
Abstract
Purpose
Patients with growth hormone deficiency (GHD) demonstrate an increased cortisol/cortisone ratio which could potentially explain the metabolic features of GHD, while GH treatment (GHT) could increase the cortisol metabolism.
Methods
In 35 children (27 M, mean age 10.1 years) with idiopathic GHD at baseline and after 12 months of GHT and in 25 controls, in addition to metabolic parameters, we assessed adrenal function by morning serum cortisol, its peak, and its area under the curve (AUCCOR) during insulin tolerance test (ITT).
Results
A cortisol peak <18 µg/dl was shown in 22 and 31% of GHD children at baseline and after GHT, respectively. At baseline, GHD children had lower fasting glucose (p < 0.001) and ISI-Matsuda (p = 0.042), with concomitant higher Homa-IR (p = 0.006) and morning cortisol (p = 0.012) than controls. Morning cortisol was negatively correlated with GH (p < 0.001), fasting glucose (p < 0.001) and ISI-Matsuda (p < 0.001) and positively with Homa-IR (p = 0.010). Both cortisol peak and AUCCOR were negatively correlated with GH (all p < 0.001) and ISI-Matsuda (p = 0.016 and p = 0.001, respectively). After 12 months of GHT, a significant increase in fasting glucose (p < 0.001), and Homa-IR (p = 0.011) was documented, with a concomitant decrease in morning cortisol (p = 0.002), AUCCOR (p = 0.038), total (p = 0.003) and LDL-cholesterol (p = 0.016). No significant correlations were found among cortisol levels and all parameters were investigated.
Conclusions
Cortisol levels correlate with GH secretion and with many metabolic parameters in GHD children, while the metabolic effects during GHT are mainly due to GHT per se and less to cortisol reduction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The relationship between the growth hormone (GH)-insulin like growth factor (IGF)-I system and the hypothalamic–pituitary–adrenal (HPA) axis is complex and not univocal. Both a stimulatory and a neutral effect of IGF-I on the HPA axis have been demonstrated in in vitro models and in healthy subjects [1, 2], and the effect of the GH on the HPA axis, and specifically on the 11beta-hydroxysteroid dehydrogenases (11beta-HSD) isozymes, is widely documented. In peripheral tissues, corticosteroid hormone action is partially determined by the activity of 11beta-HSDs, two isozymes which interconvert hormonally active cortisol and inactive cortisone. 11beta-HSD2 inactivates cortisol to cortisone in the kidney, whilst 11beta-HSD1 performs the reverse reaction, activating cortisol from cortisone in the liver and adipose tissue. Neither GH nor IGF-I seem to affect 11beta-HSD2 activity, while GH inhibits cortisone to cortisol conversion through a reduction in 11beta-HSD1 activity [3, 4]. Consequently, patients affected by growth hormone deficiency (GHD) demonstrate an increased cortisol/cortisone ratio and an alteration in 11beta-HSD1 activity in GHD could partially explain the increased cortisol production in key target tissues including liver and adipose tissue, promoting insulin resistance and visceral adiposity, which are some of the phenotypic features of GHD [5, 6].
Conversely, in GH-treated patients, the GH-mediated increase in cortisol metabolism may unmask or precipitate adrenal insufficiency (AI) in patients with partial adrenocorticotropic hormone deficiency commencing GH treatment (GHT), through the negative effect on 11beta-HSD1 activity and the reduction in cortisone to cortisol conversion. In addition, the well-known beneficial effects of GHT on cardiovascular risk factors of GHD patients could also be an indirect effect via alterations in cortisol metabolism [7,8,9]. Therefore, this effect is always to be considered in patients affected by GHD both at diagnosis and during GHT. While evaluation of integrity of the HPA axis is essential in patients with organic GHD because it is well known that these patients may have additional pituitary hormone deficits [10] and GHT may unmask a central hypoadrenal state [11], weaker and quite discordant data exist on HPA function in patients with idiopathic GHD.
The aim of this study was to evaluate (1) the HPA function in children affected by idiopathic GHD at diagnosis and its potential change during GHT; (2) the role of serum cortisol in determining the metabolic features of GHD.
Materials and methods
We prospectively enrolled 35 children (27 M, 8 F; mean age 10.1 ± 1.4 years; range 6.9–11.6) affected by idiopathic GHD consecutively admitted to the Section of Endocrinology of the University of Palermo from January 2015 to December 2016. Twenty-five healthy subjects, matched for sex (16 M, 9 F), age (mean age 9.4 ± 1.9 years; range 5–11.4), stature, and pubertal status, were recruited as a control group at baseline among children referred for short stature. In this group, screening for short stature did not reveal endocrine disease and GHD was excluded. We excluded children affected by organic GHD, already known multiple pituitary hormone deficiency or receiving other hormonal replacement treatment. All patients, including the older ones, were in the first stage of sexual development according to the criteria of Marshall and Tanner [12], to avoid any interference of puberty on the metabolic parameters analyzed, and these children maintained their prepubertal hormonal status during the observation period (i.e., FSH and LH <1 mU/ml, total testosterone and 17β-Estradiol <0.50 ng/ml and <5 pg/ml in males and females, respectively).
