
Abstract
Continuous culture techniques were developed in the early twentieth century to replace cumbersome studies of cell growth in batch cultures. In contrast to batch cultures, they constituted an open concept, as cells are forced to proliferate by adding new medium while cell suspension is constantly removed. During the 1940s and 1950s new devices have been designed—called “automatic syringe mechanism,” “turbidostat,” “chemostat,” “bactogen,” and “microbial auxanometer”—which allowed increasingly accurate quantitative measurements of bacterial growth. With these devices cell growth came under the external control of the experimenters and thus accessible for developing a mathematical theory of growth kinetics—developed mainly by Jacques Monod, Aron Novick and Leo Szilard in the early 1950s and still in use today. The paper explores the development of continuous culture devices and claims that these devices are simulators for standard cells following specific requirements, in particular involving mathematical constraints in the design and setting of the devices as well as experiments. These requirements have led to contemporary designs of continuous culture techniques realizing a specific event-based flow algorithm able to simulate directed evolution and produce artificial cells and microorganisms. This current development is seen as an alternative approach to today’s synthetic biology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In his 1949 paper on The Growth of Bacterial Culture Jacques Monod presented a mathematical model for the growth of microorganisms, today known as the “Monod equation.” Similar in form to the Michaelis–Menten equation of enzyme kinetics (Michaelis and Menten 1913), the Monod equation is commonly used in current modelling and simulation projects, either as Michaelis–Menten–Monod kinetics for modelling metabolic pathways of cell simulations in molecular biology or for modelling bacterial growth and substrate utilization in environmental engineering, e.g. for activated sludge models of sewage treatment simulations. While Leonor Michaelis and Maud Menten had derived their equation in the 1910s at the laboratory of the Urban Hospital in Berlin by studying the kinetics of invertase (saccharase), an enzyme that catalyses the hydrolysis of sucrose, Monod—inspired by André Lwoff at the Institute Pasteur in Paris as well as by an early genetics paper of Salvador Edward Luria and Max Delbrück (1943; Monod 1966)—derived his equation in the late 1940s by studying bacterial growth limited by one enzyme system. Both studies, Michalis’ and Menten’s one as well as Monod’s unveil an interesting practice of using continuous culture techniques as “simulators” for standard models of cell processes and cells, that could easily be translated in mathematical models. Therefore, Michalis and Menten “linearized” the enzymatic process in their experimental setting, while Monod designed a new device, called bactogen, which realized a specific version of so-called “continuous culture techniques.”
Analysing the effort of experimentally fitting life processes and cells to mathematics by studying the history of continuous culture techniques is the topic of this paper. The aim is to challenge the notion of “simulation.” In the current philosophical literature on modelling and simulation mathematical models are usually seen as conceptual representations of processes and entities, and simulations are interpreted as computational applications of mathematical models (e.g. Sismondo and Gissis 1999; Fox Keller 2003; Humphreys 2004; Lenhard et al. 2007; Winsberg 2010; Humphreys and Imbert 2011; Morrison 2015). For instance, in Stephan Hartmann’s basic definition, simulation is an imitation of “one process by another process […] carried out by a computer. […] More concretely, a simulation results when equations of the underlying dynamic model are solved” (1996, pp. 77, 79, 83). Therefore, it is not surprising that models and simulations are sometimes seen as fictions in philosophy of science (Suarez 2010).
However, as the case of the Monod equation will show, some continuous culture techniques can be seen as simulators. This view should not be confused with the view on simulations as instruments or machines for performing numerical experiments (Dowling 1999; Winsberg 2003; Gramelsberger 2010; Gramelsberger and Feichter 2011). Continuous culture techniques as simulators—and that is the main hypothesis of my paper—are designed in order to perform quantitative experiments that help to derive at mathematical models, while the mathematical constraints determine the design of the devices. This interdependency between continuous culture techniques and mathematical constraints is of interest. Thus, these devices have to fulfil various characteristics in order to qualify them as simulators. First, they have to be designed to provide a quantitative, not a qualitative understanding of processes and entities (quantification). Second, in order to obtain such a quantitative understanding the experimental design has to practically externalize internal parameters and make them accessible to the experimenter (parameter control). This is a common practice in physics that links model-based simulations and experiment-based measurements much closer to each other (Morrison 2015), but this practise is much more difficult to achieve for micro- and molecular biology. Therefore, they have, third, to fulfil mathematical constraints materially (mathematical constraints). Finally, external parameters have to be set up in such a way that they realize a standard model of a process or an entity (standard model). In sum, continuous culture techniques as simulators do not investigate “natural” processes or entities, but create highly artificial ones. A research strategy familiar to biologists using modified “model organisms” (e.g. Rader 2004) as well as to engineers deeply involved in “how things ought to be—how they ought to be in order to attain goals, and to function” (Simon 1996, p. 5).
What is of interest here is on the one hand the role of mathematics as a materialized practice of constraining and standardization guiding experimental design and on the other hand the linking of continuous culture techniques with theory by this practice. Exploring mathematics as a materialized practice beyond a pure representational function is a new approach to philosophy of mathematics. It expands David Baird’s concept of “thing knowledge” for the case of mathematics (Baird 2004). Perhaps, one could argue that continuous culture techniques as simulators complement Baird’s typology of instruments consisting of “models; devices that create a phenomenon; and measuring instruments” (p. 5). However, continuous culture techniques as simulators can also be seen as an intersection of the two latter subclasses. While Baird places emphasis on the “working knowledge” (p. 15) employed in instrumental epistemology and owed to the interaction of the experimenter with the material agency of the device—a kind of tacit or implicit knowledge; the “experimental simulator knowledge” is guided by explicit mathematical constraints. Nevertheless, the implementation of mathematical constrains in experimental designs is a development involving many trials and errors. Therefore, continuous culture techniques as simulators have some similarities with analogue simulators performing computations, e.g. like electromechanical pendulum simulators for modelling and solving the moment equation of motion by damped oscillation (Lange 2011).
The comparison with analogue simulators performing computations is insofar instructive as it unveils a fifth characteristic of continuous culture techniques as simulators. While computer simulations of mathematical models imitate “one process by another process” (Hartmann 1996, p. 77)—a real, observed process by a semiotically created one (mathematical model, computer code); continuous culture techniques as simulators have to be processual (flow design). Monod’s equation could not have been derived from static batch cultures, but necessarily required the experimental mastering of flow. Against this backdrop the paper outlines the experimental mastering of flow (Sect. 2), it discusses the various characteristics of continuous culture techniques as simulators by exploring the development of continuous culture techniques in detail (Sect. 3) and it concludes with some remarks on the role of continuous culture techniques as simulators in today’s molecular biology becoming fully self-controlled cybernetic simulators (Sect. 4) and some general thoughts on using them for simulating directed evolution (Sect. 5).
2 Experimental mastering of flow
2.1 Kinetic thinking
A major influence that conceptually set the stage for flow design resulted from quantitative studies of (bio)chemical reactions (Burton 1998). According to the historian of chemistry Viktor Kritsman, kinetics became a topic of chemistry in the mid-nineteenth century, when chemistry was mainly organic chemistry (1997). In 1850 Ludwig Wilhelmy studied the catalytic reaction of acid on sugar using the new method of polarimetry. Polarimetry measures the torsion angle of polarized light reflected by diluted substances, and thus determines the quantitative characteristics of the reaction process and its molecular transformations. Based on this method, Wilhelmy derived a first reaction-rate equation, depending mainly on the reaction rate constant k. As often in history of science, Wilhelmy’s results remained unnoticed until the 1880s (Kritsman 1997, p. 293). Thus, the foundations had to be rediscovered by researchers like Guldenberg and Waage (1867), Van’t Hoff (1884), Arrhenius (1889), and others.
