
Abstract
Background
The familial nonmedullary thyroid cancer (FNMTC) is suspected to be a Mendelian condition in up to 3–8% of thyroid cancers. The susceptibility chromosomal loci and genes of 95% of FNMTC cases remain to be characterized. The inheritance of FNMTC appears to be autosomal dominant with incomplete penetrance and variable expressivity. The finding of the causative gene of FNMTC and the identification of patients at risk that need genetic testing were our aim.
Methods
We analyzed by whole-exome sequencing patients and non-affected relatives of five families with at least two family members affected by papillary thyroid cancer, selecting for new or extremely rare variants with predicted pathogenic value.
Results
A family showed, in all three affected members, a new loss-of-function variant (frameshift deletion) in BROX gene at 1q41 that was absent from all internal and external databases. In a second family with three affected relatives, we found an additional new BROX variant. The smaller families presented no variants in BROX or in the other causative genes studied.
Conclusions
BROX could be a new causative gene for FNMTC. Variants in BROX may result in the haploinsufficiency of a key gene involved in the morphogenesis of MVBs, in the endosomal sorting of cargo proteins, and in EGFR. Functional studies are needed to support this result. The thorough genomic analysis by NGS in all families with three or more affected members should become a routine approach to obtain a comprehensive genetic view and find confirmative second cases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thyroid cancer occurs in 7–15% of cases presenting thyroid nodules depending on age, sex, radiation exposure, and family history. Approximately 1.1% of people present this condition at some point in their life [1]. In 2015 the number of people with thyroid cancer in the USA was approximately 63,000 with 2000 deaths [2]. In particular, papillary thyroid cancer (PTC) is the commonest histologic sub-type, accounting for at least 90% of all thyroid cancers [2]. PTC remains one of the most treatable cancers with a median 5-year survival rate of 98% [2]. On the contrary, patients with familial nonmedullary thyroid cancer (FNMTC) present PTC at an earlier age, have a more multi-focal and aggressive disease with local invasion, lymph node metastases, and increased risk of recurrence and decreased survival rates [3,4,5,6,7]. FNMTC is a rare disorder, diagnosed when two or more first-degree relatives have thyroid cancer without another familial syndrome. Based on a review of the prevalence of FNMTC from Surveillance Epidemiology and End Results (SEER) database, Mayo clinic and mathematical analysis, if the definition of having two first-degree relatives with thyroid cancer is used for FNMTC, only 31–38% of members in the family are likely to carry the familial trait, with the rest being sporadic cases. The diagnosis is probable in 96–99% of the cases if at least three family members are affected. This familial condition has been reported in 3–9% of thyroid cancer cases, and in only 5% of the syndromic forms have been identified germline mutations [2]. The advent of next-generation sequencing (NGS) technologies is providing a clear advantage compared to the conventional DNA analysis techniques, such as Sanger sequencing, by allowing a comprehensive overview of larger regions of the genome and offering high sensitivity of mutation detection and quantitative assessment of mutant alleles. Yu et al. designed a panel to capture 31 known cancer susceptive genes possibly related to FNMTC, using NGS [8]. Although they identified some candidate variants, such a targeted approach was best suited for sporadic cases and did not provide any information on all the other genes. The unbiased whole-exome sequencing (WES) approach was indeed used by Gara et al. who identified a susceptibility variant of the HABP2 gene, not shared by additional families [9,10,11,12,13]. This result supports the genetic heterogeneity of FNMTC and the difficulty for a differential diagnosis between syndromes conditions, sporadic PTC and FNMTC. Many studies that use the FNMTC criterion of two affected members might be analyzing sporadic thyroid cancer cases rather than actual FNMTC cases. We approached the study of families presenting two or more relatives affected by PTC by sequencing the whole exome (WES) with enhanced coverage of the most relevant disease-associated targets.