GHD was diagnosed by the clinical, auxological, radiological, and biochemical criteria of the GH Research Society [13]. The auxological criteria included height more than two standard deviations (SD) below the mean and a growth velocity of 1 year before diagnosis more than 1 SD below the mean for age, or a decrease in height SD of more than 0.5 over 1 year or, without severe short stature, a growth velocity more than 2 SD below the mean over 1 year or, finally, height more than 1.5 SD below the midparental height. As radiological criteria we considered a bone age delay, estimated from an X-ray of the left wrist and hand and evaluated according to the methods of Greulich and Pyle, of at least 1 year with respect to the chronological age [14]. Biochemically, GHD was diagnosed by the failure of GH to respond to glucagon stimulation test (GST) and insulin tolerance test (ITT), with GH peaks below 8 µg/l [15]. Neuroimaging, with magnetic resonance of the hypothalamic–pituitary region, was performed in all children with more severe GHD, i.e., with GH peak ≤3 μg/l, and when we suspected there was a multiple pituitary deficiency (overall in 11 children). Among them, no patient showed pituitary abnormalities.
All GHD children received GH once daily at bedtime with a pen injection system for at least 12 months. IGF-I levels and growth velocity allowed us to determine the GH dose. Specifically, the main targets were arbitrarily IGF-I levels between 0.5–1.5 SDS and growth velocity >0.5 SDS. On average, in all patients, the initial daily dose of GH was 0.025 mg/Kg and it was gradually increased to 0.027–0.028 mg/Kg from months 6 to 12, to achieve the abovementioned targets.
Study protocol
In all children at baseline, we measured body height [expressed as standard deviation, (SD)], growth velocity, body mass index (BMI), waist circumference (WC), and bone/chronological age ratio. GH secretion was evaluated by means of GST and ITT, performed on two different days. During GST, blood samples were collected at 0, 30, 60, 90, 120, 150, 180, and 240 min after the injection of 30 μg/kg (up to 1000 μg) intramuscularly of glucagon (GlucaGen, NovoNordisk, Bagsvaerd, Denmark), for measurements of GH. ITT (0.1 U/kg of body weight of human Humulin R insulin) was performed in all children as a diagnostic test for GHD at baseline and at the same time to evaluate HPA function both at diagnosis and during GHT. Blood samples for GH, cortisol, and glucose were measured during ITT at baseline and at 30, 60, 90, and 120 min after insulin administration; results were accepted with blood glucose lower than 2.2 nmol/l (40 mg/dl). The areas under the curve (AUC) of GH (AUCGH) during GST and ITT and cortisol (AUCCOR) during ITT were calculated using the trapezoidal rule.
On a different day, a blood sample was drawn after an overnight fast for the measurement of hemoglobin A1c (HbA1c) and IGF-I concentrations. This sample also served as the baseline sample for an oral glucose tolerance test (OGTT). Blood samples were collected every 30 min for 2 h for glucose and insulin measurements. AUC of glucose (AUCGLU) and insulin (AUCINS) during OGTT was calculated using the trapezoidal rule.
HPA function was assessed through the morning adrenocorticotropic hormone (ACTH) and cortisol levels and the cortisol response to ITT, by considering both the highest value during the test, i.e., cortisol peak and the AUCCOR. We considered a morning basal cortisol value of <3 µg/dl as already diagnostic for AI, while we excluded the diagnosis with a basal cortisol value >18 µg/dl. A serum cortisol increase to at least 18 µg/dl during ITT was used as a criterion for a functioning HPA.
After the diagnosis of GHD was made, in GHD children in addition to auxological parameters and IGF-I measurement we performed both ITT (for cortisol) and OGTT (for glucose and insulin) after 12 months of GHT. As surrogate estimates of insulin sensitivity, we considered the homeostasis model assessment estimate of insulin resistance (Homa-IR) [16] and the insulin sensitivity index (ISI), a composite index derived from the OGTT and validated by Matsuda and DeFronzo [17], while as estimates of insulin secretion we considered fasting insulin and AUCINS. In the control group, these evaluations were performed only at baseline.
The institutional Ethics Committee of the University of Palermo approved this study. At the time of hospitalization, an informed consent for the scientific use of the data was obtained from both the participants and their parents.
Hormone and biochemical assays
All biochemical data were collected after overnight fasting. Glucose and lipids were measured in the centralized accredited laboratories of the University of Palermo with the standard methods. Serum insulin was measured by ELISA (DRG Instruments GmbH, Germany). The sensitivity of the method was 1 IU/ml. The normal insulin range (IU/ml) was 5–19. HbA1c levels were determined by HPLC with an ion-exchange resin (HA8121, Hi-AutoA1c, Menarini, Florence, Italy). Serum GH levels were measured by means of immunoradiometric assay using commercially available kits (Radim, Italy). The sensitivity of the assay was 0.04 µg/l. The intra- and inter-assay coefficients of variation (CV) were 2.5–3.9 and 3.8–5.0%, respectively. We reported GH concentrations in µg/l of IS 98/574. Serum IGF-I levels were measured by means of a chemiluminescent immunometric assay (Immulite 2000; Diagnostic Products Corp., Los Angeles, CA, USA) using murine monoclonal anti-IGF-I antibodies. The standards were calibrated against the World Health Organization second IS 87/518. The sensitivity of the method was 1.9 µg/l. The intra- and inter-assay CVs were 2.3–3.9 and 3.7–8.1%, respectively. Values were expressed as SD according to the normative data provided by the manufacturer. Serum cortisol was evaluated using the electrochemiluminescent (ECLIA) automated method access (Beckman Coulter, Brea, CA, USA). ACTH levels were detected by electrochemiluminescence immunoassay (Elecsys, Roche, Milan).
