
Abstract
Vigna radiata L., commonly referred to as mungbean or green gram, holds significant importance as a pulse crop in India. However, its productivity is severely impacted by the combined incidence of dry root rot disease and drought stress. Dry root rot, caused by Macrophomina phaseolina, manifests as reduced yield and compromised produce quality. M. phaseolina is a necrotrophic fungus with a broad host range. Screening studies in several crops’ germplasms have shown a skewness towards susceptibility. Further, the fungus has augmented virulence and survivability in soil under low moisture and high heat. Thus, concurrent drought and dry root rot leads to significantly higher yield losses. This review highlights the status of the disease in mungbean and its future implications owing to the changing climate scenario. We also highlight the molecular and genomic studies conducted in mungbean and several other crops to elucidate the mechanisms involved in M. phaseolina resistance. The review also suggests management practices which can alleviate yield losses in dry root rot affected fields. Understanding the physiological and molecular mechanisms of dry root rot, drought, and their interaction on disease proliferation can help mitigate the challenges associated with dry root rot management and aid future research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vigna radiata L., commonly referred to as mungbean or green gram, is widely grown in East Asia, Southeast Asia, and the Indian subcontinent. Global mungbean cultivation spans approximately 7.3 million ha, with an average yield of 721 kg/ha (source: https://iipr.icar.gov.in/mungbean/). Myanmar, India, China, Indonesia, Thailand, and Kenya are the key global producers of mungbean (Nair et al., 2020).
In India, mungbean is the third most significant leguminous crop, contributing approximately 16% to the overall pulse production. According to estimates from the Government of India (averaged for 2017–18 to 2021–22), mungbean is grown on 48.5 lakh ha, with a total production of 26.5 lakh tonnes and a productivity of 546 kg/ha. Rajasthan is the major mungbean-growing state, with an acreage of 23.3 lakh ha (46% of the total area), production of 11.2 lakh tonnes (45% of the total production), and productivity of 480 kg/ha.
Other significant mungbean-growing states include Madhya Pradesh (5.1 lakh ha), Maharashtra (4.3 lakh ha), Karnataka (4.1 lakh ha), Odisha (2.4 lakh ha), Tamil Nadu (1.7 lakh ha), Bihar (1.6 lakh ha), Gujarat (1.4 lakh ha), and Andhra Pradesh (1.1 lakh ha). The productivity of mungbean in many Indian states is much lower than the national average (~ 24%). The low productivity of mungbean is mainly due to production constraints, including biotic and abiotic stresses that affect the crop throughout its life cycle. Fungal and viral pathogens pose significant threats to mungbean cultivation, with more than half of the crop losses attributed to soil-borne pathogens. The situation is exacerbated by climate change, leading to frequent occurrences of abiotic stresses such as drought and heat, which further promote the proliferation of soil-borne pathogens. Breeding for climate-resilient cultivars possessing adaptive traits that confer resistance/tolerance to the combined biotic and abiotic stresses is a viable strategy to realize higher productivity in mungbean.
Dry root rot—a globally emerging disease in mungbean
Dry root rot (DRR) is caused by the pathogen Macrophomina phaseolina and is known to affect over 500 plant species, including agricultural crops. The pathogen is also known to occasionally affect humans (Arora et al., 2012). The phytopathogenic fungus affects all stages of plant growth, from germination to maturity (Anupriya et al., 2023). In vulnerable plant species, the infection can manifest as damping-off, stem blight, stalk rot, leaf blight, or pod rot (Khan et al., 2023). The emergence of DRR has become a significant concern for mungbean cultivation worldwide (Kaur et al., 2012; Mallaiah & Rao, 2016; Pandey et al., 2021). The disease has caused 25–48% yield loss in South Asia (Bashir & Malik, 1988; Iqbal & Mukhtar, 2014). In India, the yield loss in mungbean due to M. phaseolina infection ranged from 30 to 44% (Kaur et al., 2023), up to 30% in Rajasthan (Tyagi et al., 1988; Sharma & Singh, 2000; Basandrai et al., 2021), up to 25% in Andhra Pradesh (Mallaiah & Rao, 2016), and up to 10.8% in Haryana (Tyagi et al., 1988). The disease is widely occurring in other states as well.
In addition to direct yield losses at the field level, the pathogen causes seed contamination during storage, leading to significant deterioration of seeds and consequent losses (Pandey et al., 2021), reduction in seed germination, and protein content. DRR has been reported to cause up to 36% deterioration in stored mungbean (Ashwini & Giri, 2014; Basandrai et al., 2021) and a 12.3% loss in protein content (Kaushik and Chand, 1987; Ahmad et al., 2015).
Dry root rot incidence in mungbean
DRR in mungbean is a widely prevalent disease in Asia, Africa, and Australia (Batzer et al., 2022). In Asia, the disease is a menace in Myanmar and is rapidly spreading in South Asia, particularly in India, Pakistan, and China (Iqbal & Mukhtar, 2014; Pandey et al., 2021; Zhang et al., 2011). The first report on DRR incidence in India was from Jabalpur in Madhya Pradesh (Singh et al., 2022a, 2022b). The disease has been devastating in several districts of Rajasthan (Kumar et al., 2017a) and Maharashtra (Khaire et al., 2023). A systematic survey has been conducted to assess the level of disease incidence and spread of DRR in a few mungbean-growing states of India (Fig. 1).

Spread of dry root rot incidence in major mungbean growing states of India. Extensive literature survey was conducted to mine studies/surveys conducted on mungbean DRR incidence in various locations across India. Pins represent the districts/locations where mungbean DRR incidence surveys were conducted. Colour grades of the states is based on the average DRR disease incidence in mungbean
DRR incidence ranging from 0.5 to 38% was observed in seed samples collected from 11 districts of Rajasthan (Sharma & Singh, 2000). The incidence of DRR was found in all the seed samples collected from mungbean grown in the farmers’ fields of Chamarajanagar, Mysore, and Gundlupet taluks of Karnataka (Murthy et al., 2003). Based on farmer’s field surveys, DRR incidence of 5.7–12% was reported in Chittoor district of Andhra Pradesh (Mallaiah & Rao, 2016). Similarly, about 30% disease incidence was reported in Namakkal district of Tamil Nadu, based on roving surveys conducted in Mohanur, Namakkal, Senthamangalam, and Rasipuram areas (Chandraprakash et al., 2022). In Telangana, DRR incidence was observed to the tune of 5.3 to 31.7% based on roving surveys conducted in villages of Adilabad, Warangal, Khammana, and Mahububabad districts (Avanija et al., 2023).
The emergence of DRR has also become a significant concern for the cultivation of other Vigna species. DRR incidence ranging from 16 to 33% was reported as early as 1999 in urd bean grown in rice-fallow regions of Karaikal (Rettinassababady and Ramadoss, 1999), where both mungbean and urd bean are preferred after the rice crop. The disease incidence was reported in cowpea grown in Cuddalore, Thiruvannamalai, and Vellore districts of Tamil Nadu (Mohanapriya et al., 2017), signifying the prevalence and threat of DRR in mungbean-growing areas in the state. Variations in the level of disease incidence depended on prevailing weather conditions, physiochemical attributes of the soil, inoculum load, and pathogen virulence.
The dry root rot pathogen
M. phaseolina (Tassi) Goid. is a necrotrophic, soil-borne Ascomycete phytopathogenic fungus belonging to the family Botryosphaeriaceae. The fungus exists in a sporulating and a non-sporulating stage. The sporulating stage is generally not associated with DRR and can be observed in the above ground symptom stage (generally referred to as charcoal rot). Further, in some plants such as chickpea, the sporulating stage is not reported (Irulappan et al., 2021) Morphologically, the pathogen produces hyphae that appear as thin-walled, transparent strands ranging from light to dark brown with multiple septa. Their branches typically emerge right-angled to the parent hyphae, constricting at the starting point. Initially, sclerotia are light brown and darken over time, shifting from irregular, spherical, oval, to oblong shapes. However, morphological and genetic variations exist among the isolates that determine their virulence to infect host species.