Methods
NGS procedures
Genomic DNA (at least 2 microg) was extracted from venous blood of affected and unaffected individuals by QIAamp DNA Blood Mini Kit (Qiagen). We performed whole-exome sequencing (WES) by using the Agilent SureSelect QXT Clinical Research Exome that provides a 54Mbase target, including an enhanced coverage of disease-relevant targets from HGMD, OMIM and ClinVar (Agilent Technologies, Santa Clara, CA, USA). Enriched DNA was validated and quantified by microfluidic analysis using the Bioanalyzer High Sensitivity DNA Assay kit (Agilent Technologies) and the 2100 Bioanalyzer with 2100 Expert Software. Libraries were sequenced using the NextSeq500 system performing paired-end runs covering at least 2 × 150 nt. (Illumina Inc., San Diego, CA, USA). The average exome coverage of the target bases of at least 100X with 90% of the bases covered by at least 40 reads. All possible inheritance mechanisms were considered, and we focused our attention on variants that were present at a minor allele frequency of ≤ 0.001 in SNP databases (ExAC; gnomAD, 1000 genomes, internal database of ~ 1500 Italian subjects).
Selection of genomic variants
Sequencing data were processed using in-house software for the execution of the GATK Best Practices pipeline for whole-exome sequencing variant analysis. Reads pre-processing and cleaning was performed with the following tools: TrimGalore, to cut sequence reads with Phred Score less than 20; BWA, to align the reads against the human genome (ver. Hg19); PICARD MarkDuplicates, to remove duplicated reads likely to be sequencing artifacts. GATK (ver.3.8) was used for reading base recalibration prior of variant calling with GATK HaplotypeCaller; variants were genotyped with GATK GenotypeGVCFs and filtered with GATK VariantFiltration. SNVs and INDELs were filtered with different parameters. In particular, we used a frequency threshold of 0.005 compared with ExAC, gnomAD, and internal database of 2700 Italian subjects. Genotype phasing was obtained using GATK PhaseByTransmission. Furthermore, variants were annotated with Annovar to assign frequencies in large-scale variants datasets (1000 Genomes, ExAC, gnomAD) and potential impact on protein function. Finally, a tabular output containing both the variants and their annotation was generated for downstream analyses. All the candidate variants identified by bioinformatic analysis of WES data were confirmed by capillary sequencing of both DNA strands on PCR products. Healthy controls for each family were included in the analysis.
Subjects
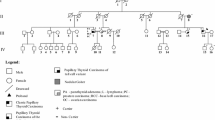
We analyzed five families in which two or more relatives received diagnosed with PTC. In particular, three families presented three relatives with PTC (Family A, B and C) (Figs. 1 and 2) and two families with two affected members (Family D and E) (Fig. 2). The families A, C, D and E were referred to our institution for evaluation of affected and nonaffected relatives. Family B had a clinical evaluation by The Division of Endocrinology of University of Siena, Italy. All the patients were affected at the time of the evaluation. Affected individuals, plus A4 as a healthy family member, were studied by whole-exome sequencing. We analyzed three kindred with PTC in family A and two affected relatives in B, C, D and E families. The affected patients were treated according to the ATA guideline [14]. All recruited patients signed an informed consent form. The study was approved by the local ethical committee of the Università della Campania “Luigi Vanvitelli” protocol n°54 del 2/5/2019 on Horizon 2020 Solve the Unsolved project. This study is in compliance with the Declaration of Helsinki.

Pedigree of families A and B. Arrow indicates patients and normal relatives studied by whole-exome sequencing

Pedigree of families C, D, and E. Arrow indicates patients and normal relatives studied by whole-exome sequencing
Results
Table 1 depicted the clinical-pathological information of the FNMTC patients of families A and B. FNMTC cases received total thyroidectomy with prophylactic central neck node dissection. PTC was confirmed by surgical pathology in all FNMTC patients. Post operatory adjuvant RAI ablation up to 100 mCI was performed and all patients were on thyroid hormone treatment. Median follow up period was 78 ± 8 months. The patients showed an excellent response, an undetectable serum thyroglobulin level and negative cervical US.