Statistical analysis
The Statistical Packages for Social Sciences SPSS version 19 was used for data analysis. Baseline characteristics were presented as mean ± SD for continuous variables (normality of distribution for the quantitative variables was assessed with the Kolmogorov–Smirnov test); rates and proportions were calculated for categorical data. The differences between groups were evaluated with the t test. Pearson’s correlation was performed among continuous variables. A p value <0.05 was considered statistically significant.
Results
Clinical and hormonal parameters
The clinical and hormonal parameters of control subjects, GHD children at diagnosis and after 12 months of GHT are shown in Table 1.
No significant difference in height and bone/chronological age ratio between GHD children at baseline and control subjects was found. Conversely, GHD children at baseline showed significantly lower growth velocity (−3.1 ± 1.8 vs. −1.1 ± 1.0 SD; p = 0.023), IGF-I levels (88.4 ± 26.7 vs. 198.4 ± 53.2 µg/l; p < 0.001 or −0.74 ± 0.31 vs. 1.30 ± 0.46 SD; p < 0.001), GH peak and AUCGH after ITT (4 ± 1.4 vs. 12.9 ± 2.8 µg/l; p < 0.001 and 308 ± 168 vs. 962 ± 297 µg/l; p < 0.001) and GH peak and AUCGH after GST (4.2 ± 3.3 vs. 11.5 ± 6.6 µg/l; p < 0.001 and 282 ± 154 vs. 712 ± 368 µg/l; p = 0.001) than controls, with concomitant higher BMI (−1.2 ± 0.9 vs. −2.1 ± 0.6 SD; p < 0.001) and WC (62.1 ± 7.5 vs. 57.7 ± 4.3 cm; p = 0.015).
In the GHD group, growth significantly increased after 12 months of GHT (height: −1.5 ± 0.7 vs. −2.2 ± 0.6 SD; p < 0.001; growth velocity: 1.6 ± 0.9 vs. −3.1 ± 1.8 SD; p < 0.001), with a concomitant increase in IGF-I (213.9 ± 122.1 vs. 88.4 ± 26.7 µg/l; p < 0.001 or 1.45 ± 0.75 vs. −0.74 ± 0.31 SD; p < 0.001) and a significant decrease in WC (59.6 ± 7.9 vs. 62.1 ± 7.5 cm; p = 0.032). No significant change was documented for BMI and bone/chronological age ratio (Table 1).
Metabolic parameters
The metabolic parameters of control subjects, GHD children at diagnosis, and after 12 months of GHT are shown in Table 1.
All GHD children at baseline had normal fasting glucose levels, although they were significantly lower than controls (3.89 ± 0.40 vs. 4.49 ± 0.41 mmol/l; p < 0.001). In addition, GHD children showed higher fasting insulin (6.3 ± 3.2 vs. 3.1 ± 2 IU/ml; p = 0.001), Homa-IR (1.19 ± 0.65 vs. 0.64 ± 0.43; p = 0.006), and lower ISI-Matsuda (11.5 ± 5 vs. 15.8 ± 7.9; p = 0.042) than controls. No significant difference was found in AUCGLU, AUCINS, HbA1c, and lipid profile between GHD children at baseline and control subjects (Table 1).
After 12 months of GHT, a significant increase in fasting glucose (4.63 ± 0.21 vs. 3.89 ± 0.40 mmol/l; p < 0.001), fasting insulin (12.1 ± 2.7 vs. 6.3 ± 3.2 IU/ml; p = 0.021), and Homa-IR (2.48 ± 0.53 vs. 1.19 ± 0.65; p = 0.011) was documented, although all children maintained normal glucose tolerance. No significant difference was found in AUCGLU, AUCINS, HbA1c, and ISI-Matsuda.
Total (3.92 ± 0.98 vs. 4.16 ± 0.85 mmol/l; p = 0.003) and LDL-cholesterol (1.97 ± 0.81 vs. 2.17 ± 0.70 mmol/l; p = 0.016) significantly decreased after 12 months of GHT, while no change in HDL cholesterol and triglycerides was documented (Table 1).
Adrenal function
The HPA evaluation of control subjects, GHD children at diagnosis and after 12 months of GHT are shown in Table 2.
Morning serum cortisol levels, AUCCOR, and cortisol peak during ITT proved to be positively and strongly correlated with one another both in the totality of children and separately in GHD children at both baseline and after GHT (all p < 0.010; data not shown).
Overall, based on morning cortisol levels and considering 3 µg/l as the cut-off value for diagnosing AI, no subject showed overt AI at both baseline and after 12 months of GHT.
Based on cortisol peak levels, a subnormal cortisol response (i.e., <18 µg/dl) to ITT was shown in 10 out of 60 children [2 out of 25 controls (8%) and 8 out of 35 GHD children (22%), with a mean cortisol peak of 17.7 and 13.8 µg/l, respectively] at baseline, while 11 out of 35 GHD children (31%) showed a cortisol peak value lower than 18 µg/l (mean cortisol peak of 12.1 µg/l) after 12 months of GHT, without any typical clinical signs or symptoms of AI.
At baseline, GHD children showed higher morning serum cortisol (11.7 ± 2.8 vs. 9.4 ± 2.8 µg/l; p = 0.012) and lower ACTH levels (18.4 ± 6.3 vs. 22 ± 6.1 pg/ml; p = 0.041), while no differences in serum sodium (p = 0.709) and potassium (p = 0.112), cortisol peak (p = 0.535) and AUCCOR (p = 0.128) during ITT were found (Table 2).