Cultural and morphological variability of M. phaseolina isolates collected from mungbean
M. phaseolina isolates from disease-affected mungbean plants from various geographical regions exhibit variations in morphology and virulence levels. As a polyphagous plant pathogen, isolates from distinct hosts also display morphological differences (Pandey et al., 2020). Climatic factors, such as temperature, seasonality, and rainfall, significantly influence the genetic variation in M. phaseolina (Ortiz et al., 2023). Common morphological variations among isolates include colony color, sclerotial color, shape, size, weight, count, diameter, and growth pattern (Iqbal & Mukhtar, 2014; Pandey et al., 2021).
A total of 56 isolates from mungbean and urd bean collected from 11 different locations representing North, South, Northeast, and Central India were grouped into six distinct categories based on morphological characterization (Prameela Devi & Singh, 1998). Furthermore, these isolates were classified as highly virulent, moderately virulent, and weakly virulent strains based on their intensity on the host and disease incidence. Iqbal and Mukhtar (2014) characterized 65 isolates from Pakistan based on sclerotial size, weight, radial growth, and observed no relationship between these morphological parameters and pathogenicity of isolates.
In an investigation of M. phaseolina isolates from 11 distinct locations (10 from India and one from Myanmar) in mungbean, Pandey et al. (2021) noted variations in the shape of sclerotia from round to oblong, with sclerotial counts ranging between 141.7 and 208.4/9 mm disc, and diameters spanning from 76.0 to 113.2 μM/9 mm disc. The sclerotial count correlated with pathogenicity, suggesting that isolates with higher sclerotial counts tend to be more pathogenic compared to those with lower counts. Additionally, sclerotia exhibited varying shades of greyish white, greyish black, blackish grey, black, dark brown greyish (Khaire et al., 2023; Mallaiah & Rao, 2016; Pandey et al., 2020, 2021). The growth pattern also fluctuates, transitioning from less feathery to moderately feathery and eventually to more feathery structures. Despite widespread cultural variation among the isolates, their morphology remains quite similar and has been used as the primary method for identification of the pathogen.
Molecular characterization of M. phaseolina isolated from mungbean
While morphological features are useful for the initial identification of the fungus, molecular tools are employed for further confirmation and strain typing. DNA marker systems, such as restriction fragment length polymorphism (RFLP), randomly amplified polymorphic DNA (RAPD), simple sequence repeats (SSR), and amplified fragment length polymorphism (AFLP), have been utilized for molecular characterization and assessing genetic variability among isolates of M. phaseolina from various host species. The RAPD technique has been instrumental in identifying and differentiating isolates of M. phaseolina infecting mungbean (Babu et al., 2010; Fuhlbohm et al., 2013). The internal transcribed sequences (ITS) of 18S rRNA have been pivotal in molecular characterization of M. phaseolina isolated from various hosts, for species-level identification. Sequence analysis has revealed a high degree of similarity among the isolates (Mahdizadeh et al., 2011; Pandey et al., 2021; Zhang et al., 2011). In addition to ITS sequences, Macrophomina-specific primers (MpKF1 and MaKR1) have also been utilized in mungbean for confirming the pathogen’s identity (Zhang et al., 2011). Next-generation sequencing (NGS) technologies have also been employed for characterizing isolates of M. phaseolina, shedding light on genetic divergence and distinct genetic clumping at the continental level (Oritz et al., 2023). Isothermal amplification techniques, such as loop-mediated isothermal amplification (LAMP), capable of fungal detection at a constant temperature within a short time with a high degree of specificity and sensitivity, are also being utilized (Notomi et al., 2015). Although the use of LAMP assay to detect M. phaseolina isolates in mungbean has not been reported to date, the technique has been employed to detect the pathogen in other hosts such as common bean (Rocha et al., 2017), soybean (Lu et al., 2015), chickpea (Ghosh et al., 2018), and strawberry (Burkhardt et al., 2018).
Mungbean-M. phaseolina interaction and disease cycle
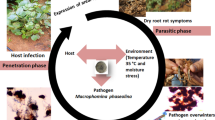
M. phaseolina can infect mungbean at almost all growth stages, with propagules penetrating the seedcoat, endosperm, and embryo, leading to a significant reduction in seed germination and viability (Buts et al., 2014). The disease cycle consists of four stages: germination, penetration, parasitic, and saprophytic phases, with microsclerotia serving as the primary inoculum. The pathogen further spreads via airborne pycnidiospores and sclerotia, facilitating secondary dissemination (Singh et al., 2022a, 2022b). Root exudates stimulate microsclerotia germination. Upon germination, microsclerotia develop germ tubes and generate appressoria that aid in penetration into host cells. During the parasitic phase, germinated microsclerotia give rise to hyphal branches, while in the saprophytic phase, infectious hyphae penetrate plant tissues through wounds or cells on the root surface (Gupta et al., 2023), producing enzymes and toxins that aid in breaking down plant cell walls, facilitating entry into root tissues (Irulappan et al., 2021). As the fungus’s hyphae grow inside plant cells, they disrupt water and nutrient transport, resulting in visible symptoms as the fungus progresses within the plant. Subsequently, neighbouring cells collapse, potentially leading to the death of heavily infected plants. The extent of invasion depends on both the plant’s defense response and the pathogen’s ability to counteract it (Marquez et al., 2021). Since disease outbreaks are severe under optimal environmental conditions, they often result in the premature death of host plants. Following the decay of dead roots and other plant parts, microsclerotia are discharged into the soil in clusters, serving as inoculum, and the disease cycle continues (Ghosh et al., 2018) (Fig. 2). Microsclerotia capable of enduring in soil and root debris for three years or longer enable the fungus to survive in unfavourable environmental conditions in the field (Marquez et al., 2021).

Disease cycle of Macrophomina phaseolina. Microsclerotia in close vicinity host roots germinate upon recognition of host root exudates. Upon contact with the root, germ tube and appressoria are formed leading to tissue penetration. Necrotic lesions begin to appear on the host roots. Vascular bundle plugging leads to premature plant death. Microsclerotia survive in host debris and in soil which serves as inoculum in the next cycle
M. phaseolina harbours several potentially virulent elements that actively interfere with the host plant’s defense mechanism. Initially, the pathogen establishes communication with the host via a class II hydrophobic protein and adheres to the root through the action of CEBL (cellulose-binding elicitor lectins) and transglutaminase-like proteins. Phytotoxins such as botrydiploidin, phaseolinon, and patulin secreted by the pathogen play a crucial role during the initial stages of pathogenesis. On perceiving the pathogen associated molecular patterns (PAMPS), the host immune system begins the first line of defense response by producing signaling molecules like salicylic acid. The pathogen counteracts this initial host defense by secreting compounds like salicylate-1-monoxygenase and penetrates the host tissues. The invasion process is regulated by cAMP dependent and mitogen activated protein (MAP) kinase pathway (Islam et al., 2012). Upon invading the host tissues, a wide repertoire of hydrolytic enzymes, cell wall-breaking enzymes, and effector proteins released by M. phaseolina are involved in the infection process, facilitating disease development, and eventually causing host cell death (Islam et al., 2012; Marquez et al., 2021) (Fig. 3). Sinha et al. (2022) identified 117 proteins, including xylanase, endoglucanase, and amylase, in the secretome of M. phaseolina. It has been observed that M. phaseolina can produce indole acetic acid (IAA), a hormone known to stimulate plant growth through several synthesis pathways (Amairani et al., 2023). This hormone plays a dual role in plant-pathogen interaction, acting both as a plant hormone that modifies host physiology to enhance susceptibility and as a microbial signal that impacts the pathogen to increase virulence (Kunkel et al., 2021). The elicitor molecules produced by the pathogen during the infection process trigger a series of signaling molecules involved in defense reactions that accumulate inhibitors, enzymes and pathogeneis related (PR) proteins in the host cells to prevent infection and disease progression (Fig. 3). One such elicitor molecule isolated from M. phaseolina was used to treat cell cultures of mungbean cultivars. The treated host cells responded to the elicitor treatment by producing increased levels of phenolics and enzymes like phenylalanine ammonia lyase and peroxidases, known to impart resistance in host plants against pathogen infection (Vidyasekaran et al., 2002).