Whole exome sequencing identified FNMTC susceptibility gene candidates
We first searched for possible gene variants in genes involved in sporadic cases. Molecular analysis by WES of the ThyroSeq v2 and v3 panel genes, comprising the most commonly detected alterations SNVs/indels in the BRAF, RAS, EIF1AX, TERT, RET, DICER1, TP53, PTEN, and PIK3CA genes, did not reveal rare or pathogenic variants in any of these genes [15,16,17]. Subsequently, we excluded the presence of significant variants of genes so far identified for familial FNMTC: HABP2, FOXE1 and MAP2K5. The complete sequencing of these genes confirmed that no variants were present in analyzed families. We thus extended the analysis to all the other variants in both known and unknown genes. In particular, we performed multiple evaluations using unbiased combinations of filters. First, we searched for loss-of-function autosomal recessive variants shared by affected family members. No shared variant was found, as expected, considering most cancer susceptibility genes so far identified recognize an autosomal dominant model. Considering FNMTC a rare disease, the causative mutation should be very rare in the general population. Moreover, since the incidences of thyroid cancer in different countries range from 1 to 12.5 per 100,000, and that FNMTC only accounts for less than 5% of thyroid cancer cases, SNPs and Indels with allele frequencies higher than 0.001 (either in the ExAC or dbSNP databases) were excluded [18,19,20,21]. We selected several shared rare heterozygous variants in genes that are also highly expressed in thyroid tissue.
With this criterion, five genes: SPOC2, G3PB1, FAM122C, AIF1L, and BROX were selected among two dozen as the best susceptibility gene candidates for FNMTC. The most interesting was the loss of function variant in BROX, while the others were missense variants. We then performed segregation analysis by genotyping.
We found a positive segregation results for BROX only, while the other genes were excluded by further genotyping in nonaffected family members.
BROX haploinsufficiency in familial nonmedullary thyroid cancer
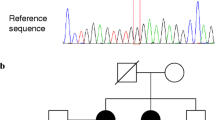
A new variant of BROX gene consisting in a frameshift deletion chr1:222892283 (NM_001288579:c.119delG:p.R40fs) was found in three members of family A and confirmed by Sanger sequencing. The predicted protein of 46 kDa should be truncated after ~ 50 residues, comprising its Bro1 domain. We confirmed the absence of this null variant in two unaffected members of the family. Interestingly, we screened this gene in the other families and found a new variant in 5′UTR region of BROX gene (chr1: 222886144 NM_144695:c.-2898C > T) in two sisters of family B (Fig. 1). We have no direct evidence of a functional effect of this second variant.
Discussion
The susceptibility chromosomal loci and genes of 95% of FNMTC cases remain to be characterized. To elucidate the molecular basis of FNMTC, we performed a high coverage whole-exome sequencing on five FNMTC families. For the first time, we describe a frameshift mutation of BROX gene. The predicted protein of 46 kDa will result truncated after ~ 50 residues, deleting its Bro1 domain. This conceivably reduces the likelihood of a gain-of-function effect. This variant should generate a null allele, considering the very small size of the protein product whenever it is expressed. Thus, a haploinsufficiency model is predicted, with the reduction of BROX expression that predisposes to disease or, alternatively, with the occurrence in the thyroid of a somatic hit that inactivates the remaining allele. This is the first report showing a possible association between BROX variants and cancer susceptibility. BROX is a protein-coding gene expressed in several tissues and with its highest mRNA levels in the thyroid gland, as demonstrated by the transcriptome analysis of 27 different human organs and tissues from 95 individuals [22]. BROX encodes for human Brox, a 46-kDa protein that has a Bro1 domain-like sequence and a C-terminal thioester-linkage site of isoprenoid lipid. Bro1 domain is necessary for the morphogenesis of multivesicular bodies (MVBs) and is involved in the endosomal sorting of cargo proteins, including integrin and EGFR degradation in lysosomes [23,24,25]. EGFR is one of the key regulators of cell survival and growth. Its aberrant expression or uncontrolled activity is directly implicated in a variety of tumours. Ultimately, BROX loss of function could induce EGFR aberrant activity and tumor growth. Our results suggest the need to address new research on the genetic basis of FNMTC, looking to different pathways, comprising the overall working mechanism of BROX regulated EGFR trafficking, which is of great interest for its direct association with cell survival and cancer signaling. A hypothesis of the possible pathogenetic mechanism of BROX haploinsufficiency in FNMTC is suggested in Fig. 3.