After 12 months of GHT, a significant decrease in morning serum cortisol (8.9 ± 2.3 vs. 11.7 ± 2.8 µg/l; p = 0.002) and AUCCOR (1489 ± 464 vs. 1868 ± 878 µg/l; p = 0.038) was documented, without significant change in serum sodium and potassium, and in cortisol peak during ITT (Table 2).
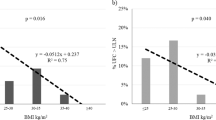
The correlations among cortisol secretion and the clinical, hormonal, and metabolic parameters in GHD children at baseline and after 12 months of GHY are shown in Table 3 and Fig. 1.

Correlations (univariate analysis) between serum morning cortisol secretion and GH peak (µg/l) during ITT, ISI-Matsuda, fasting glucose (mmol/l), and fasting insulin (IU/ml) in GHD children at baseline
At baseline, in GHD children, morning serum cortisol levels proved to be significantly and negatively correlated with GH peak (r −0.664; p < 0.001) and AUCGH (r −0.631; p < 0.001) during a/the stimulus test, fasting glucose (r −0.664; p < 0.001), and ISI-Matsuda (r −0.689; p < 0.001), while they were positively correlated with fasting insulin (r 0.483; p = 0.011) and Homa-IR (r 0.485; p = 0.010). Similarly, both cortisol peak and AUCCOR at baseline were negatively correlated with GH peak (r −0.651; p < 0.001 and r −0.722; p < 0.001, respectively), AUCGH (r −0.603; p < 0.001 and r −0.662; p < 0.001, respectively) and ISI-Matsuda (r −0.468; p = 0.016 and r −0.593; p = 0.001, respectively), while AUCCOR also positively correlated with Homa-IR (r 0.386; p = 0.047). These correlations were not confirmed when we analyzed separately the control group at baseline (data not statistically significant and not shown). No significant correlation was found between ACTH levels and the clinical, hormonal, and metabolic parameters neither in GHD nor in controls (data not shown).
After 12 months of GHT, no significant correlations were found among cortisol levels and all parameters were investigated, with the exception of a slight negative correlation between cortisol peak and fasting glucose (r −0.589; p = 0.044). No significant correlations were found between cortisol and IGF-I levels (data not shown).
Discussion
This study showed that the HPA function is strongly correlated with GH secretion and with many metabolic aspects of overt GHD. GHD children showed higher cortisol levels than controls and these levels are related to the insulin sensitivity degree. Conversely, GHT leads to a slight reduction in cortisol levels and to a loss of relationship between cortisol and metabolic parameters.
GHD patients potentially represent a population at risk of developing functional alterations of the HPA axis up to overt AI. Indeed, both GHD condition per se, due to potential concomitant multiple pituitary hormone deficiency, and GHT, due to the GH impact on cortisol metabolism, can be potentially associated with AI [10, 18]. However, there are discordant data in the literature about the HPA function in GHD and during GHT. AI is frequently observed in GHD patients with genetic or structural alterations [19, 20]. Giavoli et al. evaluated the HPA axis in patients with adult-onset GHD due to surgically treated pituitary tumors by means of morning serum cortisol levels as well as cortisol responses to stimulation test before and during GHT. The authors demonstrated central AI in 9 out of 11 patients, concluding that reassessment of HPA function in organic GHD patients during GHT is mandatory [11]. Indeed, it is widely known that GH influences cortisol metabolism by modulating the activity of 11beta-HSD1, which is the main target of cortisol modulation by GH [6, 8, 21]. However, few and sometimes discordant data about HPA axis evaluation during GHT in children with idiopathic GHD are available in the literature.
In ten consecutive children with idiopathic GHD, Giavoli et al. demonstrated that no child became AI during GHT, without differences in cortisol peak after a provocative test from baseline to a mean of 12 months of GHT. These authors concluded that probably only in patients with organic multiple pituitary hormone deficiency GHD masks the presence of a hidden central AI and that the HPA function evaluation in children with idiopathic GHD is not required during the follow-up [22]. Similarly, other authors suggested that patients with GHD without abnormalities on MRI probably do not need HPA testing. Indeed, among a relatively large number of children with idiopathic GHD whose HPA axis was evaluated, none had AI before GHT [23]. Conversely, in one of the largest series involving more than 2000 children with GHD, August et al. showed that about 11% of those with idiopathic GHD had central AI, suggesting, however, that AI is relatively uncommon in children without anatomic abnormalities [24].
Our data are partially in agreement with these studies. Indeed, we found that about 22% of GHD children at diagnosis showed a cortisol peak during ITT lower than the widely accepted cut-off (i.e., 18 µg/l), although none of the patients were symptomatic for AI, while when we considered the morning serum cortisol levels no child showed values lower than 3 µg/l.
However, it should be noted that ACTH deficiency is generally uncommon in children with idiopathic GHD and it has rarely been reported in the absence of organic lesions. In our hypothesis, despite the cortisol peak did not reach the established cut-off of 18 µg/dl for a while, the increase in cortisol levels from baseline to peak during ITT was very significant (up to 70%) and therefore it could be considered quite satisfactory both in the two controls and in the eight GHD children suspected of having AI. For these reasons, we decided not to treat these children for AI and clinically follow them. Indeed, the biochemical criteria used to diagnose AI take into account only a fixed cut-off and do not consider the size of the change of cortisol from the baseline value to the peak reached during the stimulation test, even if below the established cut-off.
