire® approach, there is a forward movement of the anterior wall of the pharynx as a byproduct of the pulling action by a vector of force on the hyoid apparatus [37]. At the time of an electrode implant, recording electrodes are placed intraorally into the genioglossus and other muscles to monitor intraoperative stimulation, but the major intraoperative outcome is observation of tongue movement.
The intention is to either stabilize or prevent backward movement to keep the tongue away from the back of the airway. Titration of the intensity (mAmp) is an empiric process, guided by experience. Infrequently an “advanced” setting is used to alter the anode and cathode configurations of the cuff electrodes. This is performed after implantation when the initial preset configuration fails in the initiation of therapy. Prior devices with stimulation electrodes on or within the muscle cause discomfort because of intramuscular pain receptors, in contrast to nerve stimulation which does not cause pain unless resultant muscle contraction is intense. When measured, the intensity of contraction that is therapeutic for the Inspire technology is considerable, > 50% of maximal voluntary force, enough to produce, or suggest muscle fatigue might occur. Infrequently, a patient will complain that snoring reappears at the end of a long sleep period.
HNS is now extensively clinically deployed since 2014 as upper airway stimulation Inspire® (Inspire Medical Systems, Golden Valley, MN, USA). Inspire® works through placement of three stimulating electrodes in a cuff placed around the CNXII along its way to the tongue. In the Inspire® system, the electrodes are activated by an implanted programmable generator (IPG). The IPG is programmed by an external tablet. A sensing lead for pressure changes placed within the chest informs the IPG when there might be a breath. This feature provides some degree of “feedback” to the IPG as to the phase of respiration (inspiration or expiration) and is used in the programing of the IPG to optimize the stimulation time across the timing of a breath, inspiration, and expiration, also called “duty cycle.” The system intended to stimulate upper airway opening at or near the point of a fall in intrathoracic pressure, i.e., the start of inspiration. Theoretically, one could save IPG power by synchronizing the pulse train with inspiration, although a comparison of intermittent vs. continuous stimulation showed equivalent outcomes at 1 week [38].
Surgery for the implant requires two or three incisions. One below the angle of the jaw in a natural crease permits a direct approach to the hypoglossal nerve for decisive placement of the cuff [39••, 40]. The three-electrode spiral cuff is placed directly around the distal, medial branch, and tongue protrusion optimized intraoperatively for tongue protrusion and/or stabilization, as empirically determined during phase II and phase III FDA trials and in post-approval studies [18, 35, 39••, 41]. The Inspire® IPG is inserted into a subcutaneous pocket ∼ 4 cm below the clavicle, much like a cardiac pacemaker. The leads from the cuff electrode and the pressure sensor are tunneled subcutaneously and connected to the IPG. The placement of the IPG for the HNS is usually within the soft tissues of the right upper chest; however, left-sided placement is preferred for those who shoot firearms right-handed. Some are reluctant to place an IPG in a person who might anticipate blows to the chest as in fire rescue personnel or those in contact sports. There is a theoretical concern about HNS implants in those with cardiac or other neural stimulation devices. A case report found a successful co-use of HNS with an implantable defibrillator [42]. However, post-implant cardioversion is reported to disrupt functioning of the IPG. A retrospective case series concluded that there is a need to counsel patients with HGNS undergoing external electrical cardioversion about the possibility of device damage and either reprogramming or operative IPG replacement. Anteroposterior placement of defibrillator pads may help prevent such mishaps.
Another component in the Inspire® system can be a sensing lead placed between the intercostal muscles in the third to fifth intercostal space [18]. The pressure sensor lead is snapped in place into the IPG. If there is good exposure, there is a modification that may result in the insertion of the sensor through the same incision for the IPG. The function of the sensor lead and the triggering of stimulation can be monitored wirelessly, and a time trace is displayed on the programmer.
Clinical Data for the Inspire® HNS Device
The stimulation therapy for apnea reduction (STAR trial: ClinicalTrials.gov Identifier: NCT01161420) became a first-in-class device in 2014. The inclusion and exclusion criteria came out of the conduct of phase II safety and efficacy study [23]. The STAR trial exclusion criteria included body mass index (BMI) > 32, AHI < 20 or > 60, or central and/or mixed apnea index present in > 20% of the AHI on polysomnogram (PSG), and a pattern of complete concentric collapse at the level of the velopharynx observed with DISE. Unless otherwise stated, the current definition here of surgical “success” or of a “responder” is the Sher’s criteria, a reduction in AHI by 50% and an AHI < 20/h, a somewhat difficult bar for those with severe disease (AHI > 30). Early on, polysomnography was required to define success, but more recently, home sleep testing is used at 6 and 12 months.
The predetermined STAR endpoints were objective (AHI and the 4% oxygen-desaturation index (ODI)) and subjective (patient-based sleepiness and sleep related quality of life). As a quasi-control, at 12 months, 46 patients who responded well were randomly assigned to either continue therapy or to a 1-week cessation of stimulation. The latter resulted a change in AHI and symptoms towards pre-treatment levels [43]. At 12 months, 66% of participants were responders by AHI Sher criteria, and 75% were responders by ODI criteria.
