
Abstract
Purpose
Germline mutations in DNA repair-related genes have been recently reported in cases with familial non-medullary thyroid carcinoma (FNMTC). A Portuguese family from the Roma ethnic group with four members affected with papillary thyroid carcinoma (PTC), and three members with multinodular goiter (MNG) was identified. The aim of this study was to investigate the involvement of DNA repair-related genes in the etiology of FNMTC in this family and in the Roma ethnic group.
Methods
Ninety-four hereditary cancer predisposition genes were analyzed through next-generation sequencing. Sanger sequencing was used for variant confirmation and screening. Twelve polymorphic markers were genotyped for haplotype analysis in the CHEK2 locus.
Results
A germline pathogenic frameshift variant in the CHEK2 gene [c.596dupA, p.(Tyr199Ter)] was detected in homozygosity in the proband (PTC) and in his brother (MNG), being heterozygous in his mother (PTC), two sisters (PTC), and one nephew (MNG). This variant was absent in 100 general population controls. The screening of the CHEK2 variant was extended to other Roma individuals, being detected in 2/33 Roma patients with thyroid cancer, and in 1/15 Roma controls. Haplotype segregation analysis identified a common ancestral core haplotype (Hcac), covering 10 Mb in the CHEK2 locus, shared by affected CHEK2 variant carriers. Analysis of 62 individuals CHEK2 wild-type indicated that none presented the Hcac haplotype. The estimated age for this variant suggested that it was transmitted by a relatively recent common ancestor.
Conclusions
We identified a founder CHEK2 pathogenic variant, which is likely to underlie thyroid cancer and other cancer manifestations in the Roma population.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Thyroid cancer is the most common neoplasia of the endocrine system, accounting for about 1–3% of all malignancies [1], and its incidence rates are threefold higher in women than in men [2]. Although the majority of these tumors are sporadic, 3–9% of all follicular cell-derived thyroid tumors are familial, being designated as familial non-medullary thyroid carcinoma (FNMTC) [3]. FNMTC is currently defined by the diagnosis of two or more first degree relatives with differentiated thyroid cancer of follicular cell origin [3]. FNMTC most common subtype is the papillary thyroid carcinoma (PTC), and family members frequently present benign lesions of the thyroid, such as multinodular goiter (MNG) [3, 4]. It is thought that the inheritance pattern of FNMTC is autosomal dominant with incomplete penetrance and variable expressivity. It may occur as a minor component of familial cancer syndromes — syndromic FNMTC, or as the predominant feature — non-syndromic FNMTC [3]. The latter accounts for approximately 95% of all FNMTC cases [3, 5]. The genetic alterations underlying syndromic forms are well defined [6] and these include several familial cancer syndromes such as Cowden syndrome, familial adenomatous polyposis/Gardner’s syndrome, Carney complex, Werner syndrome and DICER1 syndrome [7,8,9,10,11,12]. The genetic background of non-syndromic FNMTC is not as well understood. Nevertheless, several susceptibility loci [MNG1 (14q32), TCO (19p13.2), PRN (1q21), NMTC1 (2q21), FTEN (8p23.1–p22), 6q22, 8q24] for the non-syndromic forms have already been mapped [13,14,15,16,17,18]. Furthermore, the analysis of candidate genes, located in these mapped regions, and/or that had suggestive roles in thyroid physio/pathology, together with the recent advent of high-throughput technologies, such as next-generation sequencing (NGS) for whole exome and genome analyses, allowed the identification of several susceptibility genes for FNMTC, such as: NKX2.1 [19], DICER1 [20], SRGAP1 [21], FOXE1 [22], HABP2 [23], SRRM2 [24], RTFC [25], and more recently, MAP2K5 [26] and MYO1F [27]. These genes have distinct functions and participate in diverse cellular processes. In addition, recent studies in patients with NMTC (both familial and not specified for familial status), identified germline mutations in DNA repair genes, namely in BRCA1/2, ATM, and CHEK2, suggesting a role for these genes in the predisposition to familial thyroid cancer [28,29,30,31,32]. Although these findings support a high genetic heterogeneity, the etiology of FNMTC continues poorly understood, since the predisposing genes involved in this disease have not yet been identified for the great majority of the families [3, 6].
In the present study, 94 cancer predisposition genes were analyzed through NGS in a family with FNMTC. Interestingly, a pathogenic germline variant in CHEK2 was detected in homozygosity in affected members from a Portuguese Roma family, suggesting that it could be a founder variant. Therefore, the role of this variant in the etiology of FNMTC in the Roma family and in the Roma ethnic group was investigated.
Materials and methods
Patients
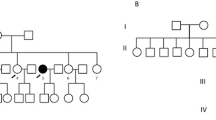
One family with FNMTC from our cohort, followed in the Endocrinology Department from Instituto Português de Oncologia de Lisboa Francisco Gentil (IPOLFG), was chosen for the present study. This family belongs to the Roma ethnic group, and benign and malignant thyroid lesions were the most common phenotypes, as follows: four family members were affected with non-medullary thyroid carcinoma (NMTC), namely with PTC (in some cases in concomitance with goiter) (II.2, III.1, III.3, and III.6), and four members affected with goiter only (I.2, III.4, IV.1, and IV.2) (Family fA, Supporting Table S1). In addition, of the four cases affected with NMTC, three of them were also affected with other types of cancer: two with breast cancer (II.2 and III.6) and one with prostate cancer (III.1). Furthermore, the proband’s father (II.1), already deceased, had lung cancer.