Molecular mechanisms underlying pathogen virulence and plant defense in the Macrophomina-host interface. During successful infection, root exudates are identified by M. phaseolina which in turn activates pathways to induce necrosis and dampen host defences. As the fungus traverses the cortex, it eventually breaches the endodermis subsequently plugging the vascular bundles with developing microsclerotia, leading to premature plant death. In an attempt to defend against M. phaseolina, the host employs several strategies such as activation of SA mediated signal cascade, Ca2+ mediated responses, cell-wall depositions, secondary metabolite synthesis, ROS signalling, and PR proteins
The pathogen infests every part of the mungbean plant, from roots and stems to branches, petioles, leaves, pods, and seeds. It severely affects the overall health of the plant, weakening it by reducing its ability to uptake water and nutrients. This leads to stunted growth, reduced yield, and in severe cases, complete crop loss. The pathogen can affect plants in both pre-emergence and post-emergence stages. Initially, it causes seed rot and kills germinating seedlings. After emergence, cotyledons may be affected due to soil or seed-borne infections (Fuhlbohm et al., 2013). Usually, the onset of symptoms coincides with the crop’s flowering period.
The fungus primarily targets the stem near ground level in one-month-old crops, forming localized, raised white cankers that expand and develop into upward-spreading brown streaks (Seethapathy et al., 2017). Infected plant leaves exhibit silvery-grey coloration of stems and lateral branching, with senesced leaves still attached to the plant (Zhang et al., 2011). Leaves may show dark green, mottled patterns and reduced size, followed by sudden wilting and drying, leading to a drastic decline in flowering and pod production (Singh et al., 2022a, 2022b). Necrotic lesions can appear on pod surfaces without specific placement. Initially, infected green pods exhibit a blue-green color, transitioning to brown or reddish color. Mature dry pods infected by this fungus appear white to grey and bear scattered or widespread black structures. The fungus penetrates both pods and grains, and affected grains either abort earlier or desiccate, resulting in emptiness (Ghosh et al., 2018). Seeds obtained from infected plants appear shrunken, brown, and sometimes have scattered black M. phaseolina sclerotia (Akhtar et al., 2011). Vertical splitting of the drying and wilt-affected plants reveals discoloration in internal tissues (Basandrai et al., 2021). The affected plants can be easily uprooted, leaving behind dried, decayed root sections in the soil. The rotten, decaying stem and root tissues show dark discoloration due to the presence of numerous small black sclerotia, hence the symptom is popularly described as charcoal rot (Singh et al., 2022a, 2022b). Additionally, deterioration of secondary roots, shredding of the cortex area in the taproot, dark brown necrotic lesions on the exterior surface of the taproot, underneath the epidermis, and pith of lower stems in wilt-affected plants are also prominent signs of infection caused by the pathogen (Fig. 4A to D). Furthermore, at 7 days post-inoculation (dpi), the in planta fungal DNA significantly increases in mungbean seedlings, implying extensive cortical colonization by M. phaseolina (Fig. 4E, Supplementary File 1).

Symptoms of dry root rot on mung bean caused by Macrophomina phaseolina (Tassi) Goid. The mung bean (Vigna radiata L.) variety TM 2000–2 was tested for its response to M. phaseolina using the blotter paper assay. Typical root necrotic lesions were observed at 7 days after inoculation (DAI). Images of the whole plant at 7 DAI: A Control, B Pathogen. Scale bars—1 cm. Stereomicroscopic observations of the roots of mung bean at 7 DAI: C Control, D Pathogen. Scale bars—250 μm. E In planta fungal DNA quantification using ITS primers specific to M. phaseolina. N = 5, significance was tested using one-way ANOVA followed by Tukey’s post-hoc test. Error bars represent the standard error of mean
Drought and heat favor dry root rot incidence
An optimal disease cycle entails a susceptible host, virulent pathogen, and conducive environmental conditions. DRR disease incidence caused by M. phaseolina across various host crops is favored by maximum ambient temperatures exceeding 30 °C and dry conditions inducing moisture stress during the plant’s reproductive stages (Seethapathy et al., 2017; Rai et al., 2022). This is attributed to M. phaseolina’s capacity to thrive and endure well in high-temperature and water-stressed environments (Chamorro et al., 2015). Although a wide temperature range of 15 to 40 °C supports the growth of M. phaseolina, optimal conditions for infection occur between 28 and 35 °C. At 20 °C and 25 °C, a reduction in the size of microsclerotia is observed compared to temperatures of 30 °C and 35 °C (Akhtar et al., 2011; Basandrai et al., 2021). Higher root rot disease incidence is observed under low moisture conditions, with maximum disease incidence recorded at 40% soil moisture content (Kumar et al., 2019; Soni et al., 2022). The low level of disease incidence at high soil moisture content is attributed to the fungal sclerotia’s inability to survive under wet soil conditions (Kumar et al., 2019). Additionally, apart from drought and heat, other abiotic factors such as soil properties also determine the incidence of DRR (Irulappan et al., 2022).
The combined effect of drought stress and M. phaseolina infection has been studied in various crops such as sorghum (Goudzrzi et al., 2011), common bean (Mayek-PÉrez et al., 2002), chickpea (Chilakala et al., 2023; Irulappan et al., 2022), cotton (Ghaffar & Erwin, 1969), and strawberry (Sanchez et al., 2019). In all these studies, observations consistently indicated that drought stress exacerbated DRR infection by altering key physiological mechanisms due to disrupted plant-water relationships. Particularly in chickpea, it was found that drought stress weakened the plant’s defense mechanisms and compromised the integrity of the endodermal barrier, accelerating the spread of the pathogen within the roots. Furthermore, in response to drought stress, the expression of genes linked to hormone regulation was differentially regulated, exacerbating DRR by affecting the plant’s innate resistance to infection (Irulappan et al., 2022).
Conversely, similar studies in mungbean (Fulbohm et al., 2013; Kaur et al., 2023, 2024) and soybean (Mengistu et al., 2018) have found no or limited association, highlighting the need to consider several factors when analysing the impact of these combined stresses. Fulbohm et al. (2013) observed increased seed infection by the pathogen in the Australian mungbean cultivar Beken during the rainy period, likely due to localized pod infection rather than systemic plant infection. In a study by Kaur et al. (2023), drought stress impeded the systemic progression of the pathogen from the root to the leaf and enhanced a better defense response in tolerant cultivars by accumulating antioxidants and lignin deposition.
Continuing this line of research, Kaur et al. (2024) screened ten-day-old seedlings of six mungbean cultivars by subjecting them to combined drought stress and Macrophomina infection in three combinations: (i) drought followed by pathogen infection and normal watering, (ii) drought followed by pathogen infection and drought again, (iii) pathogen infection followed by drought stress. The results showed that cultivars exposed to drought stress prior to Macrophomina infection performed exceptionally well in terms of yield and nutritional quality. The resistance response under drought followed by fungal infection was primarily due to callose deposition, while under fungal infection followed by drought stress, it was due to increased accumulation of proline and soluble sugars, indicating different defense strategies adapted by the genotypes in response to the order of combined stresses encountered. These findings, arising from limited research, require further elucidation and suggest that the eventual impact of drought on DRR incidence in mungbean cannot be predicted and is subject to genotypic responses as well as the sequential order, duration, and severity of stresses encountered by the host species. The morphological, physiological and biochemical changes induced in mungbean on exposure to Macrophomina infection and combined stress are presented in Table 1.
Host plant resistance to dry root rot in mungbean germplasm
Resistance or tolerance to the combined stresses of DRR and drought is crucial for improving mungbean production under climate change scenarios. Managing DRR is exceptionally challenging due to the soil-borne nature of the pathogen, its asexual reproduction, the prolonged longevity of microsclerotia, and its complex relationship with drought conditions. Additionally, the virulence of M. phaseolina isolates appears to vary significantly across locations (Bimla et al., 2016; Kumar et al., 2017a). Varieties possessing an innate ability to resist or withstand the disease under drought conditions are essential for a long-term, eco-friendly, and economically viable strategy to combat DRR.
Large-scale screening of mungbean germplasm is essential to identify resistant sources to DRR. Various phenotypic screening methods, such as the paper blot assay, sick pot technique, sick plot technique, and field evaluation, are utilized for this purpose. The paper blot assay, for instance, involves placing eight to ten-day-old seedlings with roots dipped in fungal inoculum inside paper towels, allowing disease development under controlled conditions, with scoring based on symptom severity (Nene et al., 1981; Pandey et al., 2021). Other techniques, like the sick pot and sick plot methods, involve inoculating susceptible genotypes with M. phaseolina to assess symptom development under controlled conditions (Choudhary et al., 2011; Irulappan et al., 2021).