The subjects in the 12-month FDA phase III STAR trial signed up for an 18-month follow-up; a benefit of a ~ 60% AHI and ~ 90% subjective response was found for individuals with moderate to severe OSA who had failed nasal continuous positive airway pressure [43]. Studies after re-consent were performed at 24, 36, 48, and 60 months [43,44,45,46]. At 5 years, Epworth Sleepiness Scale and quality of life were improved, with normalization of scores increasing from 33 to 78% and 15 to 67%, respectively. Objectively, a polysomnography-determined AHI surgical response (AHI < 20 events per hour and > 50% reduction) occurred in 75% (n = 71). The responder rate was estimated at ~ 65% at 5 years, with all points at all follow-ups included; subjective use was > 80% of nights (there is now implant monitoring of use). Serious device-related events all related to lead/device adjustments were reported in 6% of patients. Functional amplitudes for stimulation and thresholds for sensation were unchanged at 5 years, suggesting no deterioration of function at the electrode-nerve interface.
Post-Approval Studies
The European Union approved Inspire® in 2013 and the FDA in 2014. The FDA approval includes slightly modified criteria: BMI of 32 with a warning for those > 32 to 35, an AHI > 15 and < 65, a predominance of obstructive apneas and hypopneas, and a predominant antero-posterior pattern of collapse on a drug-induced endoscopy.
The first reports outside of a clinical trial were from three German centers with training during the phase III trial. In a prospective single-arm study, 6- and 12-month visits included Epworth Sleepiness Scale and home sleep testing AHI as objective measures. In the 60 participants, the median AHI reduced from 28 to 10/h from baseline to 12 months. Subjective outcomes improved significantly from baseline to 12 months. The average usage time was 39 h per week based on recordings by the implanted device. One patient requested a removal of the device for cosmetic reasons, and this occurred without sequelae. This study was the first to utilize a follow-up plan using home sleep testing and reported an independent cohort to indicate a safe and effective treatment option for patients with OSA in routine clinical practice [47].
A registry (ADHERE) for the Inspire® was requested by the FDA, and there are now several interim reports which suggest that implants when criteria are generally met continue to show efficacy either equivalent or better to the STAR trial [39••]. Between October 2016 and January 2018, 508 participants were enrolled from 14 centers. Median AHI by either polysomnography or home sleep testing was reduced from 34 to 7 events/h, and median ESS reduced from 12 to 7 from baseline to final visit at 12-month post-implant. For each 1-year increase in age, there was a 4% increase in odds of treatment success. For each 1-unit increase in body mass index (BMI), there was 9% reduced odds of success. Age persisted in a multivariate model as a significant predictor of treatment success, and age was directly assessed in another analytic approach [48]. The more subjective and objective use, the better the patient-centered outcome [47]. At the 1000-patient milestone, therapy effect is durable, and adherence remains similar [41].
The ADHERE trial will proceed to enroll patients who volunteer for follow-up registry for subjective and when indicated objective outcomes. Furthermore, patient experience is positive compared to prior PAP therapy and the manner of therapy with the controller and office visits and adjustments [49]. In a nested case control trial, those who do not meet the formal objective AHI metrics for success nor a robust symptomatic response, but continue to use the device as a reasonable level (< 4 h per night), still show improvement in percent time of sleep with a saturation of > 90%, reductions in daytime sleepiness, and improved quality of life [50].
For those patients with suboptimal adherence, there is a need to develop an individualized plan for improving use and effectiveness. This should be captured early on in the initiation of therapy, and personal plans for sleep education, therapy discomfort, and comorbid insomnia or circadian conditions can be instituted.
Multi-center studies occur within the registry mechanism. A double-blinded, randomized, sham-controlled, crossover trial examined the effect of Inspire® stimulation versus sham stimulation, each therapy given for 2 weeks controlling for treatment order [51]. The study randomized almost 90 participants. After 1 week, the AHI response rate was 77% with active therapy and 30% with sham therapy, a difference of 47%. Similarly, ESS had a significant difference of 4.6 × between the two groups. The crossover phase showed no carryover effect. Upper airway stimulation effectively treated both REM and NREM sleep disordered breathing. In this design which is unique in terms of technology, studies show this therapy with Inspire® reduced OSA severity, sleepiness symptoms, and improved quality of life among participants with moderate-tosevere OSA.
There is a study of potential effects of Inspire® on cardiovascular risk. Blood pressure was examined in a retrospective study in regard to the consequences of therapy [52]. Mixed-effect models were used to compare outcomes at 2 to 6 months in 201 patients matched using propensity matching. Results were adjusted for therapy adherence. PAP showed greater improvement in blood pressure, but HNS was associated with greater improvement in sleepiness symptoms. Results need to be confirmed in studies of better experimental designs.
Since the FDA trial, Inspire Medical Systems has developed a second-generation IPG for the Inspire® that has advanced features for programming. This device is compatible with MRI imaging of the brain and limbs and is being assessed for safety in MRI imaging of the chest, abdomen, and pelvis. The second-generation patient remote has improved functionality—a capacity to follow therapy use and timing and remote (cloud) reporting of data. Smaller IPG units and elimination of the sensing lead altogether are in development.
Other HNS Devices in Trials
Three other devices reported clinical trials at one level or another. None have been compared to each other in formal trials, and none have long-term safety and efficacy reports. Direct comparison of the stimulation strategies has not been done.