In 1913 Michaelis and Menten used polarimetry to conduct their study on enzymatic reactions, deriving their famous equation. Michaelis and Menten chose the invertase enzyme because “the ease of [polarimetrically] measuring its activity means that this particular enzyme offers especially good prospects of achieving the final aim of kinetic research, namely to obtain knowledge on the nature of the reaction from a study of its progress” (1913, p. 1). Various researchers like Claude S. Hudson had already arrived at mathematical descriptions, proposing that the inversion by invertase follows a simple logarithmic function (which was false, even as a first approximation), and Victor Henri, who provided a more complex model that came closer to experimental observations than Hudson’s (Henri 1903; Hudson 1908). However, as Michaelis and Menten pointed out in 1913, these studies ignored important aspects involved in enzyme kinetics, in particular the influence of the hydrogen ion concentration and the mutarotation of sugar, and therefore arrived at false results. In contrast, Michaelis and Menten developed more precise experiments, as their aim was to develop a mathematical model that fit better with experimental findings. They introduced an acetate mixture as a buffer to keep the hydrogen ion concentration constant and decided “to take samples of the inversion reaction mixture at known time intervals, to stop the invertase reaction and to wait until the normal rotation of glucose is reached before measuring the polarization angle” (p. 2). This approach avoided errors in the rate of inversion by a change in the polarization of freshly formed glucose. All in all they conducted seven experiments with varying quantities of a sucrose stock solution and took nine samples of each experiment at known time intervals, in which “every measurement [of the polarization angles] recorded in the protocol is the average of 6 individual measurements” (p. 2). Then they inferred the velocity of the inversion from the change of rotation angle per minute and developed their kinetic equation for the velocity υ of the enzyme reaction.
The ingenious idea of Michaelis and Menten was to focus on measuring the starting velocity. This was the decisive aspect of their experimental setting, because at the beginning the reaction velocity is fast and the reaction is not influenced by cleavage products or other inhibitory influences. This experimental decision reduced complexity of enzymatic processes, later called the “quasi-stationary approximation.” It means that certain reactions will reach their steady state much faster than others. Thus, short transient can be neglect. As Michaelis and Menten pointed out:
Henri has already shown that the cleavage products of sugar inversion, glucose and fructose, have an inhibitory effect on invertase action. Initially, we will not attempt to allow for this effect, but will choose experimental conditions which avoid this effect. Since the effect is not large, this is, in principle, simple. At varying starting concentrations of sucrose, we need only to follow the inversion reaction in a time range where the influence of the cleavage products is not noticeable. Thus, we will initially measure only the starting velocity of inversion at varying sucrose concentrations. The influence of the cleavage products can then be easily observed in separate experiments. (Michaelis and Menten 1913, pp. 2, 3).
Fortunately for Michaelis and Menten, invertase quickly reaches a steady state, thus the rapid equilibrium assumption was valid for their case and they could correctly conclude that maximum velocity is reached when all enzymes have been associated into an enzyme–substrate-complex. In this case velocity depends only on the concentration of the substrate as expressed in the Michaelis constant. However, they were aware of this restrictive assumption and tried to quantitatively determine the influence of the cleavage products by deriving dissociation constants, which could be added as single-valued parameters to the equation.
Although Michalis and Menten claimed that the avoided effect was not large and thus legitimated the chosen experimental conditions, the consequences of this choice were huge. By focussing on the starting velocity Michaelis and Menten were able to linearize the experimental process and to articulate a simple mathematical model. Instead of a complex model of five non-linear partial differential equations they derived at a single ordinary differential equation assuming that the medium is well mixed (using ordinary instead of partial differential equations) and that the number of each species is large (using ordinary instead of stochastical differential equations). In other words: The experimental design realized mathematical constrains which were mirrored by a simplified standard model of enzymatic reaction. Just to show the extent of simplification: A current weather or climate model is based on seven non-linear partial differential equations and is therefore mathematically not solvable. It can only be numerically simulated with the help of supercomputers (Gramelsberger and Feichter 2011). The same holds for a complex model of five non-linear partial differential equations. However, as each species involved in enzymatic reaction requires a differential equation, a model of five non-linear partial differential equations does not realistically describe metabolic pathways; not to mention Michaelis’ and Menten’s linearized kinetic model. Nevertheless, Michaelis’ and Menten’s simplification corresponds today to the Systems Biology Mark-up Language’s (SBML) attribute “fast”, used to reduce the number of equations needed to simulate metabolic pathways.
That Michaelis’ and Menten’s rapid equilibrium assumption does not hold for most of the enzymatic reactions was already pointed out by Briggs and Haldane in 1925. It is valid only if the substrate reaches equilibrium on a much faster time scale than the product, but this presents a special case, e.g. for invertase. In contrast to Michaelis and Menten’s linearized model based on the rapid equilibrium assumption, Briggs and Haldane suggested a quasi-steady-state assumption considering that enzyme-substrate-complex converts into an enzyme and a product following a rate constant. In such a model, enzymes can again associate with substrate, and products can have an inhibitory effect. Thus, they introduced feedback to their model and conceived a reaction cycle, not a reaction chain as Michaelis and Menten did. Nevertheless, the form of Briggs’ and Haldane’s equation is similar to Michaelis’ and Menten’s equation and the biological literature usually refers to it as “Michaelis–Menten kinetics” and “Michaelis–Menten–Monod kinetics,” respectively.
These early studies in enzyme kinetics have established an increasingly clear understanding of processes expressed in mathematically comprehensible terms of reaction rates, reaction equilibrium, velocity constants, and the law of mass action—stating that the rate of a reaction is directly proportional to the concentrations of the reactants. This kind of “kinetic thinking” was shaped in analogy to mechanical forces in physics. The basic idea behind such kinetic studies was, and still is, that processes and their temporal development can be observed and measured over the course of their transitioning from an unstable state to a final stable state of equilibrium or steady state. It should be noted, however, this basic idea exactly defines the mathematical constraints, which have to be implemented in the design of the continuous culture techniques as well as of the experiments.
2.2 Continuous-flow methods of (bio)chemitsry
This kinetic design of quantitative experiments has in itself an interesting history which is connected to the experimental mastering of flow. Until the 1920s experimental methods like polarimetry used by Wilhelmy (1850) and Michaelis and Menten (1913) were applied to stationary solutions in test tubes. Chemists and biologists had to use stopwatches on time-scales of seconds or minutes for studying reaction kinetics. This coarse temporal resolution, of course, limited research to studies on slow reactions. Hence, in 1923 Hamilton Hartridge and Francis Roughton developed a new method that reduced the observable to milliseconds, arguing that
the dynamical study of slow reactions has thrown so much light upon the general mechanism of chemical reactions that the value of a method for measuring the velocity of rapid reactions can hardly be questioned (Hartridge and Roughton 1923, p. 377).
Hartridge and Roughton proposed a continuous-flow method for measuring rapid reactions by homogeneous mixing of two substances in order to establish unstable initial conditions (mixing chamber). By using a movable spectroscope to measure the passage of the mixture down an observation tube with a mean length of 1–30 cm, temporal measurements could be derived from the constant rate of flow and the diameter of the tube. “Supposing the rate of flow down the observation tube to be 100 cm per second, then the first observation can be made in one-hundredth of a second after the reaction” (p. 380). Flow, in this case, converted time into length and thus allowed fine-scale measurements within the kinetic design.