Although germplasm screening for DRR began decades ago, vigorous screening has gained momentum only recently due to the dramatic rise in disease incidence (Table 2). Several studies have reported mungbean genotypes exhibiting resistance or tolerance to DRR using different screening techniques (Avanija et al., 2023; Choudhary et al., 2011; Khan & Muhammad, 2007; Pandey et al., 2020, 2021). However, identifying donor sources with consistently high levels of resistance has been challenging due to the skewed distribution curve towards susceptibility (Talekar et al., 2021). Therefore, continued disease phenotyping of unexplored germplasm accessions is necessary to identify consistent and durable resistant sources for use in breeding programmes.
Given the simultaneous occurrence of drought and DRR, evaluating genotypes under combined stress conditions would be beneficial. While investigations on the effects of combined stress on morphological, biochemical, and physiological traits have been conducted in some legume crops, such as chickpea and soybean, screening for genotypes with combined tolerance to both stresses remains in its early stages in mungbean (Kaur et al., 2023, 2024; Mengistu et al., 2018). Identifying genotypes with combined tolerance to both drought and Macrophomina infection under field conditions would facilitate their effective deployment in resistance breeding programmes.
Currently, no studies to determine the genetic loci in mungbean associated with DRR resistance have been conducted. However, research conducted in legumes and other crops can provide insights into extending similar approaches for DRR resistance breeding programmes in mungbean. Inheritance studies have indicated the role of dominant genes with epistatic interactions for dry root resistance in common bean (Hernández-Delgado et al., 2009; Vitaeri and Linnaes, 2022), while QTL mapping studies in several other crops have established the polygenic nature of the trait (Table 3). Based on their mapping study, Olaya et al. (1996) identified two RAPD markers, B386900 and B4591600, associated with DRR resistance. Using AFLP analysis, Hernández-Delgado et al. (2009) discovered a potential QTL on linkage group 1 of the common bean conferring resistance to charcoal rot using an F2 population derived from a cross between BATT477 (resistant to both charcoal rot and drought) and cvPintoUI-114 BATT477. Using the RIL population of the same cross, nine QTLs for charcoal rot and three QTLs for drought were mapped; however, no overlapping QTLs for resistance to both stresses were detected. Surprisingly, the QTL identified on LG1 by Hernández-Delgado et al. (2009) was not detected in this study. The markers BPC40M12 and BPC54M150 associated with DRR resistant QTLs located on chromosome 8 and 10 were prominent candidates for marker-assisted selection (Méndez-Aguilar et al., 2017).
Karadi et al. (2021) reported a single minor QTL (‘qDRR-8’) associated with M. phaseolina in chickpea. In soybean, da Silva et al. (2019) mapped three QTLs associated with dry root resistance, one on chromosome 15 and two on chromosome 16 using an F2:3 population derived from the cross PI567562A x PI567437 genotyped using a 6 K SNP array. The QTL on chromosome 15, which explained 29.4% of the phenotypic variance, overlapped with a patented marker Satt512 used for selection of genotypes tolerant to DRR-drought complex. However, QTLseq analysis based on genotyping by sequencing (GBS) of resistant and susceptible bulks from the same population detected QTLs on three other chromosomes, namely 5, 8, and 14. The differences in the sequencing platforms were speculated as a reason for the contradictory results (da Silva et al., 2020). Muchero et al. (2011) reported nine QTLs associated with resistance to M. phaseolina in cowpea; of which, only three QTLs co-located with drought tolerance QTLs, suggesting that responses to M. phaseolina infection and drought stress could possibly be mediated by different genetic mechanisms. Tomar et al. (2017) reported three QTLs, including a major QTL, associated with resistance to M. phaseolina in castor.
The limitations in QTL mapping strategies involving the development of mapping populations and construction of genetic linkage maps have been overcome by genome-wide association studies (GWAS). Coser et al. (2017) reported SNPs associated with M. phaseolina through GWAS in a collection of 459 accessions from the USDA Soybean germplasm core collection based on field and greenhouse screening. The SNPs detected for their association with M. phaseolina under field and greenhouse conditions were different, suggesting that the genetic mechanisms underlying resistance to M. phaseolina in soybean could be complex and influenced by the environment. GWAS and SNP-based haplotyping in soybean identified TAC and CGA haplotypes associated with markers Gm08_18909193_A_G, Gm08_44422211_T_C, and Gm19_34320762_A_C to confer resistance against charcoal rot in a panel of varieties cultivated by Brazilian farmers (Vinholes et al., 2019). Recently, Zatybekov et al. (2023) detected 11 QTLs associated with resistance to M. phaseolina in a panel of 252 accessions using the 6 K SNP array and whole-genome resequencing (WGRS) technology. GWAS based on WGRS data set in a biparental mapping population derived from the cross BAT 477/NY6020-4 in common bean identified a novel QTL governing resistance to M. phaseolina on chromosome 3. Two SNP markers strongly associated with this QTL were part of the drought-sensitive gene Phvul.003G175900 (Viteri et al., 2022). Nelson et al. (2021) reported three loci (FaRMp1, FaRMp2, FaRMp3) conferring resistance to M. phaseolina in strawberry through genome-wide SNP genotyping and pedigree-based analysis (Table 4).
Functional annotation of the resistance loci mapped using QTL and GWAS analysis has identified disease resistance gene analogues (Viteri et al., 2022) and candidate genes involved in plant defense and stress signaling pathways related to pectin metabolism (Muchero et al., 2011), calmodulin, cell wall degradation, ethylene response factor, protection from oxidative stress (Coser et al., 2017), late embryogenesis (LEA) protein synthesis (Zatybekov et al., 2023), flavonoid, and isoflavonoid biosynthesis (Adeyanju et al., 2015).
The draft genome sequences of mungbean, urdbean, and cowpea have been published (Kang et al., 2014; Ha et al., 2021; Pootakham et al., 2021; Lonardi et al., 2019). A high level of conservation at the genome level is observed among these three Vigna species, facilitating the discovery of genes and QTLs associated with valuable agronomic traits through association genetics. Although no QTLs have been mapped for DRR resistance in mungbean, the high level of synteny among the three Vigna species has been utilized to identify conserved resistance loci with similar functions in mungbean. An EST-derived SNP marker (1_10853) linked to QTL Mac-2 on chromosome 3 governing resistance to charcoal rot in cowpea was functionally annotated to code for pectin esterase inhibitor. Based on comparative genomic analysis, the SNP marker (1_10853) was mapped to genes encoding for pectin esterase inhibitor on chromosome 7 (Vradi07g27890) in mungbean and chromosome 1 in urd bean (Pootakham et al., 2021).
Host-M. phaseolina-omics and gene expression studies
Similar to the annotated genes and pathways underlying QTLs, gene expression and transcript profiling of resistant and susceptible genotypes to M. phaseolina in a few legumes like soybean, groundnut, alfalfa, and other crops such as sorghum and jute have also revealed the role of a wide array of genes encoding for pathogenesis-related compounds like chitinase, stilbene synthase, nucleotide-binding leucine-rich repeat regions, jasmonic acid, salicylic acid, flavonoid and isoflavonoid biosynthesis, secondary metabolites, auxin homeostasis and transport, and stress signaling pathways involved in plant defense against the pathogen (Biswas et al., 2014; Irulappan et al., 2022; Iwuala et al., 2020; Mah et al., 2012; Marquez et al., 2018; Sharma et al., 2014). Additionally, a study in sesame has demonstrated the existence of a biotrophy-necrotrophy switch in M. phaseolina (Chowdhury et al., 2017). Significantly higher transcript levels of BAS3 (biotrophy-associated protein 3) and NIP (necrotrophy-inducing protein) marked the onset of the necrotrophy phase. SiPCHY levels were the highest at the necrotrophy phase, indicating active lignin biosynthesis. Moreover, higher levels of SA (salicylic acid) were observed in the host during the biotrophic phase, while higher levels of JA (jasmonic acid) were accumulated during the necrotrophic phase, which correlated with the transcript levels as well. A similar trend was observed in the Arabidopsis thaliana root transcriptome post-M. phaseolina infection. The upregulation of JA, SA, and ET (ethylene) responsive genes and their mutants displayed greater susceptibility to the pathogen (Schroeder et al., 2019). Gaige et al. (2010) developed a novel model pathosystem to study the molecular interactions between host-M. phaseolina. Although they observed only a weak upregulation of the JA/ET pathway genes in Medicago truncatula roots inoculated with M. phaseolina, they showed that priming the plants with JA/ET prior to inoculation enhanced resistance (Table 5).