Almost at the same time as the Inspire® device, a device from Apnex Medical (St Paul, MN, USA) began a sequence of clinical studies towards a goal of FDA approval. The technology was based on HNS delivered to a cuff placed on the main trunk of CNXII, more proximal to that described for Inspire®, and had an IPG neurostimulator and two respiratory sensing leads (impedance technology) used to synchronize stimulation to inspiration. Selection was based on patient-derived information and the polysomnogram with predominantly obstructive hypopneas rather than apneas, and a DISE examination was not part of the inclusion profile. The stimulation profiles for the cuff electrodes are not detailed, and IPG programming is for pulse width, frequency, and current amplitude. Imaging studies suggested that the effect was to increase the oropharyngeal and retropalatal dimensions. This device looked promising in phase II safety and efficacy trials [53]. There were patients who did extremely well, and the mean fall in AHI in 31 moderate to severe patients was 45%. However, the FDA pivotal phase III trial (ClinicalTrials.gov Identifier: NCT0144660) was terminated early by the company given a likelihood that efficacy outcomes might not be met to support FDA approval.
The Genio® (NCT03868618) received FDA authorization in 2021 through the Breaththrough Device Designation for treatment of OSA in those with concentric collapse of the velopharynx. It uses bilateral implantation of electrodes at the point of insertion of the CN XII into the base of the tongue. It is externally charged and controlled by a disposable patch, worn on the patient’s chin. Genio® received a European CE mark in 2019. There are reports of safety and efficacy [54], similar to the other HNS devices.
The aura6000 by LivaNova (formerly ImThera, San Diego CA) is currently in the midst of a phase III clinical trial (THN3: ClinicalTrials.gov Identifier: NCT02263859). This device places a 6-electrode cuff on the trunk of CN XII [55, 56] and does not have a synchronizing trigger, but rather the device cycles stimulation across the different electrodes in order to activate muscles of the tongue to open the upper airway during sleep. A DISE is not required for eligibility. The device is programmed through a physician’s computer. The reported phase II trial found a reduction in AHI of 53%, selection based solely on patient information and the polysomnogram, i.e., without a DISE [57]. A case series suggested that turning off the device did not rapidly result in a return to baseline AHI levels, implying either a training effect or improvements in upper airway function [58]. These results were not replicated in the 1-week withdrawal study of the Inspire® device [44]. It is unknown whether this is a unique feature to aura6000 device.
There are two meta-analyses of all clinical data from the available HNS studies from the four devices investigating objective and subjective outcomes and side effects [59, 60••]. Each report examined data from 16 studies and 381 patients, and the methodology was common one—a comprehensive literature search of PubMed and Scopus and examination of papers meeting criteria (objective and subjective outcomes and adverse events) by two independent reviewers. In this review of the currently available data, there was shown efficacy with the mean AHI reduced by 21/h (95%CI, 16.9–25.3), mean ODI reduced by 15/h (95%CI, 12.7–17.4), mean ESS reduced by 5 (95%CI, 4.2–5.8), and mean FOSQ improved by 3 (95%CI, 2.6–3.4); all showed meaningful changes in objective and subjective domains of clinical efficacy.
Cost Considerations
For the US patient covered by insurance there may be out-of-pocket costs for deductibles and co-pays; the impact of this can be considerable. Surgical assessments and up-to-date sleep assessments (HST or PSG) prior to implantation may be needed. For the Inspire procedure, a DISE procedure is needed, yet may not result in the identification of the patient as an ideal candidate.
For the Inspire® device, the estimated lifetime incremental cost effectiveness ratio (ICER) is $39,471 per quality-adjusted life year (QALY) for patients meeting the STAR inclusion criteria [61]. This cost is less than the currently accepted cost-effectiveness threshold in the USA of $40–50 K/QALY, but more than CPAP, which has an ICER of $15,915/QALY. Relative to other implants, Nyxoah has an external power source, and the argument is that over an expected device lifetime, cost might be reduced by a third.
As for insurance coverage, neurostimulation devices are expensive compared to PAP or oral appliance. Approvals are more likely now than in 2015–2020 if the narrow selection criteria are followed; moreover, there is a patient cost to denial in terms of continued moderate to severe OSA, if left untreated. Additional costs to insurance and to the patient occur if other forms of less predictable or less durable therapy like surgical anatomic procedures are required. There are reports of gender bias in approvals and delays despite evidence that an implant will equally produce equal objective and subjective benefit [62].
Selected Clinical Populations
Prior Anatomic Surgeries
One of the first reports involved a case of a patient with persistent symptoms and findings of OSA, including an AHI > 30/h, who responded to HNS to an AHI of < 10 despite a history of several multi-level procedures, including an uvulopalatopharyngoplasty (UPPP) with revision, a genioglossus advancement, and a maxillomandibular advancement [17]. The post-award registry (ADHERE) was queried as to whether previous palate or hypopharyngeal surgery was associated with efficacy of treatment of obstructive sleep apnea [19]. Previous palate and hypopharyngeal (tongue, epiglottis, or maxillofacial) procedures were documented. Any previous surgery, previous palate surgery, and previous hypopharyngeal surgery were not clearly associated with a better treatment response. A single-center case series in Germany confirmed this lack of effect of prior surgery and post-implant efficacy of surgery in non-responders after HNS implant. The implications are that upper airway surgery should be considered in patients with persistent OSA after UAS implantation if the obstruction is identified at the level of velum and oropharynx. However, upper airway surgery during an assessment for implantation may not improve HNS outcomes [63].
Inspire® has a research-exception approval protocol for those with Downs Syndrome (DS). This group of patients and their families often do not accept positive pressure airway support devices or tracheotomy. In a series of 6 adolescents, HNS placement reduced AHI below 2 in 2 patients and > 56% in the others, accompanied by improvements in the quality of life of the child and the parent [64]. A report of 20 patients was similar and documented ~ 9.2 h of nightly use [65]. There are no reports of HNS in other OSA pediatric patients in general practice with residual AHI after adenotonsillectomy.