The continuous-flow method was improved by Glenn Millikan in order to conduct quantitative experiments with haemoglobin in 1936 and by Britton Chance for enzymatic reactions in 1943.Footnote 1 Chance’s device was of particular interest. First, because
the kinetic curve was obtained directly as a trace, which could be photographed [… Second, because] the requisite extension of the time range and economy of material were secured with an accelerated flow method and a very sensitive reaction meter with a high speed of response: a manual syringe drive gave an initial impetus to the reactants; and, the subsequent variations of flow velocity, and therefore of time interval, and the corresponding variations in the extent of reaction, measured photoelectrically at a fixed distance from the mixing chamber, were continuously recorded with a double-beam cathoderay tube. (Dalziel 1953, p. 79)
With this device, flow could be accelerated or stopped. By making use of electronic devices such as photomultipliers, oscilloscopes for rapid recording, and valve amplifiers, Chance’s stopped-flow devices made it possible to study the intermediates of the enzyme mechanisms, thus overcoming pure steady-state kinetics. Continuous-flow and stopped-flow methods became the core methods for physiological investigations in biology.
To chemists, the terms “reaction mechanism” and “catalysis” imply events that involve changes in covalent binding. To those interested in the molecular basis of enzyme action or in the transduction of energy and signals in biological systems, the study of reaction mechanisms has a much wider meaning. […] Classical enzymology has developed from the investigations into metabolism and biosynthesis. The principal interest was the synthesis and degradation of compounds in the presence of catalytic concentrations of enzymes, which are negligible compared with those of the metabolites. (Gutfreund 1999, p. 459)
2.3 Early continuous culture techniques in microbiology
Observing and measuring (bio)chemical processes in a test tube is one version of the kinetic design. The other is to study (bio)chemical processes in cells and microorganisms in order to understand the influence of the environment (nutrition medium) on their growth and development. Both are preconditions for doing microbiology. As Jacques Monod stated:
The study of the growth of bacterial cultures does not constitute a specialized subject or branch of research: it is the basic method of Microbiology. […] More precisely, we shall concern ourselves with the quantitative aspects of the method, with the interpretation of quantitative data referring to bacterial growth (Monod 1949, p. 371).
The growth rates of cultures of microorganisms became a subject of studies in the 1920s, for instance on algae (for early reviews see Bold 1942; Myers and Clark 1944; Meyers and Cramer 1948) and bacteria (Jordan and Jacobs 1944). However, these early experiments were carried out in batch cultures, where cells were grown in a fixed volume of nutrient culture medium under varying conditions, such that it was impossible to study the process of growth quantitatively. As David W. Tempest, an expert in this field, complained:
However, beside the fact that growth is such a basic aspect of microbial behaviour, little attention was paid to the principles which underlie it until the advent of continuous culture techniques. Indeed, prior to the 1940’s it was generally considered sufficient to record growth as being either evident (+) or not (−), with the occasional recourse to semiquantitative flight of fancy such as +++, ++, and ±! (Tempest 1978, p. 2)
Therefore, “a fresh approach to the problem of bacterial growth” in closed-batch cultures was required, making use of flow in cell experiments in order to keep the cell “in a rigidly controlled environment […] over a relatively long period” (Jordan and Jacobs 1944, p. 580). This new approach applied continuous culture techniques, which continuously and automatically add fresh medium while cell suspension is continuously removed to keep the culture volume constant, so that the growth rate of the cells can be controlled.Footnote 2 The very first devices of the late 1920s and early 1930s were built “for the purpose of obtaining bacterial substance in quantity” (Haddon 1928, p. 299), avoiding the arduousness of constant subculturing. But, researchers like C.E. Clifton and J.P. Cleary, by comparing “stationary and continuous flow cultures of E. coli in peptone” (Clifton and Cleary 1934, p. 541), increasingly recognized that the phases of bacterial growth play an important role in determining the characteristics of an organism. Thus, the potential of continuous cultivation for theoretical studies of cell growth was recognized and continuous culture techniques turned into important research tools for microbiology from the 1940s on. The prototypical continuous culture techniques, built in the 1940s and 1950s to grow cells at their maximum rate in order to study them quantitatively, were called “automatic syringe mechanism” (Sims and Jordan 1941, 1942), “turbidostat” (Myers and Clark 1944), “chemostat” (Novick and Szilard 1950a), “bactogen” (Monod 1950), and “microbial auxanometer” (Anderson 1953), respectively. It was Monod who predicted that continuous culture techniques would become ideal devices for the investigation of the kinetics of formation of cellular constituents such as ribonucleic acid, DNA, proteins, and enzymes (Monod 1950).
3 Continuous culture techniques as simulators
3.1 Fitting cells to mathematics
The decisive aspect of continuous culture techniques is the permanent washout of the culture and the addition of fresh medium. This “open” concept—in contrast to the “closed” concept of batch cultures (Herbert 1964)—forces cells to proliferate permanently. In batch cultures, after a lag period where cells increase in volume and mass but do not divide, cells continue to grow while their number increases exponentially by division, followed by a stationary phase in which growth and division cease. However, the problem of cell growth in batch cultures is that the size, composition, and functional characteristics of cells vary considerably during growth, “often making interpretation of results difficult” (Kubitschek 1970, p. 3). Thus, continuous cultures have been developed as “a technique which has heretofore been purely preparative in scope [… now] converted into an analytical method for studying the kinetics of cell growth” (Anderson 1953, p. 733).
This “analytical method” is based on manipulating growth patterns by controlling them such that cells are presumed to be in a constant state of logarithmic growth. That is, the cell growth is kept continually close to steady state, while cells are homogeneously dispersed through permanent stirring. This implements the mathematical constraint of perfect mixing.Footnote 3 Thus, the rates for division and growth can be more easily controlled and sustained for a longer period. Cell concentration can be maintained and set independently of growth rate, and the chemical environment can be kept constant for a longer time. Furthermore, it is the only state in the growth cycle where reliable numerical values can be obtained for quantification, as Monod explained aptly:
During the exponential phase, the growth rate is constant. It is reasonable to consider that a steady state is established, where the relative concentrations of all the metabolites and all the enzymes are constant. It is in fact the only phase of the growth cycle when the properties of the cells may be considered constant and can be described by a numeric value, the exponential growth rate, corresponding to the over-all velocity of the steady state system. (Monod 1949, p. 382)
Thus, the experimental conditions turn into externally controllable and measurable parameters; they come under the purview of the experimenter and the mathematical theory (parameter control). This is common practice in physical experiments, but challenging for microbiological ones—requiring working knowledge in the sense of Baird (2004).
In fact, “numerous difficulties have arisen. The present procedure of maintaining pure culture conditions is the result of a gradual improvement in technique” (Myers and Clark 1944, p. 108). Jack Myers and L.B. Clark refer here to their study on the photosynthetic behaviour of algal cells obtained with a device called “turbidostat with photocell.” Their motivation was clear:
It would seem that if a culture could be continuously diluted so as to be maintained always at one point on its growth curve, then the effects of changing internal conditions might be eliminated entirely. This would afford at once (1) a source of experimental material of high uniformity and/or (2) a means of stabilizing internal variables so that relation of culture conditions to photosynthetic behavior might be systematically explored. (Myers and Clark 1944, pp. 103, 104)
The turbidostat combined a culture chamber, a bubble tube for inserting a gas mixture to stir the suspension, a withdrawal tube to run out suspension, a solenoid valve which adds fresh medium when it is activated, and other details, e.g. a cotton filter, a heater, etc. (Myers and Clark 1944, pp. 104 et seq.). “In operation a sample (about 1 ml.) is withdrawn daily into a flask of glucose-peptone broth as a check against bacterial or mould contamination. Many cultures have been run for a month or more without contamination” (p. 106).