Transcriptomics of arbuscular mycorrhizal fungi (AMF) colonized roots of soybean revealed an upregulation of defense, pathogenesis-related, and secondary metabolism genes (Marquez et al., 2018). They also showed that AMF relieved the defense-growth trade-off stress in soybean. Although no clues have been gained on cross talking among a network of pathways activated under pathogen infection and/or combined stress situations, the expression of certain compounds like auxins and LEA proteins under DRR infection as well as under drought stress indicates that common pathways may be involved in plant defense against both these stresses.
Several studies have been conducted on M. phaseolina as well. The first genome sequence of M. phaseolina was published by Islam et al. (2012), and since then, genome sequences of several strains from varying hosts have been published (Shirai et al., 2023). A transcriptomic study revealed that reactive oxygen species (ROS) pathways are involved in microsclerotia formation (Liu et al., 2022). In addition to cell wall-degrading enzymes (CWDEs), several studies of gene expression of secondary metabolomic pathways have shown the role of toxins in virulence (Shirai et al., 2023). Independent M. phaseolina secretome analyses conducted by Sinha et al. (2022) and Pineda-Fretez et al. (2023) revealed 117 and 250 secreted proteins, respectively, among which several effectors, CWDEs, and peptidases were identified. These -omics studies in model pathosystems can lay a foundation for future studies on M. phaseolina-host interaction in non-model crops/plants such as mungbean.
A common defense mechanism observed in plants subjected to drought and pathogen infection is the deposition of lignin in the secondary cell walls facilitated by the cell wall catalyzed multicopper oxidase family enzymes (LACCASES) encoded by LAC genes. Short non-coding RNA molecules known as microRNAs have emerged as potential players in modulating the defense response by regulating transcriptional and post-transcriptional gene expression. In chickpea, microRNA397 was found to play a key role in regulating tolerance to DRR and drought through the root lignification process (Sharma et al., 2023). In jute, the miR-845b and miR166 superfamily regulate the Nucleotide-binding site—leucine-rich repeat (NBS-LRR) and ROS mediated defense (Dey et al., 2016). The pathways involved in all these ‘omics’ studies reveal that the host plant deploys a two-tier defense strategy against Macrophomina, with the first level primarily aimed to prevent/delay the entry of the pathogen into the host cell and the second level aimed at initiating a destructive war against the invaded pathogen.
While the role of microRNAs in regulating DRR resistance in mungbean is not yet fully understood, their role in regulating drought tolerance has been established. The microRNA Vra-miR165 contributes to drought tolerance by targeting the mungbean NAC transcription factors, which also participate in biotic stress (Tariq et al., 2022). Kumar et al. (2022), identified five potential microRNAs (Vra-miR160, Vra-miR164, Vra-miR167, Vra-miR394 and Vra-miR398) that regulate drought response in the mungbean genotype K851 by targeting auxin response factor, NAC transcription factor, serine acetyl transferase 1, and multicopper oxidase LPR 2 like genes. Two of these drought-responsive microRNAs, namely miR160 and miR398, also participate in defense against Mungbean Yellow Mosaic India Virus (MYMIV) by regulating auxin perception and NAC transcription factors (Kundu et al., 2017). Interestingly, since all these genes are also reported to be involved in biotic stress response, they may be possible targets of microRNAs regulating DRR resistance in mungbean. Elucidating the role of genetic factors regulating key signalling pathways can help understand the evolutionary dynamics during host–pathogen interaction, thereby imparting resistance in plants and virulence in pathogens.
A wide array of secretory fungal proteins produced by M. phaseolina are essential for disease pathogenesis and serve as potential virulence factors to break the host defense mechanisms. In silico prediction based on whole genome sequence analysis reveals an abundant secretion of peroxidases, oxidases, cellulolytic and hydrolytic enzymes by the pathogen to decompose plant cell walls that are barriers for its entry into the host. Among the 362 carbohydrate active enzymes (CAZymes) encoded by Macrophomina genome, about 219 belong to glycoside hydrolases (GH) which is comparatively higher than the average GH possessed by other known phytopathogenic fungi (Islam et al., 2012). Proteomic analysis revealed an arsenal of secretory proteins (ranging from 117 to 250) predominated by cell wall degrading enzymes such as glucananses, xylanase and amylases and peptidases that are involved in infection process (Sinha et al., 2022; Pinedo-Fretez et al., 2023). In addition, secretome analysis identified putative effector proteins secreted by the pathogen that help in colonising the host by manipulating and suppressing the host immune system (Pinedo-Fretez et al., 2023). Understanding the role of these secretory fungal proteins and effector molecules can help in gaining insights on the fungal proteins associated with pathogenesis and their role in host–pathogen interaction, eventually helping in devising management strategies to mitigate dry root rot.
A few omics studies such as transcriptomics and metabolomics have been conducted to study the M. phaseolina-host interactions in several crops such as soybean, sorghum and in the model pant Arabidopsis thaliana (Arafat et al., 2024; Bandara et al., 2018; Bosmaia et al., 2023; Radadiya et al., 2021; Schroeder et al., 2019; Silva et al., 2021; Singh et al., 2022a, 2022b; Yan et al., 2021). However, such omics-based approaches have not yet been utilized for gaining insights into mungbean-M. phaseolina interactions. Further, CRISPR/Cas based genome editing to confer resistance to a wide range of phytopathogens has been extensively utilized (Langner et al., 2018). However, genome editing based approaches towards engineering M. phaseolina resistance are yet to be utilized in crops, including mungbean. Moreover, certain genome edited lines can be utilized as pre-breeding material to aid conventional breeding towards resistance. Integration of various omics techniques with genome editing techniques are fundamental tools in research towards M. phaseolina resistance in mungbean.
Integrated management strategies to mitigate dry root rot
Integrated management techniques, including the use of fungicides, biocontrol agents, botanical extracts, and organic amendments, have shown promising effects in controlling and reducing DRR incidence in mungbean (Deshmukh et al., 2016; Kumari et al., 2012). Seed treatment with systemic fungicides such as carbendazim has proven effective in minimizing DRR incidence in mungbean (Kumari et al., 2015; Murthy et al., 2003).
Fungal and plant growth-promoting rhizobacteria (PGPR) such as Trichoderma viride, T. harzianum, Glomus claroideum, Bacillus subtilis, Pseudomonas fluorescens, P. putida, P. aeruginosa, and Burkholderia species exhibit antagonistic effects on M. phaseolina growth. Seed and soil treatment with these biocontrol agents, either alone or in combination, have demonstrated effectiveness in reducing mungbean disease incidence in various greenhouse and field experiments (Raguchander et al., 1993; Thilagavathi et al., 2007; Mansoor et al., 2007; Chandra et al., 2007; Satya et al., 2011; Shahid and Khan, 2016a, 2016b; Hashem et al., 2017; Choudhary & Ashraf, 2019; Ahmed & Shete, 2022; Khaire et al., 2023). These antagonists induce resistance against the pathogen by synthesizing enzymes that can lyse fungal cells, mimicking host plant defense mechanisms, and competitively colonizing host plant roots, thereby eliminating the harmful pathogen from the ecological niche. Notable increases in defense-related enzyme activities such as phenylalanine ammonia-lyase, polyphenol oxidase, superoxide dismutase, and peroxidase have been observed in mungbean genotypes treated with P. putida (Khan et al., 2023).
Noreen et al. (2019) examined the function of fluorescent Pseudomonas linked to mungbean root nodules in rhizobia-induced nodulation, highlighting the potential of beneficial microbes in disease management. Additionally, botanical extracts from mustard, neem, sesame, onion, garlic vine, Eucalyptus, Datura, Sisymbrium, Launaea and palmarosa (Cymbopogon martini) have also been used to treat mungbean for mitigating DRR incidence (Mansoor et al., 2007; Javaid and Siddique, 2011; Haseeb et al., 2013; Kalaivani et al., 2023).