DNAs from peripheral blood leukocytes from seven patients of this family were available to study. DNAs from peripheral blood leukocytes from 48 additional Roma individuals [18 with FNMTC or familial multinodular goiter (FMNG), 15 with apparently sporadic NMTC, and 15 controls] (Supporting Table S2), and from 100 non-Roma Portuguese healthy controls (67% females and 33% males, average age 63.9 years; supplied by Biobanco-iMM, Lisbon Academic Medical Center, Lisbon, Portugal) were also used.
DNA extraction from blood
DNA from peripheral blood leukocytes was extracted and purified using the Puregene® Blood Core Kit (Qiagen, Hilden, Germany), according to the manufacturer’s protocol. Extracted DNA was quantified by UV spectrophotometry (NanoDrop ND-1000, Thermo Fisher Scientific, Wilmington, DE, USA).
Next-generation sequencing (NGS)
A commercial multigene panel (TruSight Cancer Panel, Illumina, San Diego, USA) was used for NGS as an enrichment system targeted to exons (and splice site regions) of 94 genes. Libraries were subjected to cluster generation and paired-end sequencing in a MiSeq sequencer platform (Illumina). Sequence data were analyzed with the instrument software MiSeq Reporter v.2.5.1 (Illumina), and the reads were aligned against the human reference sequence GRCh37. The resulting VCF files were visualized using the VariantStudio v.3 Software (Illumina), which supplies the report of all detected sequence variants and the respective annotation. Details of bioinformatic analysis are described in the Supporting Information.
Polymerase chain reaction (PCR)
PCRs were performed using Taq DNA polymerase (InvitrogenTM, California, USA) protocol. The primers and conditions for PCR amplification are listed in Supporting Table S3.
Sanger sequencing and mutational analysis
Sanger sequencing was used to confirm selected genetic variants detected by NGS, and to analyze variant segregation in the family. Details of the methodology are described in the Supporting Information.
Microsatellite and single-nucleotide polymorphisms (SNPs) analysis
For haplotype analysis, a total of 71 DNA samples (25 individuals from nine FNMTC/FMNG families, 15 apparently unrelated NMTC patients, and 15 controls, all belonging to the Roma ethnic group, as well as 16 healthy controls from the Portuguese population) (Supporting Tables S1 and S2) were genotyped using 11 microsatellite markers and one single-nucleotide polymorphism (SNP), localized at a 16.5-Mb region encompassing CHEK2 (cen-D22S420-D22S446-D22S257-D22S421-D22S1163-D22S689-D22S570-CHEK2(rs743185)-D22S275-D22S1150-D22S273-D22S1162-tel) on chromosome 22q12.1. The markers and their chromosomal locations are represented in Supporting Fig. S1. Details of microsatellite and SNPs analyses are described in the Supporting Information.
Haplotype construction and mutation age estimation
Haplotype construction for patients from the Roma ethnic group family was performed manually based on the index case and family members genotypes. For the remaining patients and controls, there was no DNA available for study from the relatives, as such, their genotypes were phase-unknown, meaning that there was no information to assign alleles to the paternal and maternal chromosomes. With the available data, the age of the mutation was estimated from the variation accumulated in their ancestral haplotypes, as described in [33]. Details are described in the Supporting Information.
Results
Investigation of a founder effect for the CHEK2 c.596dupA variant
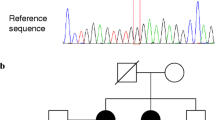
In the present study, a screening for germline mutations in 94 cancer predisposing genes was performed in the DNA of the proband from a Roma FNMTC family, through NGS analysis, using the TruSight Cancer panel. Bioinformatic analysis of the NGS data revealed a germline variant in the CHEK2 (checkpoint kinase 2) gene (NM_001005735.2:c.596dupA), which creates a premature translational stop signal [NP_001005735.1:p.(Tyr199Ter)], being expected to result in an absent or truncated protein. This variant was confirmed by Sanger sequencing (Fig. 1). This variant has not been reported in the literature, and was only described once, as a pathogenic variant, in the context of hereditary breast carcinoma [ClinVar ID: 947569; NM_007194.4:c.467dupA (p.Tyr156Ter)]. In the present study, the CHEK2 germline variant was detected in six family members, being homozygous in the proband (III.1), who presented PTC and also prostate cancer, and in his deceased brother with MNG (III.4). The variant was heterozygous in the proband’s mother (II.2), who presented PTC and breast cancer, in the sisters (III.3 and III.6), who had PTC and nodular goiter and one also had breast cancer, and in one nephew with goiter (IV.1) (Figs. 1 and 2 and Supporting Table S1). The proband’s father had died with lung cancer, thus DNA was not available to study. However, homozygosity in his offspring indicated that he was also a CHEK2 variant carrier. Since the variant was detected in homozygosity, and the proband’s parents, who belonged to Roma ethnic group, were possibly related (cousins of unknown degree), the possibility of a founder effect was put forward.