3.2 Developing a specific instrument epistemology of flow design
The intriguing aspect of the turbidostat for maintaining the culture always at one point on its growth curve was the automated and self-controlled addition of fresh medium by a photodetector.Footnote 4 Photodetectors eased and automatized the estimation of cell growth by measurements of turbidity based on determinations of transmitted or scattered light. They replaced the laborious determination of cell growth by dry weight of bacteria. In the case of the turbidostat with photocell, the control of the turbidity of the suspension by an optical sensor controlled the valve. As they described it:
An off-balance current flows through the primary photocell [P1] circuit causing the galvanometer light spot to move across the phototube. The electronic relay then actuates the solenoid valve and allows new culture medium to flow in, diluting the algae and increasing the illumination on P1 until a zero current again obtains through the galvanometer. In this way the algal suspension “grows” up the chamber. The design of the apparatus is such that conditions of illumination are independent of the total amount of the culture. (Myers and Clark 1944, p. 107)
For the turbidostat photocells from General Electric, a phototube (No. 922) and an electronic relay (No. 2051) from the Radio Corporation of America (RCA) were used to build a photometric device that could control the electromechanically operated solenoid valve for adding fresh medium. It is obvious that high sensitivity was needed for the optical detection of the turbidity of a cell culture. This required an advanced experimental design, comprised of a bridge circuit with two photodetectors:
The first to detect changes in the cell concentration, and the second as a reference detector that is directly exposed to the light source, but retains essentially constant sensitivity to changes in turbidity. A response to changes less than 5% in turbidity has been reached with some instruments. (Kubitschek 1970, p. 17)
The decisive difference between turbidostats and chemostats is the way in which growth is controlled. While in turbidostats growth is controlled internally by a photodetector to maintain constant turbidity, in chemostats it is controlled externally by limiting the supply of a critical growth factor.Footnote 5 Such a critical growth factor could be, for instance, light for cells capable of photosynthesis, a carbon source, a required amino acid or an inorganic ion. Turbidostats can be converted into chemostats by simply switching off the optical control system and supplying nutrient at a constant rate. However, the goal in using both types of devices was the same: providing constant cell concentrations and constant chemical conditions so that the continuous culture techniques could be used to study the impact of a single parameter on cell growth while other parameters were fixed. As Aron Novick and Leo Szilard pointed out for their study on spontaneous mutation of a B strain of E. coliFootnote 6:
We are interested in this stationary state in the particular case in which the growth rate of bacteria is determined by the concentration in the growth tube of a single growth factor (in our specific case tryptophane). By this we mean that the concentration of a single growth factor (tryptophane) in the growth tube is so low that a small change in it appreciably affects the growth rate of the bacteria, and at the same time the concentration of all other growth factors in the growth tube is so high that a small change in them has no appreciable effect on the growth rate of the bacteria. (Novick and Szilard 1950b, p. 710)
The same applied to the bactogen (Monod 1949, 1950), which was a device similar to the chemostat. It was used to design “the conditions of an experiment where G [total growth] is to be estimated [… in such a way] that a single limiting factor is at work” (Monod 1949, p. 379).
The decisive advantage of parameters coming under access of the experimenter, although they are partly delegated to machines, is that theoretical considerations expressed in mathematical models can guide data collection. As Monod pointed out: “Actually, the introduction around 1935 of instruments fitted with photoelectric cells has contributed to a very large degree to the development of quantitative studies of bacterial growth” (Monod 1949, p. 376). The disadvantages are the growing dependence on automatization technology and the increase in effort needed to prepare an adequate environment for the advanced devices; in other words: improving the implementation of the mathematical constraints as a decisive part of the instrument epistemology. In the case of the turbidostat with a photocell, “satisfactory operation of the entire apparatus depends upon the stability and sensitivity of the primary photocell circuit” (Myers and Clark 1944, p. 108). Therefore, it had to be insured that temperature and light were held constant and that gas mixtures were reliable, including solutions for achieving high rates of aeration by using bubbling methods to control the suspension, for cooling, for automatic control of pH, and so on. Another simultaneous advantage and disadvantage was that photodetectors effectively automatized recording, but introduced an indirect method of estimating cell growth. This required cross-checks with direct methods and the standardization of devices and records. But this was not the case in the very beginning. Monod complained that
not enough efforts have been made to check them [optical techniques] against direct estimations of cell concentrations or bacterial densities. Furthermore a variety of instruments, based on different principles, are in use. The readings of these instruments are often quoted without reference to direct estimations as arbitrary units of turbidity, the word being used in an undefined sense, or as “galvanometer deflections” which is worse. (Monod 1949, pp. 376–377)
Nevertheless, automatization of the experiments increased. In 1953, Paul Anderson introduced an advanced application of the turbidostat. His “microbial auxanometer” used photodetectors (turbidimeter) and magnetic valves like earlier turbidostats, but combined three growth tubes, each with its own turbidimeter, so that three experiments could be run in parallel. Another improvement was the automatic recording of washout rate (ω(t)) “accomplished by counting the drops of solution added in fixed, arbitrarily adjustable time intervals and registering the counts on a commercial traffic counter” (Anderson 1953, pp. 733–734).
3.3 Aiming at standard cells and mathematical unification
Due to automatization, the mathematical constraint of logarithmical growth was better achieved and the obtained quantitative data sets became increasingly larger, more accurate, more regularly collected, and more uniform than data collected by hand. Although generation time or growth rate cannot be seen as the only measures of changes in cell cultures, the main advantage of continuous cultures is clearly that “it frees the system of the variability that accompanies changes in growth rate and non steady-state environments” (James 1961, p. 42). Thus, it allowed comparative studies which directed the way from standardized cell processes to standard cells, for instance for algae (Myers and Cramer 1948). Such generalizations implied standardization of every part of the experiment, e.g. for optical density measurements indicating cell concentrations.
The data, expressed as cell concentrations, may then be considered as referring to “standard cells,” equal in size to the real bacteria observed during the exponential phase, larger than bacteria in the stationary phase and probably smaller than those in the acceleration phase (Monod 1949, p. 377).
The standard cell is an artificial cell at logarithmic growth. It refrains from changes in size during different phases of growth, but helps make results comparable. It also allowed for unification in form of a general theory of growth, but, of course, introduced simplifications. This general theory of growth was outlined as a comprehensive theory by Monod (1949, 1950) and by Novick and Szilard (1950a, b, 1951). It was presented at the 1951 Cold Spring Harbor Symposia on Quantitative Biology (Novick and Szilard 1951) and it can be summarized briefly as follows. Growth of bacteria in an undiluted system is described as
where N = number of bacteria per ml, t = time, and α = mean division/growth rate. For a continuously diluted system the equation is given as
where ω = washout or dilution rate given as the ratio of w/V (with w = rate of flow of liquid ml/h; V = volume of the culture). Thus, it is clear that for continuous culture techniques, ω, the dilution or washout rate, respectively, is the most important parameter, as the density of microorganisms is to remain constant when α equals ω. To ensure equality of growth and washout rate is precisely the task of the photodetectors in the turbidostat, which keep the organisms at the maximum growth rate possible given the particular choice of chemical and physical conditions.Footnote 7 However, the chemostat and the bactogen established a different way of stabilizing continuous cultures. Both differ from the turbidostat insofar as “the flow rate is held at some fixed value below the maximum growth rate. Furthermore the nutrient medium is composed of a large excess of all required nutrilites but one” (Novick 1955, p. 99). Thus, a single nutrilite becomes the controlling growth factor limiting the growth rate of bacteria at a certain point. Now the saturation kinetics introduced by Michaelis and Menten (1913) is applied. The growth rate increases as long as the concentration of the required substrate factor is sufficient, while the growth rate begins to decrease once it is lowered to a certain amount. “At this point the system becomes stabilized, the bacterial growth rate now remaining equal to the washing-out rate, and the bacterial density remaining constant at a value N” (Novick 1955, p. 100). Thus, for chemostat cultures the washout rate determines the cell division rate.