Agronomic practices such as crop rotation with non-host crops, fallowing fields, and soil amendments to improve soil fertility are also recommended practices to reduce M. phaseolina inoculum levels in the soil (Choudhary et al., 2010). Exposure of mungbean seeds to ultraviolet radiation for 5–20 min significantly reduces root-infecting fungi (Siddiqui et al., 2011). Given the crucial role of temperature in M. phaseolina survival, soil solarization can stimulate the temperature conditions necessary to reduce the viable population of the pathogen (Polakala et al., 2023). While these strategies help combat DRR incidence in mungbean, their cost-effectiveness and eco-friendliness on a large scale remain major limiting factors for field recommendations. Considering these factors, exploring host plant resistance is a viable strategy for developing mungbean cultivars tolerant to DRR and combined stresses.
Conclusions and future perspectives
Host plant resistance remains the most viable eco-friendly strategy to realize the potential yield and counteract virulent pathogenic strains. However, exploiting and utilizing genetic and genomic resources to combat DRR in mungbean requires significant progress. Despite the identification of limited resistant sources to date, large-scale screening of mungbean germplasm is essential to identify potential donors with durable resistance against DRR. The development of a rapid, sensitive, and reliable screening technique for high-throughput phenotyping is crucial to handle large sets of germplasm. Incorporating artificial intelligence tools and sensor-based imaging technologies that precisely capture DRR symptoms can aid in scoring disease severity accurately.
While dry and hot conditions predispose mungbean to DRR, there is no strong association to presume that selection for drought tolerance will favor DRR resistance. Therefore, selection for both traits needs to be done individually in environments displaying the combined stresses. Although mapping strategies in a few crops have indicated the polygenic nature of DRR resistance, the genetics of this trait still needs elucidation in mungbean.
The recent emergence of DRR as a globally devastating disease and challenges in identifying suitable donor sources with high levels of resistance have delayed the development of bi-parental mapping populations for QTL studies. Identification of major genomic regions associated with DRR needs to be expedited to develop marker-assisted selection programs and fast-track introgression into cultivated varieties. Co-localization of drought and DRR QTLs, as observed in legumes like soybean, can facilitate selection for tolerance against the dual complex.
With gold-standard NGS technologies and high-density SNP genotyping becoming more affordable, mungbean core and mini-core collections representing holistic diversity can be effectively utilized as association mapping panels for GWAS to identify target genomic regions governing DRR resistance. Once identified, functional annotation of these QTLs using bioinformatic tools can provide insights into genes, signaling pathways, and their interactions involved in host defense mechanisms.
The RNAseq approach to analyze transcriptomes in resistant and susceptible mungbean varieties can identify differentially regulated genes that explain genotypic responses triggered by the Mungbean-M. phaseolina interaction. Investigating the role of microRNAs in regulating tolerance towards the drought-dry root complex can help modulate the defense response in mungbean.
With the draft genome sequence available for Vigna species, synteny analysis can pave the way for identifying conserved resistance loci for DRR and drought among mungbean, urd bean, and cowpea. A pangenomics approach can capture haplotypic diversity contributing to individual and combined stress tolerance.
Despite significant research on management aspects of DRR in mungbean, there is an urgent need to prioritize and revive the DRR resistance breeding programme. Complementing conventional breeding strategies with the ‘omics’ toolbox can lead to the development of climate-resilient mungbean cultivars, ensuring food and nutritional security.
References
Adeyanju, A., Little, C., Yu, J., & Tesso, T. (2015). Genome-wide association study on resistance to stalk rot diseases in grain sorghum. G3: Genes, Genomes, Genetics, 5(6), 1165–1175. https://doi.org/10.1534/g3.114.016394.
Ahmad, A., Selim, M. M., Alderfasi, A. A., & Afzal, M. (2015). Effect of drought stress on mungbean (Vigna radiata L.) under arid climatic conditions of Saudi Arabia. In J. L. Mirallesi Garcia & C. A. Brebbia (Eds.), Ecosystem and Sustainable Development (pp. 185–193). Southampton, UK: WIT Press. https://doi.org/10.2495/ECO150171
Ahmed, M. F., & Shete, P. P. (2022). Management of dry root rot of mungbean caused by Macrophomina phaseolina using bioagents and fungicides. The Pharma Innovation Journal, 11(9), 13–26.
Akhtar, S., Khan, K. A., & Shoaib, A. (2018). Response of Vigna radiata (L.) Wilczek genotypes to charcoal rot disease. Mycopath, 14(1&2).
Akhtar, K. P., Sarwar, G., & Arshad, H. M. I. (2011). Temperature response, pathogenicity, seed infection, and mutant evaluation against Macrophomina phaseolina causing charcoal rot disease of sesame. Archives of Phytopathology and Plant Protection, 44(4), 320–330. https://doi.org/10.1080/03235400903052945
Amairani, A. R. L., Rodríguez-Castillejos, G., Garza-Cano, E., Ramírez-Quintanilla, L., Oliva-Hernández, A., Lizarazo-Ortega, C., & Hernández-Mendoza, J. L. (2023). Production of Acetic Indole Acid in Macrophomina phaseolina. International Journal of Plant & Soil Science, 35(3), 129–136. https://doi.org/10.9734/ijpss/2023/v35i32783
Anupriya Chawla, N., Ghasolia, M.K., Prahlad, Bagri, R.K., Godika, S. and Ghasolia, R.P. (2023). Management of dry root rot of mungbean caused by Macrophomina phaseolina through organic amendments, plant extracts and bio-agents. Legume Research. https://doi.org/10.18805/LR-4909.
Arafat, Md. Y., Narula, K., Kumar, M., et al. (2024). Proteo-metabolomic dissection of extracellular matrix reveals alterations in cell wall integrity and calcium signaling governs wall-associated susceptibility during stem rot disease in jute. Journal of Proteome Research. https://doi.org/10.1021/acs.jproteome.3c00781
Arora, P., Dilbaghi, N., & Chaudhury, A. (2012). Opportunistic invasive fungal pathogen Macrophomina phaseolina prognosis from immunocompromised humans to potential mitogenic RBL with an exceptional and novel antitumor and cytotoxic effect. European Journal of Clinical Microbiology & Infectious Diseases, 31, 101–107. https://doi.org/10.1007/s10096-011-1275-1
Ashwini, C., & Giri, G. K. (2014). Control of seed borne fungi in green gram and black gram through bioagents. International Journal of Applied Biology and Pharmaceutical Technology, 5(3), 168–170.
Atiq, M., Asad, S., Rafique, M., Khan, N. A., Abd-ur-Rehman, Y., & M., Shafiq, M., Ahmad, K., Bashir, N., & Khan, W.A. (2014). Identification of source of resistance in mung bean germplasm against charcoal rot disease. Pakistan Journal of Phytopathology, 26(01), 133–136.
Avanija, M., Padmaja, G., Vidyasagar, B., Hari, Y., Devi, G. U., & Hemantha, J. (2023). Screening of genotypes against dry root rot of mungbean (Vigna radiata L.) R. Wilczek. Pharma Innovation Journal, 12(8), 805–809.
Ayyanagouda, P., Bashasab, F., & Salimath, P. M. (2012). Genome-wide molecular mapping and QTL analysis, validated across locations and years for charcoal rot disease incidence traits in Sorghum bicolor (L.) Moench. Indian Journal of Genetics and Plant Breeding, 72(3), 296–302.
Babu, B. K., Reddy, S. S., Yadav, M. K., Sukumar, M., Mishra, V., Saxena, A. K., & Arora, D. K. (2010). Genetic diversity of Macrophomina phaseolina isolates from certain agro-climatic regions of India by using RAPD markers. Indian Journal of Microbiology, 50, 199–204. https://doi.org/10.1007/s12088-010-0033-x
Bandara, Y., Weerasooriya, D. K., Liu, S., & Little, C. R. (2018). The necrotrophic fungus Macrophomina phaseolina promotes charcoal rot susceptibility in grain sorghum through induced host cell-wall-degrading enzymes. Phytopathology, 108, 948–956.
Basandrai, A. K., Pandey, A. K., Somta, P., & Basandrai, D. (2021). Macrophomina phaseolina–host interface: Insights into an emerging dry root rot pathogen of mungbean and urdbean, and its mitigation strategies. Plant Pathology, 70(6), 1263–1275. https://doi.org/10.1111/ppa.13378
Bashir, M., & Malik, B. A. (1988). Diseases of major pulse crops in Pakistan—a review. International Journal of Pest Management, 34(3), 309–314.