Detection of the c.596dupA CHEK2 variant. The upper panel shows the wild-type sequence in a non-carrier of the CHEK2 variant, the middle is representative of the sequence found in a patient carrying the c.596dupA CHEK2 variant in homozygosity, and the lower panel shows the sequence found in a patient carrying the variant in heterozygosity. The red arrow indicates the position of the insertion of the additional adenine

Pedigree of a family with FNMTC from the Roma ethnic group. Results of c.596dupA CHEK2 variant screening, and haplotype segregation analysis at the CHEK2 locus, in the family members are shown. +/+, homozygous for CHEK2 variant c.596dupA; +/−, heterozygous for CHEK2 variant c.596dupA; −/−, negative for the variant. Wt, wild-type; Mut, CHEK2 variant; PA, present age; AD, age at diagnosis of thyroid disease; yr, years; *, refused fine-needle aspiration cytology (FNAC). Haplotypes H1, H2, and H3 highlighted in red, green, and blue, respectively; inferred haplotype is indicated in brackets
In order to further investigate the role of CHEK2 in the etiology of FNMTC in this family, in particular as a tumor suppressor, the CHEK2 variant identified was sequenced in thyroid tumor DNA from one affected family member who carried the variant in heterozygosity (III.6), to search for loss of heterozygosity (LOH). However, no pattern suggestive of LOH was found, because the variant was found in heterozygosity in the tumor (data not shown).
Screening of the CHEK2 c.596dupA p.(Tyr199Ter) variant in patients with thyroid cancer and in Portuguese controls
To investigate the hypothesis of a founder effect, the screening of the c.596dupA p.(Tyr199Ter) variant was extended to 48 additional Roma individuals (18 with FNMTC or FMNG, 15 with apparently sporadic NMTC, and 15 controls) (Supporting Table S2) and to 100 healthy controls from the general Portuguese population. Interestingly, the variant was detected in two additional Roma patients with apparently sporadic thyroid cancer (As.1 and As.2; Supporting Table S1), being absent in the 100 controls from the general Portuguese population. Additionally, one male control from the Roma ethnic group (Cr.1), asymptomatic at age 52, also carried the CHEK2 variant. Overall, this variant was only found in individuals originating from the Roma ethnic group, being present in 9/55 (16.4%) individuals from this ethnicity, of whom six had thyroid cancer, two had nodular goiter, and only one was apparently unaffected, thus suggesting that they may have a common ancestor.
Haplotype segregation analysis
Founder mutations reside on haplotypes that are shared by all carriers of the mutation because they are inherited from a common ancestor. Mutation carriers, whom have a shared founder, exhibit allele sharing at markers near the mutation locus by linkage disequilibrium [34].
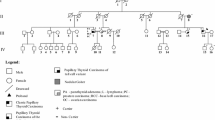
Hence, to determine whether all occurrences of the c.596dupA p.(Tyr199Ter) CHEK2 variant descended from a single ancestral mutation event or arisen independently, a haplotype segregation analysis was performed. For this purpose, a total of 71 DNA samples (described in the “Materials and methods” section) were genotyped using 11 polymorphic microsatellite markers and one SNP (Supporting Fig. S1). Whenever possible, chromosomal phasing was used to identify the alleles that were positioned on the same chromosome, and helped to determine on which of the two parental chromosomes, or haplotypes, a particular allele was present. Haplotypes were established manually based on the genotypes obtained from family members. For unphased samples, both alleles were considered to ascertain the founder haplotype status. The obtained genotypes are shown in Fig. 2 for the Roma family under study, for CHEK2 variant carriers (Table 1), and for all individuals analyzed (Supporting Table S4).
Alleles in the CHEK2 locus were phased for the individuals from the original FNMTC family from the Roma ethnic group, in which two main haplotypes linked to the CHEK2 variant were observed: H1:135-226-123-162-145-208-99-T-166-221-173-153, transmitted from the proband’s mother (and likely to be the ancestral haplotype), and H2:135-226-123-162-145-208-99-T-166-221-173-161 transmitted from the father (inferred haplotype) (Fig. 2 and Supporting Table S4). Patient III.4, besides haplotype H1, also showed another mutation linked haplotype H3:143-226-123-162-145-208-99-T-166-221-173-161, possibly resulting from a single centromeric recombination, relatively to haplotype H2. This family member, who died in 2018 (age 61), was diagnosed with MNG when he was 53 years old, but the presence of thyroid cancer could not be evaluated because he refused thyroid fine-needle aspiration cytology. The presence of other tumors was also not investigated in this case.
Haplotype analysis in the remaining thyroid cancer cases and controls, suggested that only CHEK2 variant carriers, namely As.1, As.2, and Cr.1, all from the Roma ethnic group, shared between them and with the Roma family a common region, of ~3.5 Mb, extending from marker D22S421 to D22S1150 (Table 1). The shared haplotype increases to 11.7 Mb, from marker D22S420 to D22S1150 if only the Roma patients affected with thyroid cancer are considered (excluding control Cr.1). However, although this sharing is not questionable for patient As.2, who is homozygous, for patient As.1, since she is heterozygous and in this study there was no access to DNA from her family members, there was no information to confirm the assignment of alleles to the paternal and maternal chromosomes (phase-unknown) [35].