In the ADHERE registry, an international prospective sub-study (NCT02907398) examined the natural history of an implant compared to similar patients in whom the initial insurance coverage was denied [62]. There was an ~ 50% reduction in apnea–hypopnea index in repeat sleep studies in those who underwent implant upper airway stimulation; greater improvements were noted in subjective tendency to doze using the Epworth score in the implanted patients. The implication is that moderate to severe OSA will not improve over time if implant coverage is denied.
Another is a single-center comparison of anatomic and non-anatomic surgical procedures—transoral robotic surgery vs. Inspire®. A retrospective chart review identified and compared patients with BMI and AHI criteria for HNS who either had HNS or anatomic surgery. Defined as AHI < 5, the outcomes with HNS was ~ 70% and with robotic surgery ~ 10%. Studies like this are needed to develop and evaluate treatment algorithms such as a staged approach to CPAP-intolerant patients seeking surgical management of OSA [66].
There are 2 reports of comparison to anatomic procedures. In one [67], AHI outcomes were compared between implant patients with moderate to severe OSA who underwent Inspire® (Inspire Medical Systems) and an historical cohort those who had underwent traditional airway reconstructive surgery, specifically uvulopalatopharyngoplasty (UPPP). Both traditional surgery and HNS were effective in patients with moderate to severe OSA with CPAP intolerance, but HNS to an AHI of < 5 occurred more often in Inspire®. A second study compared patients with expansion sphincter pharyngoplasty to stimulator implant. Differences in gender, age, and preoperative AHI might have contributed to surgical successes by AHI (86% with anatomic surgery and 65% with the implant), patients reaching an AHI less than 10 and 5" is incomplete. In general, the consensus is that upper airway stimulation in the selected patients with OSA shows comparable or improved outcomes to a cohort of patients undergoing expansion pharyngoplasty [68].
Age effectiveness was addressed in a matching protocol using the ADHERE database [69]. Sixty-two patients older than 64 years and who received an implant for UAS were identified, and a younger group matched for AHI, BMI, and Epworth Sleepiness Scale. While co-morbidities were significantly higher in those > 65 years, patients met the other implant criteria, and other chronic medical conditions were under stable management. In these analyses, no significant difference is found between the outcomes of study and control group for AHI and the oxygen desaturation index and subjective outcomes. Serious adverse events did not occur in both groups, and surgical implantation time did not differ. Thus, this treatment seems safe and effective in eligible older people with stable cardiopulmonary disorders.
For all devices, the most common risks are those associated with the immediate implantation [59, 60••]. If underlying health and medical conditions exist, such as those which put one at higher risk for any surgery, then, this technology might not be a good option. As with any surgery, there is a short-term risk of pain, bleeding, and healing, but usually managed without narcotics. Patients with chronic conditions like platelet disorders or immunodeficiency should be considered ineligible or proceed carefully in a case-by-case basis.
Up to this time, there have been no deaths at implant. This is attributed to the selection of medically stable patients who have not had serious illness or hospitalization for at least 6 months, and to physician training, preparation, and post-operative team management. Procedures in the USA are same-day procedures, unless surgical complications occur. There are a handful of patients who might require overnight monitoring, for instance, to observe after evacuation of a post-operative hematoma or a slow recovery from general anesthesia. Pain at the incision site is mild to moderate, mitigate in days, and when compared to UPPP is minor in nature [67].
Reported adverse events are generally minimal and not life threatening. A complication specific to the mechanism of the device is a post-operative, temporary tongue weakness reported in ~ 20%, and most resolved spontaneously within a week, the longest being 1 year. Uncommonly, activation of the device at 1 month is delayed by this phenomenon, despite no functional impairment of tongue function. Bleeding and infection in the post-operative period can be managed by those with experience with implanted devices in general. Three Inspire® devices were explanted: two due to discomfort and one due to septic arthritis. Between 12 and 48 months, two Inspire® patients required procedures to address sensing lead displacement. Longer-term risks in ADHERE related to repetitive tongue stimulation included discomfort with the electrical stimulation while awake and dry mouth upon arising [43, 44]. Pain (6.2%:0.7–16.6), tongue abrasion (11.0%:1.2–28.7), and internal (3.0%:0.3–8.4) or external device (5.8%:0.3–17.4) malfunction were adverse events found in ~ 1000 studies [59, 60••].
There are reported cases of Inspire IPG dysfunction after electrical cardioversion. At least two patients with HGNS device dysfunction had received cardioversion via anterolateral electrode pad placement. Three patients had received multiple shocks. All four patients experienced a change in device functionality or complete cessation of functionality after electrocardioversion. These patients came to attention because the device did not work after one or more shocks. Operative replacement of the Inspire IPG system in this series was the solution [70], and the system needs to tested after any such encounter.
The implant “system” can spontaneously fail or dysfunction. The handheld patient controller battery depletes and will deplete quickly especially if it is carried about during the day as activation is caused by movement. This problem is common, and a workaround is to remove or reverse an AAA battery when travelling. In < 1% of implants there is a problem with a lead. The nerve electrode would dislodge from the IPG 12 to 18 months after implant in first-generation models; however, the current device has an improved connector required close attention to tightly sit in the IPG. The sensing lead can dislodge from the extrapleural space with or without known trauma to the chest. The pacer may also fire or activate inappropriately, leading to discomfort during wakefulness, but this is usually because the device is left on or is activated by the remote if it is carried in a pocket. In one case, a patient’s device was activated by a malfunctioning automobile radio. An activation delay upon starting and the “Pause” function make it appear that the activation is spontaneous when it is not. The Inspire IPG battery will eventually fail, requiring another surgery to replace the unit. This entails a smaller incision and was done without complication in patient populations who were in phase II and III trials of Inspire®.