Hypothesis for the pathogenetic mechanism of FNMTC: BROX haploinsufficiency induces altered EGFR degradation pathway in follicular thyroid cells, with EGFR accumulation and aberrant cell growth
The BROX gene in ExAC shows a lower number of loss-of-function variants (and no homozygous subject) in comparison with the prediction. This may indicate an important role, but heterozygous individuals could be considered as healthy until the occurrence of cancer. The final demonstration of the role of BROX in FNMTC necessarily passes through the demonstration of a second family with a very similar allele. By analyzing the remaining four families, we identified a new variant (chr1: 222886144 NM_144695:c.-2898C > T) in 5′UTR region of BROX gene. This is a chromatin-rich region. The sequences of the untranslated regions (UTRs) of mRNAs play important roles in post-transcriptional regulation [26]. The regulation of gene expression can also occur through a post-transcriptional modulation of the amount of gene product and that this modulation can be mediated by 5′ untranslated exon 1, suggesting a role in dysregulation of BROX function. Although this mutation may be suggestive, data from additional families are necessary.
FNMTC is a rare manifestation of a common condition and it is characterized by a more aggressive biological behavior. Earlier age at disease onset, a higher rate of nodal metastases, extrathyroidal tumor extension and increased severity in successive generations have been described [7, 27,28,29]. A recent meta-analysis of Wang et al. reporting 12 studies with a total of 12,741 patients confirmed these findings [30]. Researches to identify candidate cancer predisposition genes in non-syndromic FNMTC have brought mainly low-to-moderate penetrance genes [31]. The finding of HABP2 G534E polymorphism, claimed to be a new susceptibility gene of FNMTC. G534E, is reported in gnomAD (https://gnomad.broadinstitute.org/) 6224 times with 32 homozygous subjects (frequency = 0.022): these numbers may be important to indicate either a very incomplete penetrance or the co-segregation by chance. Further studies support this view with the lack of an increased risk [9,10,11,12]. In a recent study from Ye et al., MAP2K5 mutation was identified as a novel susceptibility gene for FNMTC [32]. Cirello et al. did not confirm this data, and we found an intronic, non-significant, variant of MAP2K5 in affected members of two out of five studied families [33]. Despite the availability of modern NGS techniques and of huge genomic databases of hundred thousand individuals, no genetic test is yet available for FNMTC so far. Even if rare variants have been recently evidenced, the lack of a specific genetic test for FNMTC has resulted in the development of a clinical definition based on family history [34]. The most stringent definition of FNMTC requires two first-degree relatives with NMTC at the time of diagnosis of the patient in question, in the absence of a known familial syndrome. However, a clinical definition presents a number of problems. Clearly, the first family member (the index case) diagnosed with NMTC cannot be properly known as harboring familial illness, nor will the second till three cases are known.
In conclusion, we identified new variants in the BROX gene, involved in EGFR degradation pathway, in association with familial cases of FNMTC in which three relatives were diagnosed with PTC. We believe that this result may suggest new insights on the genetic basis of FNMTC. The involvement of EGFR trafficking in thyroid cancer growth will be a rational basis for future investigations to unravel the overall working mechanism of its key role in cell survival and cancer signaling. Only families where three first-degree relatives are affected can be considered to represent true familial FNMTC. Moreover, our finding can add information for the management FNMTC. The NGS results are deeply influencing both our understandings of diseases and their clinical management. This revolutionary approach needs extreme caution and expertise in the interpretation of the results, and underlines the need for data sharing in rare disease research.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Durante C, Grani G, Lamartina L, Filetti S, Mandel SJ, Cooper DS (2018) The diagnosis and management of thyroid nodules: a review. JAMA 319:914–924
Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (eds) (2017) SEER cancer statistics review, 1975–2014, National Cancer Institute. Bethesda, MD, https://seer.cancer.gov/csr/1975_2014/, based on November 2016 SEER data submission, posted to the SEER web site.