Batzer, J. C., Singh, A., Platner, E., Kandel, Y., & Mueller, D. S. (2022). Screening mungbean accessions for susceptibility to soybean fungal diseases in Iowa. Plant Health Progress, 23(4), 456–465. https://doi.org/10.1094/PHP-03-22-0026-RS
Bimla, B., Gaur, V. K., & Choudhary, K. (2016). Study of the pathogenic variability among Macrophomina phaseolina isolates in sterilized and unsterilized soil of different crops. Annals of Agri-Bio Research, 21(2), 149–152.
Biswas, C., Dey, P., Karmakar, P. G., & Satpathy, S. (2014). Next-generation sequencing and micro RNAs analysis reveal SA/MeJA1/ABA pathway genes mediated systemic acquired resistance (SAR) and its master regulation via production of phased, trans-acting siRNAs against stem rot pathogen Macrophomina phaseolina in a RIL population of jute (Corchorus capsularis). Physiological and Molecular Plant Pathology, 87, 76–85. https://doi.org/10.1016/j.pmpp.2014.07.003
Bosmaia, T. C., Agarwal, P., Dangariya, M. et al (2023) Transcriptomic analysis towards identification of defence-responsive genes and pathways upon application of Sargassum seaweed extract on tomato plants infected with Macrophomina phaseolina. Biotech 13, 179.
Burkhardt, A., Ramon, M. L., Smith, B., Koike, S. T., & Martin, F. (2018). Development of molecular methods to detect Macrophomina phaseolina from strawberry plants and soil. Phytopathology, 108(12), 1386–1394. https://doi.org/10.1094/PHYTO-03-18-0071-R
Buts, A. K., & Ranvir, S. (2014). Study of seed-borne mycoflora of mung bean (Phaseolus aureus Roxb.) treated with potassium nitrate during storage. Advances in Applied Science Research, 5(6), 11–13.
Chamorro, M., Miranda, L., Domínguez, P., Medina, J. J., Soria, C., Romero, F., López-Aranda, J. M., & De los-Santos, B. (2015). Evaluation of biosolarization for the control of charcoal rot disease (Macrophomina phaseolina) in strawberry. Crop Protection, 67, 279–286. https://doi.org/10.1016/j.cropro.2014.10.021
Chandra, S., Khare, V., & Kehri, H. K. (2007). Evaluation of arbuscular mycorrhizal fungi against Macrophomina phaseolina causing dry root-rot of urd and mung bean. Indian Phytopathology, 60(1), 42–47.
Chandraprakash, S., Rubini, R., Jebalin, V. S., Rohini, M., Parwin, S. S., & Monicaa, R. (2022). In vitro evaluation of different plant extracts and bio-agents against dry root rot of greengram caused by Macrophomina phaseolina. The Pharma Innovation Journal, 11(8), 11–15.
Chilakala, A. R., Pandey, P., Durgadevi, A., Kandpal, M., Patil, B. S., Rangappa, K., Reddy, P. C. O., Ramegowda, V., & Senthil-Kumar, M. (2023). Drought attenuates plant responses to multiple rhizospheric pathogens: A study on a dry root rot-associated disease complex in chickpea fields. Field Crops Research, 298, 108965. https://doi.org/10.1016/j.fcr.2023.108965
Choudhary, A., & Ashraf, S. (2019). Utilizing the combined antifungal potential of Trichoderma spp. and organic amendments against dry root rot of mungbean. Egyptian Journal of Biological Pest Control. https://doi.org/10.1186/s41938-019-0187-8.
Choudhary, A. K., Sharma, O. P., & Choudhary, S. (2011). Screening of mungbean (Vigna radiata) genotypes to identify source of resistance to dry root rot. Journal of Food Legumes, 24(2), 117–119.
Choudhary, S., Pareek, S., & Saxena, J. (2010). Management of dry root rot of greengram (Vigna radiata) caused by Macrophomina phaseolina. The Indian Journal of Agricultural Sciences, 80(11), 988–992.
Chowdhury, S., Basu, A., & Kundu, S. (2017). Biotrophy-necrotrophy switch in pathogen evoke differential response in resistant and susceptible sesame involving multiple signaling pathways at different phases. Scientific Reports, 7, 17251. https://doi.org/10.1038/s41598-017-17248-7
Coser, S. M., Chowda Reddy, R. V., Zhang, J., Mueller, D. S., Mengistu, A., Wise, K. A., Allen, T. W., Singh, A., & Singh, A. K. (2017). Genetic architecture of charcoal rot (Macrophomina phaseolina) resistance in soybean revealed using a diverse panel. Frontiers in Plant Science, 8, 1626. https://doi.org/10.3389/fpls.2017.01626
da Silva, M. P., Klepadlo, M., Gbur, E. E., Pereira, A., Mason, R. E., Rupe, J. C., Bluhm, B. H., Wood, L., Mozzoni, L., & Chen, P. (2019). QTL mapping of charcoal rot resistance in PI 567562A soybean accession. Crop Science, 59(2), 474–479. https://doi.org/10.2135/cropsci2018.02.0145
da Silva, M. P., Zaccaron, A. Z., Bluhm, B. H., Rupe, J. C., Wood, L., Mozzoni, L., Mason, R. E., Yingling, S., & Pereira, A. (2020). Bulked segregant analysis using next-generation sequencing for identification of genetic loci for charcoal rot resistance in soybean. Physiological and Molecular Plant Pathology, 109, 101440. https://doi.org/10.1016/j.pmpp.2019.101440
Deshmukh, M. A., Gade, R. M., Belkar, Y. K., & Koche, M. D. (2016). Efficacy of bioagents, biofertilizers and soil amendments to manage root rot in greengram. Legume Research-An International Journal, 39(1), 140–144. https://doi.org/10.18805/lr.v0iOF.6772.
Dey, P., Biswas, C., & Karmakar, P. G. (2016). Identification and characterization of differentially expressed novel miRNAs (21–24 nt) in a Macrophomina phaseolina resistant RIL line of jute (Corchorus capsularis L.). Physiological and Molecular Plant Pathology, 94, 62–66. https://doi.org/10.1016/j.pmpp.2016.04.005
Fuhlbohm, M. J., Ryley, M. J., & Aitken, E. A. B. (2013). Infection of mungbean seed by Macrophomina phaseolina is more likely to result from localized pod infection than from systemic plant infection. Plant Pathology, 62(6), 1271–1284. https://doi.org/10.1111/ppa.12047
Gaige, A. R., Ayella, A., & Shuai, B. (2010). Methyl jasmonate and ethylene induce partial resistance in Medicago truncatula against the charcoal rot pathogen Macrophomina phaseolina. Physiological and Molecular Plant Pathology, 74(5), 412–418. https://doi.org/10.1016/j.pmpp.2010.07.001
Ghaffar, A., & Erwin, D. C. (1969). Effect of soil water stress on root rot of cotton caused by Macrophomina phaseoli. Phytopathology, 59, 795–797.
Ghosh, T., Biswas, M. K., Guin, C., & Roy, P. (2018). A review on characterization, therapeutic approaches and pathogenesis of Macrophomina phaseolina. Plant Cell Biotechnology and Molecular BiolOgy, 19(3&4), 72–84.
Goudarzi, A., Banihashemi, Z., & Maftoun, M. (2011). Effect of salt and water stress on root infection by Macrophomina phaseolina and ion composition in shoot in sorghum. Iranian Journal of Pathology, 47(3), 236–237.
Gupta, R. N., & Niwas, R. (2023). Molecular tools for detection and management of Macrophomina phaseolina. In Macrophomina phaseolina (pp. 155–163). Academic Press.
Ha, J., Satyawan, D., Jeong, H., Lee, E., Cho, K. H., Kim, M. Y., & Lee, S. H. (2021). A near-complete genome sequence of mungbean (Vigna radiata L.) provides key insights into the modern breeding program. The Plant Genome, 14(3), e20121. https://doi.org/10.1002/tpg2.20121
Haseeb, H. A., Sahi, S. T., Ali, S., & Fiaz, M. (2013). Response of different mung bean varieties against Macrophomina phaseolina (Tassi) Goid and in-vitro studies of plant extracts against pathogen. Pakistan Journal of Phytopathology, 25(1), 78–83.