Other Neuroceutical Approaches for OSA
Alternative approaches in the neurostimulation space for OSA are beginning to gain traction based on the physiology of breathing as an active brain-driven process; however, the hints came from applied basic studies in animals and increasing understandings of upper airway neuromechanical function. For instance, studies of the effects of lung volume on upper airway compliance led to the recent studies of how caudal pharyngeal traction with sternothyroid muscle contraction via ansa cervicalis stimulation can stabilize the pharynx. Stimulation of the medial branch of the right hypoglossal nerve with and without transient ultrasound-guided fine-wire stimulation of the branch of the ansa cervicalis nerve plexus innervating the right sternothyroid muscle was tested during drug-induced sleep endoscopy [71]. Observed airway cross-sectional area, and expiratory airflow, significantly increased with each stimulation alone. Combining ansa cervicalis stimulation with HNS increased retropalatal cross-sectional area and increased expiratory airflow, suggesting decreases in pharyngeal collapsibility. This line of work of stimulation of the ansa cervicalis directed towards the sternothyroid muscle shows improved upper airway size and/or stiffness, or it may augment hypoglossal stimulation efficacy [72].
Conclusions
HNS can significantly reduce AHI in moderate-to-severe OSA patients, and produce symptom relief, given certain inclusion and exclusion criteria. Reports from trials of three devices appear to provide support that this line of therapy works in selected patients. In the largest (Inspire®) cohorts, those intolerant of CPAP with a lower BMI (~ 32 or less), an AHI < 65, and a favorable anterior–posterior pattern of velopharyngeal collapse on DISE do better, but a more distal, and bilateral, implantation may address this phenotype of closure. Despite strict inclusion criteria, up to one-third of CPAP-intolerant patients do not meet the “success” criteria for HNS therapy by Sher AHI criteria, yet the remainder use the therapy > 90% of the time compared to the 20% or less adherence to PAP therapy. Adverse events are not serious, often limited to post-operative healing, and post-implant profiles suggest a reasonable risk benefit for patients in whom CPAP therapy is not used.
Individual centers have published protocols in which there is a multi-disciplinary and multi-step approach to the management of obstructive sleep apnea [73••]. The surgical consensus is that the knowledge and skills for neurostimulation are embedded in the field now and for the future [27].
Limitations to widespread adoption of HNS include the invasiveness, cost, and the pre-implantation evaluations. In comparison, CPAP and oral appliances when used are relatively cost-effective and non-invasive and initially intuitive in application. For these reasons, HNS is not currently considered a first-line treatment option.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Benjafield AV, et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: a literature-based analysis. Lancet Respir Med. 2019;7(8). https://doi.org/10.1016/S2213-2600(19)30198-5.
Drager LF, Togeiro SM, Polotsky VY, Lorenzi-Filho G. Obstructive sleep apnea: a cardiometabolic risk in obesity and the metabolic syndrome. J Am Coll Cardiol. 2013;62(7). https://doi.org/10.1016/j.jacc.2013.05.045.
Weaver TE, Sawyer AM. Adherence to continuous positive airway pressure treatment for obstructive sleep apnoea: implications for future interventions. Indian J Med Res. 2010;131:245–58.
Morsy NE, et al. Obstructive sleep apnea: personal, societal, public health, and legal implications. Rev Environ Health. 2019;34(2). https://doi.org/10.1515/reveh-2018-0068.
Sawyer AM, Gooneratne NS, Marcus CL, Ofer D, Richards KC, Weaver TE. A systematic review of CPAP adherence across age groups: clinical and empiric insights for developing CPAP adherence interventions. Sleep Med Rev. 2011;15(6). https://doi.org/10.1016/j.smrv.2011.01.003.
Sunwoo BY, Light M, Malhotra A. Strategies to augment adherence in the management of sleep-disordered breathing. Respirology. 2020;25(4). https://doi.org/10.1111/resp.13589.
Sher AE, Schechtman KB, Piccirillo JF. The efficacy of surgical modifications of the upper airway in adults with obstructive sleep apnea syndrome. Sleep. 1996;19(2). https://doi.org/10.1093/sleep/19.2.156.
Sommer JU, et al. Tonsillectomy with uvulopalatopharyngoplasty in obstructive sleep apnea. Dtsch Arztebl Int. 2016;113(1–2):1–8. https://doi.org/10.3238/arztebl.2016.0001
Ramar K, et al. Clinical practice guideline for the treatment of obstructive sleep apnea and snoring with oral appliance therapy: an update for 2015. J Clin Sleep Med. 2015;11(7). https://doi.org/10.5664/jcsm.4858.
Ng JH, Yow M. Oral appliances in the management of obstructive sleep apnea. Sleep Med Clin. 2019;14(1). https://doi.org/10.1016/j.jsmc.2018.10.012.
Strohl KP, Cherniack NS, Gothe B. Physiologic basis of therapy for sleep apnea. Am Rev Respir Dis. 1986;134(4). https://doi.org/10.1164/ajrccm/136.2.526.