If meaningful results are to be achieved by this method, assumptions on the rate of cell division are required with regard to its dependence upon the concentration of a limiting substrate. Based on these assumptions an equation for culture generation time and cell generation time can be obtained, as well as an equation for the specific growth rate of continuous cultures—finally leading to the fundamental equation of growth in continuous cultures:
where S0 = substrate concentration supplied, S = residual substrate concentration, and Y = cell mass produced per unit substrate at the washout rate ω. However, this equation has no general solution for Y, as the cell division rate depends on the concentration of a limiting substrate. Specific solutions can be inferred under certain restrictive assumptions. Such a specific solution was first given by Monod 1942, assuming for Y that
Spelled out, it means that “the growth rate constant αm is the asymptotic value of α as substrate concentration is allowed to increase indefinitely, […] and the saturation constant K is the value of s for which α = αm/2″ (Kubitschek 1970, p. 28). This specific dependence (0.5 αm) was found empirically for sugar (Monod 1942) and later for amino acids.
Having arrived at a general theory of growth, continuous culture techniques were now used to study certain quantitative characteristics of the growth cycle for manifold species under various chemical and physical conditions. This led to the discovery by Novick, Szillard and Monod that, besides the diversity of species and conditions, three growth constants could be identified: total growth, exponential growth rate, and growth lag. As they claimed:
That these definitions [of growth] are not purely arbitrary and do correspond to physiologically distinct elements of the growth cycle is shown by the fact that, under appropriately chosen conditions, the value of any one of the three constants may change widely without the other two being significantly altered. The accuracy, the ease, the reproducibility of bacterial growth constant determinations is remarkable and probably unparalleled, so far as biological quantitative characteristics are concerned. (Monod 1949, p. 392)
4 Simulators in today’s biology
4.1 Critique of the emerging quantitative style
The kinetic design of quantitative experiments in (bio)chemical processes and in cell growth realized mathematical constraints leading to material realizations of artificial states of standard cells. This shift was not appreciated by all biologists, because it gave
one the impression that bacteria and other cells are considered to be rather fixed entities that can be dealt with much as a physicist deals with an atom or a molecule. […] The tendency has been to regard the cell as a model unit whose functions are invariant in time. To some degree, the use of continuous culture techniques tends to perpetuate this simplification by virtue of the necessity of reducing such studies to the kinetics of numbers and populations. (James 1961, p. 34)
Another critique addressed the underlying idea that the relationship between growth rate and generation time is too simple (Powell 1956). On the basis of this idea, age and generation time distributions were widely neglected and the assumption that maximum viability is given in continuous cultures could be doubted.
Most important was the fundamental critique on the mathematical constraints required for obtaining valid data: The “assumption of ‘perfect’ mixing–of instantaneous and homogeneous dispersal of the ingoing medium–greatly simplifies the theoretical and experimental analysis of continuous cultures, […] but it is not at all obvious that an adequate approximation to ‘perfect’ mixing is practically attainable” (Powell 1956, p. 509). However, the method of continuous cultures is inevitably based on this precondition, because the growth and washout rate are not strictly equal unless mixing is perfect. Powell also pointed out that two implicit assumptions were routinely made:
(a) that growth rate responds instantly to changes in medium concentration; (b) that growth is not inhibited or promoted by products of metabolism or by imbalance in the medium caused by the organisms themselves. Failure of either assumption can demonstrably lead to sustained oscillations in population density. (Powell 1956, p. 509)
Despite his critique, Powell experimentally validated Monod’s heavily criticized assumption that no matter what the initial values, the system will always adjust itself to the steady state. “It is this automatic self-adjusting property of the system that makes continuous culture a readily feasible possibility” (Herbert et al. 1956, p. 606). However, the critique made clear that further refinements in the continuous culture techniques as simulators were needed, although the overall opinion was that, besides all these simplifications, the basic principles of the theory were sound. The continuous culture theory as established in the 1950s still applies to current research (Marlière and Mutzel 2000; Marlière et al. 2011), but the effect of deposition of bacteria (biofilms/wall growth) turned out to be a far more challenging problem during the past decades than was clear in the literature of the 1940s and 1950s. However, “the theory of homogeneous continuous culture depends on the concept of perfectly mixed reactors” (Solomons 1972, p. 219).
4.2 Road to fully self-controlled cybernetic simulators
The automatization of continuous culture techniques with analog devices such as photodetectors, magnetic valves, and traffic counters increasingly merged with embedded computers. It paved the way to digitalization, as G. L. Solomons reported:
We have now [from 1960s on] reached the stage when the process variables are computer controlled and in some circumstances the computer can be used as part of a feedback loop to mathematically analyse and then vary the system to achieve optimisation of the process (Solomons 1972, p. 217).
As digital computers were quite expensive, and their use required specialized knowledge, in the beginning only a few devices were equipped with digital computer technology in the laboratories. But the road to digitalization was the precondition for the optimization of growth processes and this, in turn, supported the use of continuous culture techniques for industrial purposes. So-called “fermenters”—based either on chemostat or turbidostat methods–became increasingly employed in the emerging biotechnology industry, for instance in the production of beer, yeast, and drugs.Footnote 8 These industry fermenters were much larger than the lab devices and introduced new problems.Footnote 9 Fermenters became the enabling devices for the technological promises of today’s synthetic biology (Carlson 2010; Gramelsberger et al. 2013).Footnote 10
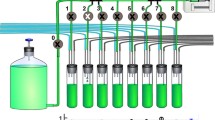
However, further refinements in the design of continuous cultures techniques for lab research directed them closer to devices for which it is not easy to decide if they are simulators or computers that perform simulations. Today’s continuous culture techniques like the GM3 device, developed by Philippe Marlière from the Institute Pasteur in Paris and Rupert Mutzel from the Freie Universität in Berlin (2000; see Fig. 1), perfect the automated proliferation of cells in suspension. GM3 is a recent version of the bacterial auxanometer, a fully self-controlled device which can maintain a bacterial culture for years by operating autonomously. The GM3 combines a growth chamber equipped with an optical sensor and integrated in a net of pipes, to dilute new medium (controlled by the optical sensor), to stir the culture with air bubbles for a homogeneous distribution of cells, to convey the overflow/washout of the culture (syphon), and to transfer the culture to a second growth chamber every 6–24 h, depending on cell generation time, to clear the other chamber. The last of these improvements turned out to be decisive for maintaining a bacterial culture over a long period, avoiding the effect of adhesion—a result of more than 10 years of development.Footnote 11 Thus, “no cultivated variant can escape dilution and selection for faster growth through the formation of biofilms” (Marlière et al. 2011, p. 7247).

GM3 device developed by Philippe Marlière (Institute Pasteur Paris) and Rupert Mutzel (Freie Universität Berlin) (photographed by Gabriele Gramelsberger in 2012)
Thus, continuous cultures techniques have turned into fully self-controlled cybernetic simulators, constituting an autonomous biological-technological circuit: The bacterial growth controls the technical system that records the impact of the bacterial growth by counting and analysing it, which, in turn, optimizes the growth rate by subtle changes in adding substrates and diluting bio mass. Of course, this kind of self-controlled cybernetic device requires digital computers that can accomplish control and analyses automatically and in real time. All in all, a fine tuning of the experiment can be achieved, which is outstanding and which cannot be accomplished by human experimenters. The biological-technological circuit replaces the experimenter, as no human experimenter can dilute a continuous culture every 10 min, 7 days a week, over a period of months. However, it is not just a matter of capacity, but also of accuracy. The differences in the opening time of valves to add growth medium are in the realm of milliseconds. Only a fully self-controlled device can conduct these fine-grained, long-term experiments.