Grossman RF, Tu SH, Duh QY, Siperstein AE, Novosolov F, Clark OH (1995) Familial nonmedullary thyroid cancer. An emerging entity that warrants aggressive treatment. Arch Surg 130:892–897
Loh KC, Lo JC, Greenspan FS, Miller TR, Yeo PP (1997) Familial papillary thyroid cancer: a case report. Ann Acad Med Singapore 26:503–506
Alsanea O, Wada N, Ain K, Wong M, Taylor K, Ituarte PH, Treseler PA, Weier HU, Freimer N, Siperstein AE, Duh QY, Takami H, Clark OH (2000) Is familial non-medullary thyroid carcinoma more aggressive than sporadic thyroid cancer? A multicenter series. Surgery 128:1043–1050
Uchino S, Noguchi S, Kawamoto H, Yamashita H, Watanabe S, Yamashita H, Shuto S (2002) Familial nonmedullary thyroid carcinoma characterized by multifocality and a high recurrence rate in a large study population. World J Surg 26:897–902
Lee YM, Yoon JH, Yi O, Tae-Yon Sung TY, Chung KW, Kim WB, Hong SJ (2014) Familial history of non-medullary thyroid cancer is an independent prognostic factor for tumor recurrence in younger patients with conventional papillary thyroid carcinoma. J Surg Oncol 109:168–173
Yu Y, Dong L, Li D, Chuai S, Wu Z, Zheng X, Cheng Y, Han L, Yu J, Gao M (2015) Targeted DNA sequencing detects mutations related to susceptibility among familial non-medullary thyroid cancer. Sci Rep 5:16129
Gara SK, Jia L, Merino MJ, Agarwal SK, Zhang L, Cam M, Patel D, Kebebew E (2013) Germline HABP2 mutation causing familial nonmedullary thyroid cancer. N Engl J Med 373:448–455
Sahasrabudhe R, Stultz J, Williamson J, TCUKIN et al (2016) The HABP2 G534E variant is an unlikely cause of familial non-medullary thyroid cancer. J Clin Endocrinol Metab 10:1098–1103
Alzahrani AS, Murugan AK, Qasem E, Al-Hindi H (2016) HABP2 Gene mutations do not cause familial or sporadic non-medullary thyroid cancer in a highly inbred middle eastern population. Thyroid 26:667–671
Weeks AL, Wilson SG, Ward L, Goldblatt J, Hui J, Walsh JP (2016) HABP2 germline variants are uncommon in familial nonmedullary thyroid cancer. BMC Med Genet 17:60
Peiling Yang S, Ngeow J (2016) Familial non-medullary thyroid cancer: unraveling the genetic maze. Endocr Relat Cancer 23:577–595
Haugen BR, Alexander EK, Bible KC et al (2015) (2016) American thyroid association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer. Thyroid 26:1–133
Nikiforov YE, Carty SE, Chiosea SI, Coyne C, Duvvuri U, Ferris RL, Gooding WE, Hodak SP, LeBeau SO, Ohori NP, Seethala RR, Tublin ME, Yip L, Nikiforova MN (2014) Highly accurate diagnosis of cancer in thyroid nodules with follicular neoplasm/suspicious for a follicular neoplasm cytology by ThyroSeq v2 next-generation sequencing assay. Cancer 120:3627–3634
Nikiforova MN, Mercurio S, Wald AI, Barbi de Moura M, Callenberg K, Santana-Santos L, Gooding WE, Yip L, Ferris RL, Nikiforov YE (2018) Analytical performance of the ThyroSeq v3 genomic classifier for cancer diagnosis in thyroid nodules. Cancer 124:1682–1690
Agrawal N, Akbani R, Aksoy BA, on behalf of Cancer Genome Atlas Research Network (2014) Integrated genomic characterization of papillary thyroid carcinoma. Cell 159:676–690
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J (2016) Cancer statistics in China, 2015. CA Cancer J Clin 66:115–132
Morris LG, Sikora AG, Tosteson TD, Davies L (2013) The increasing incidence of thyroid cancer: the influence of access to care. Thyroid 23:885–891
Vriens MR, Suh I, Moses W, Kebebew E (2009) Clinical features and genetic predisposition to hereditary nonmedullary thyroid cancer. Thyroid 19:1343–1349
Khan A, Smellie J, Nutting C, Harrington K, Newbold K (2010) Familial nonmedullary thyroid cancer: a review of the genetics. Thyroid 20:795–801