Decker MJ, Haaga J, Arnold JL, Atzberger D, Strohl KP. Functional electrical stimulation and respiration during sleep. J Appl Physiol. 1993;75(3). https://doi.org/10.1152/jappl.1993.75.3.1053.
Decker M, Yamauchi M, Strohl KP. Keep the airway open and let the brain sleep. Am J Respir Crit Care Med. 2014;190(11). https://doi.org/10.1164/rccm.201410-1939ED.
Camacho M, et al. Thirty-five alternatives to positive airway pressure therapy for obstructive sleep apnea: an overview of meta-analyses. Expert Rev Respir Med. 2018;12(11). https://doi.org/10.1080/17476348.2018.1522253.
Strohl KP, Butler JP, Malhotra A. Mechanical properties of the upper airway. Compr Physiol. 2012;2(3). https://doi.org/10.1002/cphy.c110053.
•• Eckert DJ, White DP, Jordan AS, Malhotra A, Wellman A. Defining phenotypic causes of obstructive sleep apnea: identification of novel therapeutic targets. Am J Respir Crit Care Med. 2013;188(8). https://doi.org/10.1164/rccm.201303-0448OC. Important as a summary of the causal pathways in OSA and the relative role of muscle stimulation to the anatomic and ventilatory control contributions to repetitive apneas during sleep.
Strohl M, Strohl K, Palomo JM, Ponsky D. Hypoglossal nerve stimulation rescue surgery after multiple multilevel procedures for obstructive sleep apnea. Am J Otolaryngol Head Neck Med Surg. 2016;37(1). https://doi.org/10.1016/j.amjoto.2015.08.008.
Heiser C, Thaler E, Boon M, Soose RJ, Woodson BT. Updates of operative techniques for upper airway stimulation. Laryngoscope. 2016;126. https://doi.org/10.1002/lary.26158.
Kezirian EJ, et al. Previous surgery and hypoglossal nerve stimulation for obstructive sleep apnea. Otolaryngol Head Neck Surg. 2019;161(5). https://doi.org/10.1177/0194599819856339.
Eichler C, Sommer JU, Stuck BA, Hörmann K, Maurer JT. Does drug-induced sleep endoscopy change the treatment concept of patients with snoring and obstructive sleep apnea? Sleep Breath. 2013;17(1). https://doi.org/10.1007/s11325-012-0647-9.
De Vito A, et al. European position paper on drug-induced sedation endoscopy (DISE). Sleep Breath. 2014;18(3). https://doi.org/10.1007/s11325-014-0989-6.
De Vito A, et al. European position paper on drug-induced sleep endoscopy: 2017 Update. Clin Otolaryngol. 2018;43(6).https://doi.org/10.1111/coa.13213
Van De Heyning PH, et al. Implanted upper airway stimulation device for obstructive sleep apnea. Laryngoscope. 2012;122(7). https://doi.org/10.1002/lary.23301.
Vanderveken OM, et al. Evaluation of drug-induced sleep endoscopy as a patient selection tool for implanted upper airway stimulation for obstructive sleep apnea. J Clin Sleep Med. 2013;9(5). https://doi.org/10.5664/jcsm.2658.
Vroegop AVMT, Vanderveken OM, Van de Heyning PH, Braem MJ. Effects of vertical opening on pharyngeal dimensions in patients with obstructive sleep apnoea. Sleep Med. 2012;13(3). https://doi.org/10.1016/j.sleep.2011.08.005.
Steffen A, Frenzel H, Wollenberg B, König IR. Patient selection for upper airway stimulation: is concentric collapse in sleep endoscopy predictable? Sleep Breath. 2015;19(4). https://doi.org/10.1007/s11325-015-1277-9.
Suurna MV, et al. Improving outcomes of hypoglossal nerve stimulation therapy: current practice, future directions, and research gaps. Proceedings of the 2019 International Sleep Surgery Society Research Forum. J Clin Sleep Med. 2021;17(12). https://doi.org/10.5664/jcsm.9542.
Borek RC, Thaler ER, Kim C, Jackson N, Mandel JE, Schwab RJ. Quantitative airway analysis during drug-induced sleep endoscopy for evaluation of sleep apnea. Laryngoscope. 2012;122(11). https://doi.org/10.1002/lary.23553.
Vroegop AV, et al. Drug-induced sleep endoscopy in sleep-disordered breathing: Report on 1,249 cases. In Laryngoscope. 2014;124(3). https://doi.org/10.1002/lary.24479.
Remmers JE, DeGroot WJ, Sauerland EK, Anch AM. Pathogenesis of upper airway occlusion during sleep. J Appl Physiol Respir Environ Exerc Physiol. 1978;44(6). https://doi.org/10.1152/jappl.1978.44.6.931.
Popratiloff AS, et al. Hypoglossal and reticular interneurons involved in oro-facial coordination in the rat. J Comp Neurol. 2001;433(3). https://doi.org/10.1002/cne.1145.
Streppel M, et al. Morphological connections between the hypoglassal and facial nerve in the brain stem of the rat. HNO. 2000;48(12):911–6. https://doi.org/10.1007/s001060050687.
Ateş S, Karakurum E, Dursun N. Origin, course and distribution of the hypoglossal nerve in the new zealand rabbit (Oryctolagus cuniculus L). J Vet Med Ser C Anat Histol Embryol. 2011;40(5). https://doi.org/10.1111/j.1439-0264.2011.01080.x.