For GM3 an extrinsic form of digitalization can be distinguished from an intrinsic one. The extrinsic digitalization is given by employing computer chip-based tools for controlling and recording. The GM3 is equipped with a central processing unit (CPU) and a data logger (see Fig. 1). The CPU processes the signal of the turbidity control (which transforms the qualitative turbidity data via a threshold setting into discrete 1/0 decisions) to the valve relay that controls the valves, diluting the relaxing or the stressing medium, respectively. Furthermore, the CPU steers the other valves at regular time intervals, thus operating as a clock. The data logger records the opening time of valve 1, which provides the relaxing medium. The decisive information results from the opening time of valve 1, which allows calculation of the dilution rate, while every other parameter is fixed and has been calibrated beforehand, including the air pressure for stirring the culture, turbidity threshold and the delivery rate of the valves (ml/sec). Thus, the actual opening times of valve 1 provide all information necessary for analysing the experiment’s course of cell growth due to the outlined theory of continuous culture techniques.
Intrinsic digitalization is established by the fluidic format of the GM3 device as a conditional pulse-feed regime. The temporal choreography of the various valves schedules the biological system. Nutrient pulses every 10 min allow generation times of 2 h, while 20-min pulses lead to 4 h generation times, thus shaping the various experiments. In other words, E. coli is trained to follow a specific rhythm of on/off feeding/starvation and to survive and adapt in a scheduled environment. Furthermore, an “if/then else-logic” is introduced into the experimental design as known from computer algorithms: if population density falls below a fixed threshold (controlled by the optical sensor) then pulses of the relaxing medium are provided, else pulses of the stressing medium are given. Thus, the self-controlled and self-optimizing cybernetic simulator, establishing a biological-technological circuit, introduces an event-based flow algorithm to biological cultures (personal communication with Rupert Mutzel 2012).Footnote 12
4.3 Simulating evolution
An device like the GM3 opens up the possibility to conduct directed evolution experiments. Directed/experimental evolution is not a new field. Back in (1892), Henri de Varigny proposed long-term evolution experiments in his book Experimental Evolution. In the early twentieth-century evolution experiments were conducted with Drosophila and later with E. coli. In 1988 Richard Lenski, a pioneer of experimental evolution, started to grow twelve E. coli strains in flasks on a daily rhythm by hand—an overwhelmingly laborious task, culminating in 50,000 generations of the long-term lines in February 2010. “Each day, including weekends and holidays, someone in my group withdraws 0.1 ml from a culture and transfers that into 9.9 ml of fresh medium. The bacteria grow until the glucose is depleted, and then sit there until the same process is repeated the next day” (Lenski 2011, p. 32). However, automatization with continuous culture techniques offers directed/experimental evolution experiments in a much more convenient way. The GM3 is the first continuous culture technique that provides a fluidic format on laboratory scale that allows long-term experiments to be carried out automatically up to several months and years. Furthermore, the if/then else-logic of GM3 experiments (event-based flow algorithm) actively forces microorganisms to adapt.
For instance, in 2010 the GM3 was used to create a DNX version of E. coli by metabolic selection of a genetically modified E. coli (THY1) strain (Marlière et al. 2011). In this experiment, the THY1 culture was connected to two nutrient reservoirs: a relaxing medium containing the canonical nutrients (valve 1) and a stressing medium containing 5-chlorouracil (valve 2). Depending on the turbidity, which indicates the state of the adapting cells, at regular intervals the culture received liquid pulses of the relaxing medium (if population density fell below a fixed threshold) or of the stressing medium (if the density was equal or higher than the threshold)—thus establishing a conditional pulse-feed regime. The incorporation of thymine analogues, for instance 5-chlorouracil, can be manipulated in vivo more easily than other nucleotides, because it’s metabolism is disentangled from RNA biosynthesis. By disrupting the thyA gene for thymidylate synthase in E. coli, thymine starvation causes a rapid loss of viable cell titre and thus forces the cells to incorporate exogenous thymine analogues like 5-chlorouracil (a detailed description of the modified metabolism of THY1 is given by Marlière et al. 2011). After 23 days the resilience of both cultures to 5-chlorouracil was sufficient enough to implement a harsher regime by increasing the concentration of 5-chlorouracil. After more than 140 further days the adapted cells consumed only the stressing medium. DNA extraction and enzymatic hydrolysis revealed the massive substitution of thymine by 5-chlorouracil.
5 Conclusion
Continuous cultures techniques as simulators of cell growth and division have turned into cybernetic devices for simulating evolution and creating even DNX-organisms. Already in the 1940s, Monod and others used continuous cultures techniques to study spontaneous mutations. “Using a strain of Escherichia coli mutabile […] we showed that an apparently spontaneous mutation was allowing these originally ‘lactose-negative’ bacteria to become ‘lactose-positive’” (Monod 1966, p. 475). Thus, Monod was able to explain a specific phenomenon of enzyme adaption, he had called “diauxie” (1942), meaning that bacteria adapt to different nutrition sources they are exposed to in the growth phases. “Monod, like others, hoped to use enzymatic adaptation as a model of the controlled switching of cell identity in differentiation. From the beginning, this program was supposed to provide a general account of regulation at the cellular level” (Burian and Gayon 1999, p. 329). However, before the discovery of the structure of the DNA it was difficult to explain this finding. The quantitate approach was the “escape from the old tradition of morphological description” (Loison 2013, p. 175) and the continuous culture techniques not only offered such a quantitate approach, but also allowed to study unicellular organisms in a given and controllable milieu. This was achieved by implementing mathematical constraints materially in the design of the devices. Of course, as critics complained, this came along with simplification: “[…] it is a model built through neglect to attack the total problem” (James 1961, p. 34). Or, in other words, as Laurent Loison in his telling study on “Monod before Monod: Enzymatic Adaptation, Lwoff, and the Legacy of General Biology” pointed out:
Monod became fascinated by the experimental possibilities allowed by the exponential phase of growth of bacterial culture […]. He liked to compare such a system to a perfect gas. Individual peculiarities did not matter, and only population characteristics were relevant in order to establish scientific laws of nature. Through a new quantitative approach, Monod’s ultimate goal was indeed to physicalize biology (2013, p. 176).
However, it took more than 60 years until Monod’s general account of regulation at the cellular level became reality in form of directed evolution guiding spontaneous mutation. Continuous culture techniques are no longer used as simulators of standard processes leading to standard cells, but of evolutionary processes creating artificial cells. The mastering of flow, leading to today’s fully self-controlled fluidic format, wires up cells and microorganisms, respectively, with the device—both quantitatively steering each other and constituting a singular cybernetic circuit comparable to current flow in digital computer circuitry. Thus, these self-controlled cybernetic devices like GM3 are on the one hand simulators for artificial states of cells and microorganisms, but on the other also simulation programs for directed evolution. They constitute an alternative to the engineering of biology paradigm of synthetic biology. Instead of engineering biosynthetic circuits, biological principles (mutation, adaptation, fitness) can be utilized to achieve useful organism in so far as microorganisms can be coached to adapt to desired behaviours by mutation. Rather than creating a highly technicized version of life as synthetic biology aims at, knocking out all disturbing features like evolvability, adaptivity, and multifunctionality (Gramelsberger et al. 2013), everything can be left to the “robustness of biochemical machineries” in continuous culture techniques (Marlière et al. 2011, p. 7247). However, continuous cultures techniques are pulse-feed devices and thus turn the coaching of microorganisms into something akin to operating flight simulators; and molecular biology, by analogy, turns into a behavioural science for coaching microorganisms and cells. Of course not in order to study the natural behaviour of cells, but to study the ability of cells to adapt to pulse-feed training programs implemented by fully self-controlled fluidic formats and to “learn” new behaviours like including 5-chlorouracil.