Taken together, haplotype analysis suggested that the ancestral haplotype, in which the mutation probably occurred was H1, as it was found in the Roma family and in patient As.2 in homozygosity, and that a common ancestral core haplotype (Hcac), from D22S446 to D22S273 (10.2 Mb), was shared by all affected and CHEK2 variant carriers with phased alleles (Fig. 2 and Table 1).
Haplotypes H1, H2, H3, and Hcac were detected, respectively, in 45% (4/9 carriers), 45% (4/9), 11% (1/9), and 78% (7/9) of these Roma mutation carriers (Table 2).
The most parsimonious relationships among flanking haplotypes are presented as a phylogenetic network in Fig. 3.

Phylogenetic network. The network shows the most parsimonious relationships among flanking short tandem repeat based haplotypes in FNMTC families/individuals carrying the c.596dupA CHEK2 pathogenic variant. Circle sizes are proportional to the number of individuals with the haplotype that is written within these, and diamonds indicate recombination events
In order to further investigate whether the linkage of specific haplotypes to the CHEK2 variant was indeed the result of a founder effect, the genotypes for the whole set of markers were also determined in the remaining 62 individuals (FNMTC/FMNG, NMTC, and controls), who were non-carriers of the CHEK2 variant (Supporting Table S4). This analysis suggested that none of these individuals presented the H1, H2, or H3 haplotypes, and neither the Hcac haplotype. In the general population controls, a random population frequency of ~1/139,620,524, 1/17,452,566, 1/6,544,712, and 1/1,636,178 was estimated for haplotypes H1, H2, H3, and Hcac, respectively; and specifically in Roma controls it was estimated to be: 1/15,066,964, 1/3,766,741, 1/33,900,670, and 1/301,339, respectively. These are approximate frequencies as some alleles were not found in any of the controls. These data suggest that the likelihood of finding any of these haplotypes in the Portuguese population and Roma ethnic group is very low.
Estimation of the CHEK2 mutation age
Since there was no access to DNAs from relatives of some individuals included in this study, recombinants could not be identified unambiguously, which would be necessary to estimate the age of this founder CHEK2 variant by likelihood-based methods, as these take into account the variation accumulated in the ancestral haplotypes and include both recombination and mutation rates in the generation of variation [33, 36, 37]. Nevertheless, assuming that alleles were phased in case As.2 (who is homozygous for all markers), it was possible to identify an ancestral haplotype (H1), co-segregating with the CHEK2 variant in patient As.2 and in some patients from the original Roma family with FNMTC. Two additional haplotypes linked to the variant were identified, H2 and H3, but in the latter case it is debatable if it should be considered for age estimation because it was identified in the same family, in a first degree relative. Still, if H3 haplotype is considered, and assuming a generation time of 25, the estimated age for this mutation would be 109 ± 63 years (Table 3). Taken together, these results suggest that this mutation in CHEK2 may have occurred at approximately four generations ago, and was transmitted by a relatively recent common ancestor.
Discussion
In this study, a germline CHEK2 pathogenic variant [c.596dupA, p.(Tyr199Ter)] was identified in the proband and in additional five members of a Roma FNMTC family affected with (multi)nodular goiter and/or PTC.
CHEK2 gene encodes a serine/threonine protein kinase, which is required for checkpoint-mediated cell cycle arrest, activation of DNA repair and apoptosis in response to the presence of DNA double-strand breaks [38]. This gene has 22 exons, and the c.596dupA pathogenic variant, located in exon 5, leads to the change of a tyrosine residue to a premature stop codon, being expected to encode a truncated protein, lacking part of the forkhead-associated domain and the entire serine/threonine protein kinase catalytic domain. It is, however, possible that the mutant mRNA is not translated, due to nonsense-mediated decay (NMD). Zhao et al. showed that a CHEK2 (p.Tyr139Ter) heterozygous germline mutation, which also introduces a premature stop codon, resulted in a strong decrease in mutant mRNA, in patients from a family with PTC, indicating that the NMD pathway may be triggered, and that some CHEK2 mutants may contribute to tumorigenesis via the haploinsufficiency mechanism due to low CHK2 protein levels [32].
Tumor initiation and progression mechanisms were also investigated in the present family. A putative second hit in CHEK2 gene inactivation process was evaluated in the PTC from one affected member of the family. However, no LOH was suggested by this analysis. As regards to tumor progression, our group previously demonstrated that somatic BRAF, HRAS, NRAS, and TERT promoter mutations are likely to have a role in the development and aggressiveness of FNMTC [39, 40]. However, it is noteworthy that in that earlier study [40], no alterations of these genes were detected in the PTC from the proband of the Roma family here reported.