Mediano O, et al. Obstructive sleep apnea: emerging treatments targeting the genioglossus muscle. J Clin Med. 2019;8(10). https://doi.org/10.3390/jcm8101754.
Heiser C, Knopf A, Hofauer B. Surgical anatomy of the hypoglossal nerve: A new classification system for selective upper airway stimulation. Head Neck. 2017;39(12). https://doi.org/10.1002/hed.24864.
Eastwood PR, et al. Bilateral hypoglossal nerve stimulation for treatment of adult obstructive sleep apnoea. Eur Respir J. 2020;55(1). https://doi.org/10.1183/13993003.01320-2019.
Elshebiny T, Venkat D, Strohl K, Hans MG, Alonso A, Palomo JM. Hyoid arch displacement with hypoglossal nerve stimulation. Am J Respir Crit Care Med. 2017;196(6). https://doi.org/10.1164/rccm.201612-2521LE.
Steffen A, et al. Changes in breath cycle sensing affect outcomes in upper airway stimulation in sleep apnea. Laryngoscope Investig Otolaryngol. 2020;5(2). https://doi.org/10.1002/lio2.334.
•• Heiser C, et al. Post-approval upper airway stimulation predictors of treatment effectiveness in the ADHERE registry. Eur Respir J. 2019;53(1). https://doi.org/10.1183/13993003.01405-2018. This is a recent report on the outcomes in the post-approval ADHERE of HNS and will have prior references and discussions of what has been learned now that the technology is deployed in the US and Europe.
Weidenbecher M, Tamaki A, Cabrera C, Strohl K. Improved exposure of the hypoglossal branches during hypoglossal nerve stimulator implantation: clinical outcomes of twenty patients at a single institution. Clin Otolaryngol. 2019;44(1). https://doi.org/10.1111/coa.13221.
Thaler E, et al. Results of the ADHERE upper airway stimulation registry and predictors of therapy efficacy. Laryngoscope. 2020;130(5). https://doi.org/10.1002/lary.28286.
Ong AA, O’Brien TX, Nguyen SA, Gillespie MB. Implantation of a defibrillator in a patient with an upper airway stimulation device. Laryngoscope. 2016;126(2). https://doi.org/10.1002/lary.25683.
Woodson BT, et al. Upper airway stimulation for obstructive sleep apnea: 5-year outcomes. Otolaryngol Head Neck Surg. 2018;159(1). https://doi.org/10.1177/0194599818762383.
Woodson BT, et al. Randomized controlled withdrawal study of upper airway stimulation on OSA: short- and long-term effect. Otolaryngol Head Neck Surg. 2014;151(5). https://doi.org/10.1177/0194599814544445.
Woodson BT, et al. Three-year outcomes of cranial nerve stimulation for obstructive sleep apnea: the STAR trial. Otolaryngol Head Neck Surg. 2016;154(1). https://doi.org/10.1177/0194599815616618.
Soose RJ, et al. Upper airway stimulation for obstructive sleep apnea: self-reported outcomes at 24 months. J Clin Sleep Med. 2016;12(1). https://doi.org/10.5664/jcsm.5390.
Hasselbacher K, Hofauer B, Maurer JT, Heiser C, Steffen A, Sommer JU. Patient-reported outcome: results of the multicenter German post-market study. Eur Arch Otorhinolaryngol. 2018;275(7). https://doi.org/10.1007/s00405-018-5017-1.
Withrow K, Evans S, Harwick J, Kezirian E, Strollo P. Upper airway stimulation response in older adults with moderate to severe obstructive sleep apnea. Otolaryngol Head Neck Surg. 2019;161(4). https://doi.org/10.1177/0194599819848709.
Hofauer B, Steffen A, Knopf A, Hasselbacher K, Heiser C. Patient experience with upper airway stimulation in the treatment of obstructive sleep apnea. Sleep Breath. 2019;23(1). https://doi.org/10.1007/s11325-018-1689-4.
Coca KK, et al. Hypoglossal nerve stimulation usage by therapy nonresponders. Otolaryngol Head Neck Surg. 2021. https://doi.org/10.1177/01945998211036867.
Heiser C, et al. Effect of upper airway stimulation in patients with obstructive sleep apnea (Effect): a randomized controlled crossover trial. J Clin Med. 2021;10(13). https://doi.org/10.3390/jcm10132880.
Walia HK, et al. Upper airway stimulation vs positive airway pressure impact on BP and sleepiness symptoms in OSA. Chest. 2020;157(1). https://doi.org/10.1016/j.chest.2019.06.020.
Kezirian EJ, et al. Hypoglossal nerve stimulation improves obstructive sleep apnea: 12-month outcomes. J Sleep Res. 2014;23(1). https://doi.org/10.1111/jsr.12079.
Sommer JU, Hörmann K. Innovative surgery for obstructive sleep apnea: nerve stimulator. Adv Otorhinolaryngol. 2017;80. https://doi.org/10.1159/000470880.
Zaidi FN, Meadows P, Jacobowitz O, Davidson TM. Tongue anatomy and physiology, the scientific basis for a novel targeted neurostimulation system designed for the treatment of obstructive sleep apnea. Neuromodulation. 2013;16(4). https://doi.org/10.1111/j.1525-1403.2012.00514.x.
Friedman M, et al. Targeted hypoglossal nerve stimulation for the treatment of obstructive sleep apnea: six-month results. Laryngoscope. 2016;126(11). https://doi.org/10.1002/lary.25909.