Is this kind of bio-materialized simulation different from the computer-based simulations of computational systems biology like the Japanese E-Cell, the U.S.-American VirtualCell and the Whole-Cell, the French SyntheCell, the Dutch SiliconCell, and the German Virtual Liver Project? Not conceptually as they are all artificial in the sense that their relation to an ontologically understood “nature” is dubious, but of course ontologically as there remains a difference between bio-materialized and in silico artificial cells. However, both types of artificial cells—in particular as “standard units”—incorporate the same mathematical constraints, equations, and fluidic formats changing the traditional division of labour between theorists and experimentalists. Simulations as well as simulators inform scientific practice by processually generating quantitative access to externalized parameters of a system in order to arrive at a standardized, simplified and hence artificial system (cells/microorganisms in a medium) by controlling and manipulating these parameters. Thus, simulations/simulators are less epistemic tools for understanding real cell processes, and ever more practical devices for designing standardized cells and making artificial ones in biology.
Notes
Probably the first computer-based simulation in biology was carried out by Britton Chance, reported in a paper entitled The mechanism of catalase action. II. Electric analog computer studies in (1952).
It is also a story of transforming life into technology, comparable to the development Hannah Landecker revealed for tissue culture in medical research. “The life form of the cultured cell is a manifestly technological one: It is bound by the vessels of laboratory science, fed by the substances in the medium in which it is bathed, and manipulated internally and externally in countless ways” (2007, p. 3).
“The most important [requirement] is that the cultures should be constantly mixed, homogenized, and in equilibrium with the gas phase. This is achieved either by shaking or by bubbling air (or other gas mixtures) or both. Bubbling is often found inefficient unless very vigorous, when it may provoke foaming which should be avoided” (Monod 1949, p. 378).
Photomultipliers were used as counters in biology from the 1940s on, for instance for liquid scintillation, “in which the human eye was replaced by a highly sensitive, fast-responding photoelectric device for detecting and counting scintillations” (Rheinberger 2001, p. 148).
“It has been suggested that the difference between the two systems described above is that in the Chemostat or the Bactogen one is dealing with starved bacteria, while in the photocell controlled apparatus the bacteria are not starved” (Novick 1955, p. 101).
Novick and Szilard were interested in spontaneous mutation of a B strain of E. coli (B/1), “which is resistant to the bacterial virus T1, sensitive to the bacterial virus T5, and which requires tryptophane as a growth factor” (Novick and Szilard 1950b, p. 709).
“As the control system corrects the flow rate to maintain constant density, there will be variations in flow rate whose magnitude will depend on the sensitivity of the density-detecting system. The mean flow rate, however, will precisely equal the growth rate since the density N tends, in a properly adjusted machine of this type, neither to consistently increase or decrease” (Novick 1955, p. 99).
“The extent of application of continuous cultures in industry today is difficult to determine because this industrial information is not readily available in print […] Nevertheless, the available listing is impressive. Continuous cultures are used in the production of beer, sake, alcohol, and acetic and lactic acids; of vitamins (B12) and drugs (alkaloids); and of antibiotics (penicillin and chlortetracycline). Continuous cultures are also used for steroid conversion and the production of vaccines” (Kubitschek 1970, p. 9).
This delayed the introduction of continuous culture techniques to industry until the late 1950 s (Herbert et al. 1956).
An important improvement in the 1950s and 1960s for both domains, research as well as industry, was the maintenance of the artificial steady state for cells up to 4 months (Herbert et al. 1956). But the increase in duration came along with a serious problem, the selection of adhesive variants: so-called biofilms (wall growth). Biofilms are problematic because they falsify measurements of turbidity and prevent cells from dividing and evolving in long-term experiments. Thus, adventurous techniques were developed to avoid deposition in the culture chamber, among these a kind of “windshield wiper” (Anderson 1953, p. 734).
According to personal communication with Rupert Mutzel, the GM3 is currently the only device which succeeds in maintaining bacterial cells exclusively in suspension for such a long period.
The term “event-based algorithm” refers to a programming paradigm of information science in which the flow of the program is determined by events, e.g. inputs of sensors (Faison 2006). It is closely related to queuing theory, which has recently been used in synthetic biology “to investigate how ‘waiting lines’ can lead to correlations between protein ‘customers’ that are coupled solely through a downstream set of enzymatic ‘servers’” (Cookson et al. 2011, p. 1).
References
Anderson, P. (1953). Automatic recording of the growth rates of continuously cultured microorganisms. The Journal of General Physiology, 36, 733–737.
Arrhenius, S. (1889). Ueber die Reaktionsgeschwindigkeit bei der Inversion von Rohrzucker durch Säuren. Zeitschrift für Physikalische Chemie, 4, 226–248.
Baird, D. (2004). Thing knowledge. A philosophy of scientific instruments. Orlando: University of California Press.
Bold, H. C. (1942). The cultivation of algae. Botanical Review, 2, 70–138.
Briggs, G. E., & Haldane, J. B. S. (1925). A note on the kinetics of enzyme action. Biochemical Journal, 19(2), 338–339.
Britton, C. (1943). The kinetics of the enzyme-substrate compound of peroxidase. Journal of Biological Chemistry, 151, 553–577.
Britton, C. (1952). The mechanism of catalase action. II. Electric analog computer studies. Archives of Biochemistry and Biophysics, 37(2), 322–339.
Burian, R. M., & Gayon, J. (1999). The French school of genetics: From physiology and polulation genetics to regulatory molecular genetics. Annual Review of Genetics, 33, 313–349.
Burton, R. F. (1998). Biology by numbers: An encouragement to quantitative thinking. Cambridge, MA: Cambridge University Press.
Carlson, R. H. (2010). Biology is technology. Cambridge, MA: Harvard University Press.
Clifton, C. E., & Cleary, J. P. (1934). Oxidation–reduction potentials and ferricyanide reducing activities in glucose-peptone cultures and suspensions of Escherichia coli. Journal of Bacteriology, 28(6), 561–569.
Cookson, N. A., Mather, W. H., Danino, T., et al. (2011). Queueing up for enzymatic processing: correlated signaling through coupled degradation. Molecular Systems Bioology, 7(561), 1–9.
Dalziel, K. (1953). An apparatus for the spectrokinetic study of rapid reactions. Biochemical Journal, 55(1), 79–90.
de Varigny, H. (1892). Experimental evolution. London: Macmillan.
Dowling, D. C. (1999). Experimenting on theories. Science in Context, 12, 261–274.
Faison, T. (2006). Event-based programming: Taking events to the limit. New York: Springer.
Fox Keller, E. (2003). Models, simulation, and computer experiments. In H. Radder (Ed.), The philosophy of scientific experimentation (pp. 198–215). Pittsburgh: University of Pittsburgh Press.
Gramelsberger, G. (2010). Computerexperimente. Zum Wandel der Wissenschaft im Zeitalter des Computers. Bielefeld: Transcript.
Gramelsberger, G., & Feichter, J. (Eds.). (2011). Climate change and policy. The calculability of climate change and the challenge of uncertainty. Heidelberg: Springer.
Gramelsberger, G., Knuuttila, T., & Gelfert, A. (2013). Philosophical perspectives on synthetic biology [Special issue]. Studies in the history and philosophy of biological and biomedical sciences, 44(2).
Guldenberg, C., & Waage, P. (1867). Etudes sur les affinitis chimiques, Programme de I’Universite pour le ler semestre 1867. Christiania: Booggcr and Christlc.
Gutfreund, H. (1999). Rapid-flow techniques and theircontributions to enzymology. Trends in Biochemical Sciences, 11, 457–460.
Haddon, E. C. (1928). Apparatus for obtaining a continuous bacterial growth. Transactions of the Royal Society of Tropical Medicine and Hygiene, 21(4), 299–300.