Here, in order to investigate the possibility of a founder effect, the screening of the CHEK2 variant was extended to other individuals from the Roma ethnic group. This analysis led to the identification of this variant in three additional individuals (two with apparently sporadic thyroid cancer and one control), totalizing nine CHEK2 c.596dupA positive Roma individuals. Deepening the clinical history of the two apparently sporadic cases revealed that one had a sister with unspecified thyroid disease, and the other had a follicular lymphoma and a family history of other neoplasias. Genotyping of the nine carriers allowed us to identify three different haplotypes, in the variant carriers with phased alleles, and a common ancestral core haplotype (Hcac) spanning about 10.2 Mb in the CHEK2 locus. Haplotype analysis in the 62 CHEK2 wild-type Roma patients and controls indicated that none presented any of the four mentioned haplotypes. These evidences strongly suggested that the CHEK2 c.596dupA variant was linked to a specific and rare haplotype and that all carriers shared a common ancestry. The large size of this core haplotype suggests a recent founder effect in the Roma ethnic group, which is in agreement with the CHEK2 mutation estimated age of 109 ± 63 years. The difficulty in obtaining more samples of cases and controls from the Roma ethnic group was a limitation in this study. Hence, the accuracy of the mutation age estimation might be improved if further data regarding clinical history and genotypes from first degree relatives of the CHEK2 variant carriers As.1, As.2, and Cr.1 become available.
The Gypsies are described as a population with Indian origins, that has subsequently spread throughout Europe by the fifteenth century [41]. The Roma ethnic group has a strong history of consanguineous unions, and a common ancestry is a well-known feature of this population. There are nearly 40,000 Roma individuals in Portugal [42]. Although in this study most patients were from Central/Southern Portugal, it is very likely that the CHEK2 variant can be found in individuals from the North region, where the Roma community is also well represented [42].
CHEK2 mutations are presently known to increase breast, colon, prostate, and kidney cancer risk, being considered a multiorgan cancer susceptibility gene [43], with moderate penetrance [44]. Accordingly, the Roma family analyzed in this study had members with other neoplasias besides thyroid cancer, such as breast, lung, and prostate cancer. Noteworthy, this variant was described once in the ClinVar database, in the context of hereditary breast carcinoma. Interestingly, it has been reported that a prior history of thyroid cancer is a risk factor for breast cancer and vice versa [45,46,47]. Furthermore, and in line with our findings, it has been reported that CHEK2 mutations predispose to thyroid cancer, and possibly to familial aggregation of breast and thyroid cancer [48]. There are already some well-studied CHEK2 variants, with low to relatively frequent occurrence in the populations, either truncating (c.444 + 1G > A, p.(?) and c.1100del, p.Thr367fs) or missense (c.470T > C, p.Ile157Thr), found to be differentially associated with the risk of prostate, breast, and thyroid cancers [43, 48,49,50,51,52]. Different mutations in the CHEK2 gene, and distinct zygosities (homo or hetero) may lead to different cancer risks [49]. In thyroid cancer, it was seen that the c.470T > C (p.Ile157Thr) substitution in the CHEK2 gene was associated with an almost twofold increase in the risk of PTC in the Great Poland population, and that in homozygous women, it increased the risk of PTC almost 13-fold [50]. As regards to the consequence of the germline variant type, truncating or missense, different studies showed that prostate, breast, and thyroid cancers were associated with both types of variants [43, 51, 52]. Strikingly, the frequency of the truncating CHEK2 alleles was particularly high (3.5%) in patients with thyroid cancer, being the highest among all the cancers studied [43]. In line with those findings and with the present study, the association of rare germline CHEK2 variants with thyroid cancer predisposition has recently been reported in two families [31, 32], and they also had truncating mutations.
Unlike in the case of an unquestionably high-risk gene, a CHEK2 pathogenic variant should be cautiously interpreted as a part of the explanation in a family history of thyroid cancer. Future studies may help to define all genetic players in cancer risk in these families.
In the present study, evidence of strong linkage disequilibrium of a common ancestral core haplotype and CHEK2 c.596dupA pathogenic variant, in all phased carriers, whom were all from the Roma ethnic group, and the absence of that haplotype in non-carriers, suggests that these patients descend from a common founder. Thus, screening of this variant should be extended to Roma families with thyroid cancer, and also to those who have breast cancer. The identification of founder mutations can greatly facilitate the molecular diagnosis of hereditary cancer syndromes by allowing targeted gene analysis as the first step of the genetic testing strategy. Having access to further information regarding the other carriers of this CHEK2 pathogenic variant will help to clarify the risk for several cancers in high-risk individuals and possibly to modify the incidence and mortality by cancer in these families. The present work also highlights DNA repair deficiency as a potential mechanism driving FNMTC susceptibility.