Our data are in agreement with another study which showed subnormal cortisol response to ITT in 10 out of 25 children (40%) with idiopathic GHD [25]. Similarly, Lange et al. reported in a group of adults previously treated for idiopathic childhood-onset GHD a percentage of 44% of asymptomatic AI. The authors concluded that HPA axis evaluation at regular intervals is essential in these patients and that GHD may represent a predisposing factor for AI [18].
The discrepancy between these studies is probably due to the heterogeneity of the diagnostic criteria and of the tests used to assess HPA function. Indeed, although the diagnosis of overt AI is generally simple, the diagnosis of central AI, particularly in subjects with partial deficiency, or the identification of asymptomatic patients with subtle AI, is still a challenge. Patients with slight dysfunctions are at risk of developing adrenal crisis since their HPA does not appropriately react to stress. Therefore, identification of patients potentially at risk of AI is mandatory. There is considerable debate regarding the best test of the diagnosis of AI. Optimally, a screening test would be economic, convenient, and safe and it would have high sensitivity and specificity. Unfortunately, to date, no test for AI diagnosis meets all of these criteria. Response to ITT is considered the gold standard in the evaluation of the integrity of the HPA axis, although its validity has been questioned [26,27,28] and in patients with idiopathic multiple pituitary hormone deficiencies the low-dose short adrenocorticotropin test seemed to be equivalent to ITT [29,30,31] or even superior to the standard-dose corticotrophin test in the diagnosis of central AI [32]. Specifically, the reliability of the test used to evaluate HPA function in GHD patients was tested by Maghnie et al. The authors concluded that none of the tests can be considered completely reliable for establishing the presence of adrenal insufficiency, although ITT seems to be the most sensitive for detecting subclinical or partial AI [33]. In agreement, we used ITT to evaluate the HPA axis in GHD children. Although we showed good correlation between morning serum cortisol and its peak during ITT, it is widely demonstrated that the baseline morning serum cortisol concentration has limited predictive power in differentiating between normal and impaired HPA function [27, 28, 34]. In agreement with these observations, our data showed that morning cortisol evaluation cannot be considered a reliable indicator of AI in children with GHD because the capacity of morning cortisol to diagnose AI differs from that of the cortisol peak during ITT; that is to say, with the cortisol peak we can diagnose a percentage of potential AI that cannot be diagnosed with morning cortisol. Indeed, all GHD children who at baseline showed a cortisol peak less than 18 µg/dl had a baseline cortisol value greater than 3 µg/dl. In addition, when we calculated the AUC of cortisol during ITT, although it showed good direct correlation with both morning cortisol and cortisol peak during ITT, it did not prove to have greater utility than morning serum cortisol or cortisol peak. Notably, morning cortisol levels were higher in GHD children than in controls, but in the GHD group we found a higher prevalence of cortisol peak less than the established cut-off of 18 µg/dl. This finding, confirms the lack of utility of basal cortisol levels as a diagnostic test for AI.
All three of these cortisol parameters in GHD children at diagnosis demonstrated a good negative correlation with GH secretion (both GH peak and AUCGH during stimulation test) and with insulin sensitivity. Specifically, cortisol levels seem to be higher in concomitance with lower GH levels. Indeed, GHD children showed significantly higher morning serum cortisol levels and higher, although not statistically significant, peak and AUC of cortisol levels than controls. Conversely, we found a lack of correlation between cortisol and GH values in controls. We could speculate that the lower fasting glucose levels secondary to GHD, although still within the normal range, may lead to a rise in cortisol levels. Indeed, a negative correlation between morning fasting cortisol levels and fasting glucose was demonstrated at baseline in GHD children and not in controls. These data can further confirm and explain what was previously stated about the lack of reliability of morning serum cortisol as diagnostic test for AI if compared to cortisol peak in GHD.
In turn, the higher cortisol levels in GHD children may determine an increase in fasting insulin and a decrease in insulin sensitivity (as demonstrated by the higher Homa-IR and lower ISI-Matsuda in GHD than controls). The latter two are typical features of GHD, in concomitance with the evidence of higher BMI and WC, probably secondary to GHD per se, which in turn can worsen insulin sensitivity reduction. Indeed, overt GHD shows many features of metabolic syndrome [35], including abdominal obesity and insulin resistance, which are also typical of the conditions of cortisol excess [36]. This finding could lead us to speculate that higher cortisol levels, secondary to an increased activity of 11beta-HSD1 in GHD patients, may play a role in the development of the abovementioned clinical features.
However, if an overt metabolic syndrome is more easily associated with GHD in adult patients, probably because of the diagnostic and therapeutic delay, the metabolic alterations are less manifest in children and just a trend to a worse metabolic profile, such as lower insulin sensitivity degree or greater visceral obesity, may happen and the metabolic syndrome may be not yet clearly manifested, as shown in our study.
Though our data showed a correlation among cortisol levels and many metabolic aspects of overt GHD, this relationship seems to be lost after GHT. Indeed, controversial data exist in the literature on the effects of GHT on HPA function. Weaver et al. studied 19 adult hypopituitary patients who were receiving conventional hydrocortisone replacement during a 6-month period of GHT. The authors demonstrated that GHT in hypopituitary adults is associated with an apparent reduction in availability of administered hydrocortisone and concluded that the changes in cortisol metabolism suggest that GH may directly or indirectly modulate the activity of 11beta-HSD1 [7]. Recently, in a multicenter study that evaluated the HPA axis before and during 6 months of GHT therapy in 72 GHD patients, cortisol levels significantly decreased in patients already having multiple pituitary hormone deficiencies at baseline, and it decreased, although it remained within the normal range, in patients with isolated GHD [37].