Mwenge GB, Rombaux P, Dury M, Lengele B, Rodenstein D. Targeted hypoglossal neurostimulation for obstructive sleep apnoea: a 1-year pilot study. Eur Respir J. 2013;41(2). https://doi.org/10.1183/09031936.00042412.
Rodenstein D, Rombaux P, Lengele B, Dury M, Mwenge GB. Residual effect of THN hypoglossal stimulation in obstructive sleep apnea: a disease-modifying therapy. Am J Respir Crit Care Med. 2013;187(11). https://doi.org/10.1164/rccm.201211-2129LE.
Certal VF, et al. Hypoglossal nerve stimulation in the treatment of obstructive sleep apnea: a systematic review and meta-analysis. Laryngoscope. 2015;125(5). https://doi.org/10.1002/lary.25032.
•• Kompelli AR, Ni JS, Nguyen SA, Lentsch EJ, Neskey DM, Meyer TA. The outcomes of hypoglossal nerve stimulation in the management of OSA: a systematic review and meta-analysis. World J Otorhinolaryngol Head Neck Surg. 2019;5(1). https://doi.org/10.1016/j.wjorl.2018.04.006. An independent assessment of the state of the art in 2019.
Pietzsch JB, Liu S, Garner AM, Kezirian EJ, Strollo PJ. Long-term cost-effectiveness of upper airway stimulation for the treatment of obstructive sleep apnea: a model-based projection based on the STAR trial. Sleep. 2015;38(5). https://doi.org/10.5665/sleep.4666.
Mehra R, et al. Upper airway stimulation versus untreated comparators in positive airway pressure treatment-refractory obstructive sleep apnea. Ann Am Thorac Soc. 2020;17(12). https://doi.org/10.1513/AnnalsATS.202001-015OC.
Steffen A, Abrams N, Suurna MV, Wollenberg B, Hasselbacher K. Upper-airway stimulation before, after, or without uvulopalatopharyngoplasty: a two-year perspective. Laryngoscope. 2019;129(2). https://doi.org/10.1002/lary.27357.
Diercks GR, et al. Hypoglossal nerve stimulation in adolescents with down syndrome and obstructive sleep apnea. JAMA Otolaryngol Head Neck Surg. 2018;144(1). https://doi.org/10.1001/jamaoto.2017.1871.
Caloway CL, et al. Update on hypoglossal nerve stimulation in children with down syndrome and obstructive sleep apnea. Laryngoscope. 2020;130(4). https://doi.org/10.1002/lary.28138.
Yu JL, Mahmoud A, Thaler ER. Transoral robotic surgery versus upper airway stimulation in select obstructive sleep apnea patients. Laryngoscope. 2019;129(1). https://doi.org/10.1002/lary.27487.
Shah J, Russell JO, Waters T, Kominsky AH, Trask D. Uvulopalatopharyngoplasty vs CN XII stimulation for treatment of obstructive sleep apnea: a single institution experience. Am J Otolaryngol Head Neck Med Surg. 2018;39(3). https://doi.org/10.1016/j.amjoto.2018.03.003.
Huntley C, Chou DW, Doghramji K, Boon M. Comparing upper airway stimulation to expansion sphincter pharyngoplasty: a single university experience. Ann Otol Rhinol Laryngol. 2018;127(6). https://doi.org/10.1177/0003489418771395.
Zhu Z, et al. Selective upper airway stimulation in older patients. Respir Med. 2018;140. https://doi.org/10.1016/j.rmed.2018.06.002.
Vasconcellos AP, Huntley CT, Schell AE, Soose RJ, Boon MS. Dysfunctional hypoglossal nerve stimulator after electrical cardioversion: a case series. Laryngoscope. 2019;129(8). https://doi.org/10.1002/lary.27488.
Kent DT, Scott WC, Zealear D, Schwartz AR. Ansa cervicalis stimulation increases pharyngeal patency in patients with obstructive sleep apnea. J Appl Physiol. 2021;131(2). https://doi.org/10.1152/japplphysiol.00076.2021.
Kent DT, Zealear D, Schwartz AR. Ansa cervicalis stimulation: a new direction in neurostimulation for OSA. Chest. 2021;159(3). https://doi.org/10.1016/j.chest.2020.10.010.
•• Jain N, Rodin J, Boon MS, Huntley CT. A systematic approach to the evaluation and management of obstructive sleep apnea: the Jefferson protocol. Am J Otolaryngol Head Neck Med Surg. 2021;42(2). https://doi.org/10.1016/j.amjoto.2020.102866. This report describes an example of a multidisciplinary, team approach in which surgical referral processes are discussed in relation to other options in CPAP intolerant patients.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Approval
All authors have reviewed and approved the manuscript.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Conflict of Interest
Yee-Hsee Hsieh, Amy Schell, Eric Yeh, Madeleine Strohl, and Thomaz Fleury each declare no competing interests. Kingman P. Strohl was a site PI in the Inspire Phase III trial and in the ADHERE registry; he is a consultant to Sommetrics, 7 Dreamers, Merck Pharmaceuticals; and provides editorial content for Up-to-Date, Merck Manual, and Medscape.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Sleep and Technology
This is original work and has not been published or submitted elsewhere.
Rights and permissions
About this article

Cite this article
Hsieh, YH., Schell, A.E., Yeh, E. et al. Neurostimulation in the Management of Obstructive Sleep Apnea. Curr Sleep Medicine Rep 8, 168–179 (2022). https://doi.org/10.1007/s40675-022-00233-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40675-022-00233-6