References
Q.T. Nguyen, E.J. Lee, M.G. Huang, Y.I. Parkn, A. Khullar, R.A. Plodkowski,Diagnosis and treatment of patients with thyroid cancer. Am. Health Drug Benefits. 8(1), 30–40 (2015)
R.L. Siegel, K.D. Mille, A. Jemal, Cancer Statistics, 2017. CA Cancer J. Clin. 67(1), 7–30 (2017)
C. Sturgeon, O.H. Clark, Familial nonmedullary thyroid cancer. Thyroid 15(6), 588–593 (2005)
A.E. Pinto, G.L. Silva, R. Henrique, F.D. Menezes, M.R. Teixeira, V. Leite, B.M. Cavaco, Familial vs sporadic papillary thyroid carcinoma: a matched-case comparative study showing similar clinical/prognostic behaviour. Eur. J. Endocrinol. 170, 321–327 (2014)
S.P. Yang, J. Ngeo, Familial non-medullary thyroid cancer: unraveling the genetic maze. Endocr. Relat. Cancer 23(12), R577–R595 (2016)
J. Klubo-Gwiezdzinska, Y. Kushchayeva, S.K. Gara, E. Kebebew. Familial non-medullary thyroid cancer. In Practical Management of Thyroid Cancer, ed. by U.K. Mallick, C. Harmer (Springer, Cham, 2018), pp. 241–270
J.A. Carney, C. Lyssikatos, R.R. Seethala, P. Lakatos, A. Perez-Atayde, H. Lahner et al. The spectrum of thyroid gland pathology in carney complex: the importance of Follicular Carcinoma. Am. J. Surg. Pathol. 42(5), 587–594 (2018)
J. Dotto, V. Nosé, Familial thyroid carcinoma: a diagnostic algorithm. Adv. Anat. Pathol. 15(6), 332–349 (2008)
J.E. Hall, D.J. Abdollahian, R.J. Sinard, Thyroid disease associated with Cowden syndrome: a meta-analysis. Head Neck 35(8), 1189–1194 (2013)
C.D. Malchoff, D.M. Malchoff, The genetics of hereditary nonmedullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 87(6), 2455–2459 (2002)
C.D. Malchoff, M. Sarfarazi, B. Tendler, F. Forouhar, G. Whalen, V. Joshi et al. Papillary thyroid carcinoma associated with papillary renal neoplasia: genetic linkage analysis of a distinct heritable tumor syndrome. J. Clin. Endocrinol. Metab. 85(5), 1758–1764 (2000)
M. Rohaizak, A.Y. Jasmi, M.A.M. Ismail, M.J.J. Munchar, F.A. Meah, Thyroid carcinoma in patients with familial adenomatous polyposis. Asian J. Surg. 26(3), 183–185 (2003).
G.R. Bignell, F. Canzian, M. Shayeghi, M. Stark, Y.Y. Shugart, P. Biggs, J. Mangion, R. Hamoudi, J. Rosenblatt, P. Buu et al. Familial nontoxic multinodular thyroid goiter locus maps to chromosome 14q but does not account for familial nonmedullary thyroid cancer. Am. J. Hum. Genet. 61(5), 1123–1130 (1997)
F. Canzian, P. Amati, H.R. Harach, J.L. Kraimps, F. Lesueur, J. Barbier et al. A gene predisposing to familial thyroid tumors with cell oxyphilia maps to chromosome 19p13.2. Am. J. Hum. Genet. 63(6), 1743–1748 (1998)
J.D. McKay, F. Lesueur, L. Jonard, A. Pastore, J. Williamson, L. Hoffman et al. Localization of a susceptibility gene for familial nonmedullary thyroid carcinoma to chromosome 2q21. Am. J. Hum. Genet. 69(2), 440–446 (2001)
B.M. Cavaco, P.F. Batista, L.G. Sobrinho, V. Leite, Mapping a new familial thyroid epithelial neoplasia susceptibility locus to chromosome 8p23.1-p22 by high-density single-nucleotide polymorphism genome-wide linkage analysis. J. Clin. Endocrinol. Metab. 93(11), 4426–4430 (2008).