Our data are partially in agreement with these studies. Indeed, we showed a significant decrease in morning serum cortisol and in AUCCOR during 12 months of GHT, with a trend to decrease, although not significant, in cortisol peak, and without change in serum sodium and potassium. In addition, despite the slight decrease in cortisol peak observed, after 12 months of GHT a higher percentage of children than baseline (31 vs. 22%) showed a cortisol peak less than 18 µg/l, without any typical signs or symptoms of AI. Probably, also in the absence of hypothalamic-pituitary organic pathologies, as in our cohort of children, the modulation exerted by GHT leads only to a slight decrease in cortisol levels by reducing the activity of 11beta-HSD1, without clinical consequences. Probably, the possible effect in decreasing cortisol binding globulin (CBG) levels cannot be ruled out for cortisol reduction during GHT, although this effect was not always confirmed [11]. However, despite the reduction in basal cortisol and AUCCOR during GHT and the increased prevalence of suspected AI, even in this case, as already noted at baseline, the increase in cortisol levels from baseline to peak during ITT was very pronounced (up to 90%) even in the 11 children suspected of having AI and we decided to keep patients in follow-up without initiating glucocorticoid treatment. However, it is to be noted that the lack of significant change in ACTH levels after GHT (lack of feed-back) could lead us to hypothesize that there may be a partial ACTH deficiency which have influenced GH peak at diagnosis and it could potentially explain the higher prevalence of AI in GHD than controls. These data reinforce the awareness that diagnosis of central AI, particularly in children with suspected partial deficiency, or the identification of asymptomatic patients with subtle AI, is difficult and still a challenge.
In our study, GHT led to an improvement in auxological parameters and IGF-I levels with a concomitant decrease in WC, as expected. In addition, we found higher fasting glucose and insulin levels with higher insulin resistance, as demonstrated by the increase in Homa-IR and the trend to decrease of ISI-Matsuda. This trend to impairment in glucose metabolism during GHT has often been previously demonstrated [38, 39]. Indeed, it is widely known that GHT leads to a decrease in insulin sensitivity and alteration in insulin secretion even without overt changes in glucose tolerance, and the current study confirms this finding, as demonstrated by the unchanged HbA1c levels. In addition, a slight improvement in lipid profile was demonstrated after 12 months of GHT and this finding is in agreement with our previous studies [15, 40]. Overall, in our opinion, the metabolic changes which occur after GHT are likely secondary to the counter-regulating effects of GH (the increase in glucose and insulin levels and the reduction in insulin sensitivity) and to its lipolytic effect (demonstrated by the improvement in lipid profile and reduction in WC). In addition, the impact of GHT on a series of adipokines known to have a metabolic role [41], could explain the lack of a direct relationship between cortisol levels and metabolic parameters during GHT. In concomitance with a slight increase in glucose levels, and mediated by the known GH effect on cortisol metabolism, cortisol levels tended to decrease, although without determining overt AI, leading to a loss of its relationship with metabolic parameters. Therefore, the metabolic effect observed during GHT must be considered to be independent of any change in cortisol metabolism [7,8,9]. These data are in agreement with the study performed by Yuen et al., who demonstrated no correlation between the change in metabolic parameters and cortisol levels in adult GHD patients during GHT [42].
In our opinion, the strength of our study was the use of the same test (i.e., ITT) in all patients in concomitance with full metabolic evaluation. Conversely, the limits were certainly represented by the relatively small sample size, the limited period of observation and the lack of data for the control group after 12 months of follow-up. Indeed, we cannot exclude with certainty that the cortisol change after 12 months are due to some other factor, in addition to GHT. However, in our opinion, it is very difficult to perform a full metabolic and hormonal evaluation, through OGTT and ITT, in healthy children during a follow-up of 12 months.
In conclusion, cortisol levels correlate well with GH secretion and with many metabolic parameters, so they could have a potential role in the development of metabolic alterations associated with GHD in children. Conversely, the metabolic effects during GHT are mainly due to GHT per se and less to cortisol reduction. GHT may potentially lead to a reduction in cortisol levels also in children with idiopathic GHD and endocrinologists should be mindful of this phenomenon when starting treatment with GH. Indeed, most children with multiple hormone deficiency or with etiology other than idiopathic GHD are frequently subject to a full endocrine evaluation, whereas wrongly this is not the case for patients with idiopathic GHD. Larger studies, with longer periods of follow-up of GHT, will be able to confirm the results of this study.