Hartmann, S. (1996). The World as a process. In R. Hegselmann, U. Müller, & K. G. Troitzsch (Eds.), Modelling and simulation in the social sciences from the philosophy of science point of view (pp. 77–100). Dordrecht: Kluwer Academics Publisher.
Hartridge, H., & Roughton, F. J. W. (1923). A method of measuring the velocity of very rapid chemical reactions. Proceedings of the Royal Society of London, Series A, 104, 376–394.
Henri, V. (1903). Lois générales de l’action des diastases. Zeitschrift für Anorganische Chemie, 35(1), 82.
Herbert, D. (1964). Multi-stage continuous culture. In I. Malek, K. Beran, & J. Hospodka (Eds.), Continuous cultivation of microorganisms (pp. 23–44). Prague: Czechoslovak Academy of Science.
Herbert, D., Elsworth, R., & Telling, R. C. (1956). The continuous culture of bacteria; A theoretical and experimental, study. Journal of General Microbiology, 14, 601–622.
Hudson, C.S. (1908). Inversion of sticrose by invertase. Journal of the American Chemical Society 30, 1160–1166 and 1564–1583.
Humphreys, P. (2004). Extending ourselves. Computational sciences, empiricism, and scientific method. Oxford: Oxford University Press.
Humphreys, P., & Imbert, C. (Eds.). (2011). Models, simulations, and representations. New York: Routledge.
James, T. W. (1961). Continuous culture of microorganisms. Annual Review of Microbiology, 15, 27–46.
Jordan, R. C., & Jacobs, S. E. (1944). The growth of bacteria with a constant flood supply. I. Preliminary observations on Bacterium coli. Journal of Bacteriology, 48(5), 579–598.
Kritsman, V. A. (1997). Ludwig Wilhelmy, Jacobus H. van’t Hoff, Svante Arrhenius und die Geschichte der chemischen Kinetik. Chemie in unserer Zeit, 6(97), 291–300.
Kubitschek, H. (1970). Introduction to research with continuous cultures. Englewood Cliffs, NJ: Prentice-Hall.
Landecker, H. (2007). Culturing life: How cells became technologies. Cambridge, MA: Harvard University Press.
Lange, T. (2011). Computer simulation in the V2 rocket development. In: Gramelsberger, G. (ed.). From science to computational sciences. Studies in the history of computing and its influence on today’s sciences (pp. 85–96). Zurich, Berlin: diaphanes 2011 (distributed by The Chicago University Press 2015).
Lenhard, J., Küppers, G., & Shinn, T. (Eds.). (2007). Simulation: pragmatic construction of reality. (Sociology of the Sciences 25). Dordrecht: Springer.
Lenski, R. E. (2011). Evolution in action: A 50,000-generation salute to Charles Darwin. Microbe, 6, 30–33.
Loison, L. (2013). Monod before Monod: Enzymatic adaptation, Lwoff, and the legacy of general biology. History and Philosophy of the Life Sciences, 35(2), 167–192.
Luria, S. E., & Delbrück, M. (1943). Mutations of bacteria from virus sensitivity to virus resistance. Genetics, 28(6), 491–511.
Marlière, P., & Mutzel, R. (2000). Method and device for selecting accelerated proliferation of living cells in suspension. PCT/EP99/09422, Patent No. WO 00/34433, awarded 15 June 2000.
Marlière, P., Patrouix, J., Döring, V., Herdewijn, P., Tricot, S., Cruveiller, S., et al. (2011). Chemical evolution of a bacterium’s genome. Angewandte Chemie, 123, 7247–7252.
Michaelis, L., & Menten, M.L. (1913). Die Kinetik der Invertinwirkung. Biochemische Zeitschrift, 49, 333–369. (S. Roger Goody, K. A. Johnson, Trans. The Kinetics of Invertase Action, p. 34, 2011).
Millikan, G. A. (1936). The kinetics of muscle haemoglobin. Proceedings of the Royal Society of London, Series B, 120, 366.
Monod, J. (1942). Recherches sur la Croissance des Cultures Bacteriennes. Paris: Hermann and Co.
Monod, J. (1949). The growth of bacterial cultures. Annual Review of Microbiology, 3, 371–394.
Monod, J. (1950). La technique de culture continue, theorie et applications. Annales de l’Institut Pasteur, 79, 390–410.
Monod, J. (1966). From enzymatic adaptation to allosteric transition. Science, 154(3748), 475–483.
Morrison, M. (2015). Reconstructing reality. Models, mathematics, and simulations. Oxford: Oxford University Press.
Myers, J., & Clark, L. B. (1944). Culture conditions and the development of the photosynthetic mechanism, II. An apparatus for the continuous culture of chlorella. Journal of General Physiology, 28, 103–112.
Myers, J., & Cramer, M. (1948). Metabolic conditions in chlorella. Journal of General Physiology, 32, 103–110.
Novick, A. (1955). Growth of bacteria. Annual Review of Microbiology, 9, 97–110.
Novick, A., & Szilard, S. (1950a). Description of the chemostat. Science, 15, 715–716.
Novick, A., & Szilard, S. (1950b). Experiments with the chemostat on spontaneous mutations of bacteria. PNAS Proceedings of the National Academy of Sciences, 36(12), 708–719.
Novick, A., & Szilard, L. (1951). Experiments on spontaneous and chemical induced mutations of bacteria growing in the chemostat. Cold Spring Harbor Symposia on Quantitative Biology, 16, 337–343.
Powell, E. O. (1956). Growth rate and generation time of bacteria, with special reference to continuous culture. Journal of General Physiology, 15, 492–511.
Rader, K. A. (2004). Making mice: Standardizing animals for American biomedical research, 1900–1955. Princeton: Princeton University Press.
Rheinberger, H.-J. (2001). Putting isotopes to work: Liquid scintillation counters, 1950–1970. In B. Joerges & T. Shinn (Eds.), Instrumentation between science, state and industry (pp. 143–174). Dordrecht: Kluver Academic Publishers.
Simon, H. A. (1996). The sciences of the artificial. Cambridge, MA: The MIT Press.
Sims, A. L., & Jordan, R. C. (1941). A simple bacterial colony counting device. Journal of Scientific Instruments, 18, 243.
Sims, A. L., & Jordan, R. C. (1942). An accurate automatic syringe mechanism. Journal of Scientific Instruments, 19, 58–61.
Sismondo, S., & Gissis, S. (Eds. 1999). Practices of modeling and simulation [Special issue]. Science in Context, 12(2).
Solomons, G. L. (1972). Improvements in the design and operation of the chemostat. Journal of Chemical Technology and Biotechnology, 22, 217–228.
Suarez, M. (2010). Fictions in science: Philosophical essays on modeling and idealization. London: Routledge.
Tempest, D. W. (1978). Dynamics of microbial growth. Australia: Wiley.
Van’t Hoff, J. (1884). Etudes de dynamique chimique. Amsterdam: F. Muller.
Wilhelmy, L. (1850). Ueber das Gesetz, nach welchem die Einwirkung der Säuren auf den Rohrzucker stattfindet. Poggendorffs Annalen der Physik und Chemie, 81, 499–526.
Winsberg, E. (2003). Simulated experiments: Methodology for a virtual world. Philosophy of Science, 70, 105–125.
Winsberg, E. (2010). Science in the age of computer simulation. Chicago: The Chicago University Press.
Acknowledgements
I would like to thank Rupert Mutzel from the Freie Universität Berlin for supporting my study on continuous culture techniques.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gramelsberger, G. Continuous culture techniques as simulators for standard cells: Jacques Monod’s, Aron Novick’s and Leo Szilard’s quantitative approach to microbiology. HPLS 40, 23 (2018). https://doi.org/10.1007/s40656-017-0182-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40656-017-0182-x