I. Suh, S. Filetti, M.R. Vriens, M.A. Guerrero, S. Tumino, M. Wong et al. Distinct loci on chromosome 1q21 and 6q22 predispose to familial nonmedullary thyroid cancer: a SNP array-based linkage analysis of 38 families. Surgery. 146(6), 1073–1080 (2009)
H. He, R. Nagy, S. Liyanarachchi, H. Jiao, W. Li, S. Suster et al. A susceptibility locus for papillary thyroid carcinoma on chromosome 8q24. Cancer Res. 69(2), 625–631 (2009)
E.S.W. Ngan, B.H.H. Lang, T. Liu, C.K.Y. Shum, M.T. So, D.K.C. Lau et al. A germline mutation (A339V) in thyroid transcription factor-1 (TITF-1/NKX2.1) in patients with multinodular goiter and papillary thyroid carcinoma. J. Natl Cancer Inst. 101(3), 162–175 (2009)
T. Rio Frio, A. Bahubeshi, C. Kanellopoulou, N. Hamel, M. Niedziela, N. Sabbaghian et al. DICER1 mutations in familial multinodular goiter with and without ovarian Sertoli-Leydig cell tumors. JAMA 305(1), 68–77 (2011)
H. He, A. Bronisz, S. Liyanarachchi, R. Nagy, W. Li, Y. Huang et al. SRGAP1 is a candidate gene for papillary thyroid carcinoma susceptibility. J. Clin. Endocrinol. Metab. 98(5), 973–980 (2013)
J.S. Pereira, J.G. da Silva, R.A. Tomaz, A.E. Pinto, M.J. Bugalho, V. Leite et al. Identification of a novel germline FOXE1 variant in patients with familial non-medullary thyroid carcinoma (FNMTC). Endocrine 49(1), 204–214 (2015)
S.K. Gara, L. Jia, M.J. Merino, S.K. Agarwal, L. Zhang, M. Cam et al. Germline HABP2 mutation causing familial nonmedullary thyroid cancer. N. Engl. J. Med. 373(5), 448–455 (2015)
J. Tomsic, H. He, K. Akagi, S. Liyanarachchi, Q. Pan, B. Bertani et al. A germline mutation in SRRM2, a splicing factor gene, is implicated in papillary thyroid carcinoma predisposition. Sci. Rep. 5(1), 1–13 (2015)
C. Liu, Y. Yu, G. Yin, J. Zhang, W. Wen, X. Ruan et al. C14orf93 (RTFC) is identified as a novel susceptibility gene for familial nonmedullary thyroid cancer. Biochem. Biophys. Res. Commun. 482(4), 590–596 (2017)
F. Ye, H. Gao, L. Xiao, Z. Zuo, Y. Liu, Q. Zhao et al. Whole exome and target sequencing identifies MAP2K5 as novel susceptibility gene for familial non-medullary thyroid carcinoma. Int. J. Cancer 144(6), 1321–1330 (2018)
C. Diquigiovanni, C. Bergamini, C. Evangelisti, F. Isidori, A. Vettori, N. Tiso et al. Mutant MYO1F alters the mitochondrial network and induces tumor proliferation in thyroid cancer. Int. J. Cancer. 143(7), 1706–1719 (2018)
S. Fahiminiya, L. de Kock, W.D. Foulkes, Biologic and clinical perspectives on thyroid cancer. N. Engl. J. Med. 375(11), 1054–1067 (2016)
A. Srivastava, A. Kumar, S. Giangiobbe, E. Bonora, K. Hemminki, A. Försti et al. Whole genome sequencing of familial non-medullary thyroid cancer identifies germline alterations in MAPK/ERK and PI3K/AKT signaling pathways. Biomolecules 9(10), 605 (2019)
Y. Yu, L. Dong, D. Li, S. Chuai, Z. Wu, X. Zheng et al. Targeted DNA sequencing detects mutations related to susceptibility among familial non-medullary thyroid cancer. Sci. Rep. 5, 16129 (2015)
Y. Wang, S. Liyanarachchi, K.E. Miller, T.T. Nieminen, D.F. Comiskey Jr, W. Li, H. He, A. de la Chapelle et al. Identification of rare variants predisposing to thyroid cancer. Thyroid 29(7), 946–955 (2019)
Y. Zhao, T. Yu, L. Chen, D. Xie, F. Wang, L. Fu, C. Cheng, Y. Li, X. Zhu, G. Miao, A germline CHEK2 mutation in a family with papillary thyroid cancer. Thyroid. 30(6), 924–930 (2020)
S. Martins, F. Calafell, C. Gaspar, V.C. Wong, I. Silveira, G.A. Nicholson et al. Asian origin for the worldwide-spread mutational event in Machado-Joseph disease. Arch. Neurol. 64(10), 1502–1508 (2007)
J.H. Choi, R. Balasubramanian, P.H. Lee, N.D. Shaw, J.E. Hall, L. Plummer, R. Quinton, Expanding the spectrum of founder mutations causing isolated gonadotropin-releasing hormone deficiency. J. Clin. Endocrinol. Metab. 100(10), E1378–E1385 (2015)
T. Strachan, A. Read. Human Molecular Genetics, 4th edn (Garland Science, Taylor & Francis Group, Madison Avenue, New York, 2010)
P.M. Machado, R.D. Brandão, B.M. Cavaco, J. Eugénio, S. Bento, M. Nave, F. Vaz, Screening for a BRCA2 rearrangement in high-risk breast/ovarian cancer families: evidence for a founder effect and analysis of the associated phenotypes. J. Clin. Oncol. 25(15), 2027–2034 (2007)
M. Pinheiro, I. Francisco, C. Pinto, A. Peixoto, I. Veiga, B. Filipe, R. Santos, The nonsense mutation MSH2 c. 2152C> T shows a founder effect in Portuguese Lynch syndrome families. Genes, Chromosomes Cancer. 58(9), 657–664 (2019)
S. Matsuoka, M. Huang, S.J. Elledge, Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 282(5395), 1893–1897 (1998)
B.M. Cavaco, P.F. Batista, C. Martins, A. Banito, F. do Rosário, E. Limbert, L.G. Sobrinho, V. Leite, Familial non-medullary thyroid carcinoma (FNMTC): analysis of fPTC/PRN, NMTC1, MNG1 and TCO susceptibility loci and identification of somatic BRAF and RAS mutations. Endocr. Relat. Cancer. 15(1), 207–215 (2008)