References
Le Roy C, Li JY, Stocco DM, Langlois D, Saez JM (2000) Regulation by adrenocorticotropin (ACTH), angiotensin II, transforming growth factor-beta, and insulin-like growth factor I of bovine adrenal cell steroidogenic capacity and expression of ACTH receptor, steroidogenic acute regulatory protein, cytochrome P450c17, and 3beta-hydroxysteroid dehydrogenase. Endocrinology 141(5):1599–1607
Gianotti L, Ramunni J, Lanfranco F, Maccagno B, Giordano R, Broglio F, Maccario M, Muller EE, Ghigo E, Arvat E (2001) Recombinant human IGF-I does not modify the ACTH and cortisol responses to hCRH and hexarelin, a peptidyl GH secretagogue, in humans. J Endocrinol Invest 24(2):67–71
Ricketts ML, Verhaeg JM, Bujalska I, Howie AJ, Rainey WE, Stewart PM (1998) Immunohistochemical localization of type 1 11beta-hydroxysteroid dehydrogenase in human tissues. J Clin Endocrinol Metab 83(4):1325–1335
Seckl JR, Chapman KE (1997) Medical and physiological aspects of the 11beta-hydroxysteroid dehydrogenase system. Eur J Biochem 249(2):361–364
Stewart PM, Toogood AA, Tomlinson JW (2001) Growth hormone, insulin-like growth factor-I and the cortisol-cortisone shuttle. Horm Res 56(Suppl 1):1–6
Gelding SV, Taylor NF, Wood PJ, Noonan K, Weaver JU, Wood DF, Monson JP (1998) The effect of growth hormone replacement therapy on cortisol-cortisone interconversion in hypopituitary adults: evidence for growth hormone modulation of extrarenal 11 beta-hydroxysteroid dehydrogenase activity. Clin Endocrinol (Oxf) 48(2):153–162
Weaver JU, Thaventhiran L, Noonan K, Burrin JM, Taylor NF, Norman MR, Monson JP (1994) The effect of growth hormone replacement on cortisol metabolism and glucocorticoid sensitivity in hypopituitary adults. Clin Endocrinol (Oxf) 41(5):639–648
Walker BR, Andrew R, MacLeod KM, Padfield PL (1998) Growth hormone replacement inhibits renal and hepatic 11 beta-hydroxysteroid dehydrogenases in ACTH-deficient patients. Clin Endocrinol (Oxf) 49(2):257–263
Paulsen SK, Pedersen SB, Jørgensen JO, Fisker S, Christiansen JS, Flyvbjerg A, Richelsen B (2006) Growth hormone (GH) substitution in GH-deficient patients inhibits 11beta-hydroxysteroid dehydrogenase type 1 messenger ribonucleic acid expression in adipose tissue. J Clin Endocrinol Metab 91(3):1093–1098
Maghnie M, Triulzi F, Larizza D, Preti P, Priora C, Scotti G, Severi F (1991) Hypothalamic-pituitary dysfunction in growth hormone-deficient patients with pituitary abnormalities. J Clin Endocrinol Metab 73(1):79–83
Giavoli C, Libé R, Corbetta S, Ferrante E, Lania A, Arosio M, Spada A, Beck-Peccoz P (2004) Effect of recombinant human growth hormone (GH) replacement on the hypothalamic-pituitary-adrenal axis in adult GH-deficient patients. J Clin Endocrinol Metab 89(11):5397–5401
Marshall WA, Tanner JM (1969) Variations in pattern of pubertal changes in girls. Arch Dis Child 44:291
Growth Hormone Research Society (2000) Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research Society. J Clin Endocrinol Metab 85:3990–3993
Anderson M (1971) Use of the Greulich-Pyle ‘radiographic atlas of skeletal development of the hand and wrist’ in a clinical context. Am J Phys Anthropol 35(3):347–352
Ciresi A, Cicciò F, Amato MC, Giordano C (2015) Revaluation of the clinical and metabolic behavior of children with isolated growth hormone deficiency during GH treatment according to newly proposed note 39 of the Italian Medicines Agency (AIFA). J Endocrinol Invest 38(12):1301–1307
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419
Matsuda M, DeFronzo RA (1999) Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22:1462–1470
Lange M, Feldt-Rasmussen U, Svendsen OL, Kastrup KW, Juul A, Müller J (2003) High risk of adrenal insufficiency in adults previously treated for idiopathic childhood onset growth hormone deficiency. J Clin Endocrinol Metab 88(12):5784–5789
Böttner A, Keller E, Kratzsch J, Stobbe H, Weigel JF, Keller A, Hirsch W, Kiess W, Blum WF, Pfäffle RW (2004) PROP1 mutations cause progressive deterioration of anterior pituitary function including adrenal insufficiency: a longitudinal analysis. J Clin Endocrinol Metab 89(10):5256–5265
di Iorgi N, Secco A, Napoli F, Tinelli C, Calcagno A, Fratangeli N, Ambrosini L, Rossi A, Lorini R, Maghnie M (2007) Deterioration of growth hormone (GH) response and anterior pituitary function in young adults with childhood-onset GH deficiency and ectopic posterior pituitary: a two-year prospective follow-up study. J Clin Endocrinol Metab 92(10):3875–3884
Moore JS, Monson JP, Kaltsas G, Putignano P, Wood PJ, Sheppard MC, Besser GM, Taylor NF, Stewart PM (1999) Modulation of 11beta-hydroxysteroid dehydrogenase isozymes by growth hormone and insulin-like growth factor: in vivo and in vitro studies. J Clin Endocrinol Metab 84(11):4172–4177
Giavoli C, Bergamaschi S, Ferrante E, Ronchi CL, Lania AG, Rusconi R, Spada A, Beck-Peccoz P (2008) Effect of growth hormone deficiency and recombinant hGH (rhGH) replacement on the hypothalamic-pituitary-adrenal axis in children with idiopathic isolated GH deficiency. Clin Endocrinol (Oxf) 68(2):247–251