Fagerberg L, Hallström BM, Oksvold P et al (2014) Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics 13:397–406
Pradhan-Sundd T, Verheyen EM (2014) The role of Bro1- domain-containing protein Myopic in endosomal trafficking of Wnt/Wingless. Dev Biol 392:93–107
McCullough J, Fisher RD, Whitby FG, Sundquist WI, Hill CP (2008) ALIX-CHMP4 interactions in the human ESCRT pathway. Proc Natl Acad SciUSA 105:7687–7691
Mu R, Dussupt V, Jiang J, Sette P, Rudd V, Chuenchor W, Bello NF, Bouamr F, Xiao TS (2012) Two distinct binding modes define the interaction of Brox with the C-terminal tails of CHMP5 and CHMP4B. Structure 20:887–898
Matoulkova E, Michalova E, Vojtesek B, Hrstka R (2012) The role of the 3' untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol 95:563–576
Klubo-Gwiezdzinska J, Yang L, Merkel R, Patel D, Nilubol N, Merino MJ, Skarulis M, Sadowski SM, Kebebew E (2017) Results of screening in familial non-medullary thyroid cancer. Thyroid 27:1017–1024
Capezzone M, Marchisotta S, Cantara S, Busonero G, Brilli L, Pazaitou-Panayiotou K, Carli AF, Caruso G, Toti P, Capitani S, Pammolli A, Pacini F (2008) Familial non-medullary thyroid carcinoma displays the features of clinical anticipation suggestive of a distinct biological entity. Endocr Relat Cancer 15:1075–1081
Capezzone M, Secchi C, Fralassi N, Cantara S, Brilli L, Ciuoli C, Pilli T, Maino F, Forleo R, Pacini F, Castagna MG (2019) Should familial disease be considered as a negative prognostic factor in micropapillary thyroid carcinoma? J Endocrinol Invest 42:1205–1213
Wang X, Cheng W, Li J, Su A, Wei T, Liu F, Zhu J (2015) Endocrine tumours: familial nonmedullary thyroid carcinoma is a more aggressive disease: a systematic review and meta-analysis. Eur J Endocrinol 172:253–262
Hińcza K, Kowalik A, Kowalska A (2019) Current knowledge of germline genetic risk factors for the development of non-medullary thyroid cancer. Genes (basel) 10:482
Ye F, Gao H, Xiao L et al (2019) Whole exome and target sequencing identifies MAP2K5 as novel susceptibility gene for familial non-medullary thyroid carcinoma. Int J Cancer 144:1321–1330
Cirello V, Colombo C, Persani L, Fugazzola L (2019) Absence of the MAP2K5 germline variants c.G961A and c.T1100C in a wide series of familial nonmedullary thyroid carcinoma Italian families. Int J Cancer. 145:600
Wang X, Cheng W, Li J, Su A, Wei T, Liu F, Zhu J (2019) Identification of rare variants predisposing to thyroid cancer. Thyroid 29:946–955
Acknowledgements
We thank the study participants, their families, and all the investigators and study staff members. VN Supported by grants from Telethon 2015 “Medicina Traslazionale in Oncologia: Dalla Ricerca alla Terapia PON01_02418”, Telethon 2016–2018 “Telethon Undiagnosed Disease Program”, 2018-22 EU Research Funding H2020-HEALTH-SC1-2017-RTD: “SOLVE RD”.
Funding
VN Supported by grants from Telethon 2015 “Medicina Traslazionale in Oncologia: Dalla Ricerca alla Terapia PON01_02418”, Telethon 2016–2018 “Telethon Undiagnosed Disease Program”, 2018-22 EU Research Funding H2020-HEALTH-SC1-2017-RTD: “SOLVE RD”.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have nothing to disclose.
Ethics approval
The study was approved by the local ethical committee of the Università della Campania “Luigi Vanvitelli” protocol n°54 del 2/5/2019 on Horizon 2020 Solve the Unsolved project.
Informed consent
The informed consent was obtained from the patients included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pasquali, D., Torella, A., Accardo, G. et al. BROX haploinsufficiency in familial nonmedullary thyroid cancer. J Endocrinol Invest 44, 165–171 (2021). https://doi.org/10.1007/s40618-020-01286-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-020-01286-6