I.J. Marques, M.M. Moura, R. Cabrera, A.E. Pinto, J. Simões-Pereira, C. Santos, F.D. Menezes, D. Montezuma, R. Henrique, M. Rodrigues Teixeira, V. Leite, B.M. Cavaco, Identification of somatic TERT promoter mutations in familial nonmedullary thyroid carcinomas. Clin. Endocrinol. 87(4), 394–399 (2017)
M. Fiore, X. Pillois, P. Nurden, A.T. Nurden, F. Austerlitz, Founder effect and estimation of the age of the French Gypsy mutation associated with Glanzmann thrombasthenia in Manouche families. Eur. J. Hum. Genet. 19(9), 981–987 (2011)
M. Cabral, A.V. Neves. Caracterização das condições de habitação das Comunidades Ciganas residentes em Portugal (Instituto da Habitação e da Reabilitação Urbana, I.P, Lisboa, 2014) pp. 1–28
C. Cybulski, B. Górski, T. Huzarski, B. Masojć, M. Mierzejewski, T. Dębniak et al. CHEK2 is a multiorgan cancer susceptibility gene. Am. J. Hum. Genet. 75(6), 1131–1135 (2004)
C. Stanislaw, Y. Xue, W.R. Wilcox, Genetic evaluation and testing for hereditary forms of cancer in the era of next-generation sequencing. Cancer Biol. Med. 13(1), 55–67 (2016)
R. Vassilopoulou-Sellin, L. Palmer, S. Taylor, C.S. Cooksley, Incidence of breast carcinoma in women with thyroid carcinoma. Cancer. 85(3), 696–705 (1999)
A.Y. Chen, L. Levy, H. Goepfert, B.W. Brown, M.R. Spitz, R. Vassilopoulou-Sellin, The development of breast carcinoma in women with thyroid carcinoma. Cancer. 92(2), 225–231 (2001)
C.I. Li, M.A. Rossing, L.F. Voigt, J.R. Daling, Multiple primary breast and thyroid cancers: role of age at diagnosis and cancer treatments (United States). Cancer Causes Control. 11(9), 805–811 (2000)
M. Siołek, C. Cybulski, D. Gąsior-Perczak, A. Kowalik, B. Kozak-Klonowska, A. Kowalska et al. CHEK2 mutations and the risk of papillary thyroid cancer. Int. J. Cancer. 137(3), 548–552 (2015)
M.A. Adank, M.A. Jonker, I. Kluijt, S.E. van Mil, R.A. Oldenburg, W.J. Mooi, A.W. van der Vaart, CHEK2* 1100delC homozygosity is associated with a high breast cancer risk in women. J. Med. Genet. 48(12), 860–863 (2011)
M. Kaczmarek-Ryś, K. Ziemnicka, S.T. Hryhorowicz, K. Górczak, J. Hoppe-Gołębiewska, M. Skrzypczak-Zielińska, I. Siatkowski, The c. 470T>C CHEK2 missense variant increases the risk of differentiated thyroid carcinoma in the Great Poland population. Hered Cancer Clin. Pract. 13(1), 8 (2015)
P. Apostolou, F. Fostira, V. Mollaki, A. Delimitsou, M. Vlassi, G. Pentheroudakis, I. Konstantopoulou, Characterization and prevalence of two novel CHEK2 large deletions in Greek breast cancer patients. J. Hum. Genet. 63(8), 877–886 (2018)
E. Złowocka, C. Cybulski, B. Górski, T. Dębniak, M. Słojewski, D. Wokołorczyk, R.J. Scott, Germline mutations in the CHEK2 kinase gene are associated with an increased risk of bladder cancer. Int. J. Cancer. 122(3), 583–586 (2008)
Acknowledgements
The authors are grateful to the patients and their families for their cooperation.
Funding
This work was funded by Liga Portuguesa Contra o Cancro — Núcleo Regional do Sul (LPCC-NRS), Televisão Independente (TVI), Instituto Português de Oncologia de Lisboa Francisco Gentil (IPOLFG), iNOVA4Health — UIDB/04462/2020 (a program financially supported by Fundação para a Ciência e Tecnologia/Ministério da Educação e Ciência), and Associação de Endocrinologia Oncológica.
Author information
Authors and Affiliations
Contributions
C.P.: investigation, methodology, formal analysis, data analysis, writing—original draft, writing—review and editing; I.J.M.: investigation, methodology, formal analysis, data analysis, writing—review and editing; D.D.: investigation, clinical data analysis, writing—review and editing; A.S.: methodology, data analysis, writing—review and editing; V.L.: investigation, clinical data analysis, writing—review and editing; B.M.C.: investigation, conceptualization, supervision, formal analysis, data analysis, funding acquisition, writing—review and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
This study was approved by the Ethical Committee of Instituto Português de Oncologia de Lisboa Francisco Gentil, and is in accordance with the ethical standards from the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Informed consent
The collection of biological samples from all subjects involved in this study was performed after written informed consent.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Pires, C., Marques, I.J., Dias, D. et al. A pathogenic variant in CHEK2 shows a founder effect in Portuguese Roma patients with thyroid cancer. Endocrine 73, 588–597 (2021). https://doi.org/10.1007/s12020-021-02660-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-021-02660-x