Walvoord EC, Rosenman MB, Eugster EA (2004) Prevalence of adrenocorticotropin deficiency in children with idiopathic growth hormone deficiency. J Clin Endocrinol Metab 89(10):5030–5034
August GP, Lippe BM, Blethen SL, Rosenfeld RG, Seelig SA, Johanson AJ, Compton PG, Frane JW, McClellan BH, Sherman BM (1990) Growth hormone treatment in the United States: demographic and diagnostic features of 2331 children. J Pediatr 116(6):899–903
Savas Erdeve S, Ocal G, Berberoglu M, Siklar Z, Hacihamdioglu B (2011) Is adrenocorticotropic hormone deficiency really rare in patients with idiopathic growth hormone deficiency and normal thyroid function tests? Horm Res Paediatr 75(3):200–205
Nieman LK (2003) Dynamic evaluation of adrenal hypofunction. J Endocrinol Invest 26(7 Suppl):74–82
Erturk E, Jaffe CA, Barkan AL (1998) Evaluation of the integrity of the hypothalamic-pituitary-adrenal axis by insulin hypoglycemia test. J Clin Endocrinol Metab 83(7):2350–2354
Grinspoon SK, Biller BM (1994) Clinical review 62: laboratory assessment of adrenal insufficiency. J Clin Endocrinol Metab 79(4):923–931
Weintrob N, Sprecher E, Josefsberg Z, Weininger C, Aurbach-Klipper Y, Lazard D, Karp M, Pertzelan A (1998) Standard and low-dose short adrenocorticotropin test compared with insulin-induced hypoglycemia for assessment of the hypothalamic-pituitary-adrenal axis in children with idiopathic multiple pituitary hormone deficiencies. J Clin Endocrinol Metab 83(1):88–92
Rasmuson S, Olsson T, Hagg E (1996) A low dose ACTH test to assess the function of the hypothalamic-pituitary-adrenal axis. Clin Endocrinol (Oxf) 44(2):151–156
Ambrosi B, Barbetta L, Re T, Passini E, Faglia G (1998) The one microgram adrenocorticotropin test in the assessment of hypothalamic-pituitary-adrenal function. Eur J Endocrinol 139(6):575–579
Kazlauskaite R, Maghnie M (2010) Pitfalls in the diagnosis of central adrenal insufficiency in children. Endocr Dev 17:96–107
Maghnie M, Uga E, Temporini F, Di Iorgi N, Secco A, Tinelli C, Papalia A, Casini MR, Loche S (2005) Evaluation of adrenal function in patients with growth hormone deficiency and hypothalamic-pituitary disorders: comparison between insulin-induced hypoglycemia, low-dose ACTH, standard ACTH and CRH stimulation tests. Eur J Endocrinol 152(5):735–741
di Iorgi N, Napoli F, Allegri A, Secco A, Calandra E, Calcagno A, Frassinetti C, Ghezzi M, Ambrosini L, Parodi S, Gastaldi R, Loche S, Maghnie M (2010) The accuracy of the glucagon test compared to the insulin tolerance test in the diagnosis of adrenal insufficiency in young children with growth hormone deficiency. J Clin Endocrinol Metab 95(5):2132–2139
Qing L, Wei R, Chan L, Xiaoya Z, Xin X (2017) Sensitivity of various body indices and visceral adiposity index in predicting metabolic syndrome among Chinese patients with adult growth hormone deficiency. J Endocrinol Invest 40(6):653–661
Feelders RA, Pulgar SJ, Kempel A, Pereira AM (2012) The burden of Cushing’s disease: clinical and health-related quality of life aspects. Eur J Endocrinol 167(3):311–326
Wang L, Wang Q, Li G, Liu W (2015) Dynamic changes in the hypothalamic-pituitary-adrenal axis during growth hormone therapy in children with growth hormone deficiency: a multicenter retrospective study. J Pediatr Endocrinol Metab 28(9–10):975–979
Capalbo D, Mattace Raso G, Esposito A, Di Mase R, Barbieri F, Meli R, Bruzzese D, Salerno M (2013) Cluster of cardiometabolic risk factors in children with GH deficiency: a prospective, case-control study. Clin Endocrinol (Oxf) 80(6):856–862
Ciresi A, Amato MC, Giordano C (2015) Reduction in insulin sensitivity and inadequate β-cell capacity to counteract the increase in insulin resistance in children with idiopathic growth hormone deficiency during 12 months of growth hormone treatment. J Endocrinol Invest 38(3):351–359
Ciresi A, Amato MC, Criscimanna A, Mattina A, Vetro C, Galluzzo A, D’Acquisto G, Giordano C (2007) Metabolic parameters and adipokine profile during GH replacement therapy in children with GH deficiency. Eur J Endocrinol 156(3):353–360
Ciresi A, Pizzolanti G, Leotta M, Guarnotta V, Teresi G, Giordano C (2016) Resistin, visfatin, leptin and omentin are differently related to hormonal and metabolic parameters in growth hormone-deficient children. J Endocrinol Invest 39(9):1023–1030
Yuen KC, Roberts CT Jr, Frystyk J, Rooney WD, Pollaro JR, Klopfenstein BJ, Purnell JQ (2014) Short-term, low-dose GH therapy improves insulin sensitivity without modifying cortisol metabolism and ectopic fat accumulation in adults with GH deficiency. J Clin Endocrinol Metab 99(10):E1862–E1869
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
At the time of hospitalization, an informed consent for the scientific use of the data was obtained from individual participants included in the study and their parents.
Rights and permissions
About this article

Cite this article
Ciresi, A., Radellini, S., Vigneri, E. et al. Correlation between adrenal function, growth hormone secretion, and insulin sensitivity in children with idiopathic growth hormone deficiency. J Endocrinol Invest 41, 333–342 (2018). https://doi.org/10.1007/s40618-017-0747-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-017-0747-2