Hashem, A., Abd-Allah, E. F., Alqarawi, A. A., Radhakrishnan, R., & Kumar, A. (2017). Plant defense approach of Bacillus subtilis (BERA 71) against Macrophomina phaseolina (Tassi) Goid in mung bean. Journal of Plant Interactions, 12(1), 390–401. https://doi.org/10.1080/17429145.2017.1373871
Hernandez-Delgado, S., Reyes-Valdés, M. H., Rosales-Serna, R., Mayek-Perez, N., & Narro A. (2009). Molecular markers associated with resistance to Macrophomina phaseolina (Tassi) Goid. in common bean. Journal of Plant Pathology, 163–170. https://doi.org/10.4454/jpp.v91i1.637.
Iqbal, U., & Mukhtar, T. (2014). Morphological and pathogenic variability among Macrophomina phaseolina isolates associated with mungbean (Vigna radiata L.) Wilczek from Pakistan. The Scientific World Journal. https://doi.org/10.1155/2014/950175
Irulappan, V., Kandpal, M., Saini, K., Rai, A., Ranjan, A., Sinharoy, S., & Senthil-Kumar, M. (2022). Drought stress exacerbates fungal colonization and endodermal invasion and dampens defense responses to increase dry root rot in chickpea. Molecular Plant Microbe Interactions, 35(7), 583–591. https://doi.org/10.1094/MPMI-07-21-0195-FI
Irulappan, V., Mali, K. V., Patil, B. S., Manjunatha, H., Muhammad, S., & Senthil-Kumar, M. (2021). A sick plot–based protocol for dry root rot disease assessment in field-grown chickpea plants. Applied Plant Sciences, 9, e11445. https://doi.org/10.1002/aps3.11445
Islam, M. S., Haque, M. S., Islam, M. M., Emdad, E. M., Halim, A., Hossen, Q. M. M., Hossain, M. Z., Ahmed, B., Rahim, S., & Rahman, M. S. (2012). Tools to kill: Genome of one of the most destructive plant pathogenic fungi Macrophomina phaseolina. BMC Genomics, 13, 493. https://doi.org/10.1186/1471-2164-13-493
Iwuala, E., Odjegba, V., Unung, O., & Alam, A. (2020). Expression of stress responsive β-1, 3-glucanase and chitinase genes in Arachis hypogaea seedlings against Macrophomina phaseolina. Gene Reports, 20, 100693. https://doi.org/10.1016/j.genrep.2020.100693
Javaid, A., & Saddique, A. (2011). Management of Macrophomina root rot of mungbean using dry leaves manure of Datura metel as soil amendment. Spanish Journal of Agricultural Research, 9(3), 901–905. https://doi.org/10.5424/sjar/20110903-394-10
Kalaivani, K., Senthil-Nathan, S., Stanley-Raja, V., & Vasantha-Srinivasan, P. (2023). Physiological and biochemical alterations in Vigna radiata L. triggered by sesame derived elicitors as defense mechanism against Rhizoctonia and Macrophomina infestation. Scientific Reports, 13(1), 13884, https://doi.org/10.1038/s41598-023-39660-y.
Kang, Y. J., Kim, S. K., Kim, M. Y., Lestari, P., Kim, K. H., Ha, B.-K., Jun, T. H., Hwang, W. J., Lee, T., Lee, J., Shim, S., Yoon, M. Y., Jang, Y. E., Han, K. S., Taeprayoon, P., Yoon, N., Somta, P., Tanya, P., Kim, K. S., … Lee, S.-H. (2014). Genome sequence of mungbean and insights into evolution within Vigna species. Nature Communications, 5(1), 5443. https://doi.org/10.1038/ncomms6443
Karadi, A., Samineni, S., Sajja, S., Sharma, M., Thudi, M., Mallikarjuna, B. P., Viswanatha, K. P., Varshney, R. K., & Gaur, P. M. (2021). Molecular mapping of dry root rot resistance genes in chickpea (Cicer arietinum L.). Euphytica, 217(6), 123. https://doi.org/10.1007/s10681-021-02854-4
Kaur, S., Dhillon, G. S., Brar, S. K., Vallad, G. E., Chand, R., & Chauhan, V. B. (2012). Emerging phytopathogen Macrophomina phaseolina: Biology, economic importance and current diagnostic trends. Critical Reviews in Microbiology, 38(2), 136–151. https://doi.org/10.3109/1040841x.2011.6409771
Kaur, S., Kumari, N., Parmar, P., & Sharma, V. (2023). Simultaneous exposure of contrasting Vigna radiata cultivars to Macrophomina phaseolina infection and drought stress: Morpho-physiological and biochemical implications. Plant Biosystems-an International Journal Dealing with All Aspects of Plant Biology, 157(5), 1100–1113. https://doi.org/10.1080/11263504.2023.2243942
Kaur, S., Kumari, N., & Sharma, V. (2024). Interplay of stress responses in mungbean cultivars subjected to combined exposure to Macrophomina phaseolina infection and drought stress. Plant Stress. https://doi.org/10.1016/j.stress.2024.100376
Kaushik, C. D., & Chand, J. N. (1987). Seedborne nature of Rhizoctonia bataticola causing leaf blight of mung bean. Indian Journal of Mycology and Plant Pathology, 17(2), 154–157.
Khaire, P. B., Hingole, D. G., Holkar, S. K., Ghante, P. H., & Mane, S. S. (2023). Management of Macrophomina phaseolina (Tassi.) Goid. causing leaf blight disease in mung bean in Maharashtra State. Environment and Ecology, 41(2B), 1141–1148. https://doi.org/10.60151/envec/HTUE5755.
Khan, S. H., & Muhammad, S. (2007). Identification of sources of resistance in mung bean (Vigna radiata L.) against charcoal rot Macrophomina phaseolina (Tassi) Goid. In 8th African Crop Science Society Conference, El-Minia, Egypt, 27–31 October 2007 (pp. 2101–2102). African Crop Science Society.
Khan, A., Bano, A., Khan, R. A., & Khan, N. (2023). Role of PGPR in suppressing the growth of Macrophomina phaseolina by regulating antioxidant enzymes and secondary metabolites in Vigna radiata (L.) R. Wilczek. South African Journal of Botany, 158, 443–451. https://doi.org/10.1016/j.sajb.2023.05.040
Khan, M. R., Haque, Z., Rasool, F., Salati, K., Khan, U., Mohiddin, F. A., & Zuhaib, M. (2019). Management of root-rot disease complex of mungbean caused by Macrophomina phaseolina and Rhizoctonia solani through soil application of Trichoderma spp. Crop Protection, 119, 24–29. https://doi.org/10.1016/j.cropro.2019.01.014
Khan, K. A., Shoaib, A., & Akhtar, S. (2016). Response of Vigna radiata (L.) Wilczek genotypes to charcoal rot disease. Mycopath, 14(1&2)
Kumar, P., Gaur, V. K., & Meena, A. K. (2017a). Screening of different Macrophomina phaseolina isolates on susceptible (RMG-62) variety of mungbean. International Journal of Pure & Applied Bioscience, 5(6), 698–702. https://doi.org/10.13140/RG.2.2.27108.91521.
Kumar, M., Kumhar, D. R., Kumar, P., & Choudhary, K. (2019). Studies on bio-chemical changes in dry root rot (Macrophomina phaseolina) infected plants of mungbean (Vigna radiata L.). International Journal of Current Microbiology and Applied Sciences, 8(1), 2401–2407. https://doi.org/10.20546/ijcmas.2019.801.253
Kumar, B. H., Bhat, S., Borphukan, B., & Fakrudin, B. (2017b). Association analysis of charcoal rot disease component traits in sorghum minicore germplasm with EST-SSR markers. Indian Journal of Genetics and Plant Breeding, 77(01), 74–82.
Kumar, S., Das, M., Sadhukhan, A., & Sahoo, L. (2022). Identification of differentially expressed mungbean miRNAs and their targets in response to drought stress by small RNA deep sequencing. Current Plant Biology, 30, 100246. https://doi.org/10.1016/j.cpb.2022.100246
Kumari, R., Shekhawat, K. S., Gupta, R., & Khokhar, M. K. (2012). Integrated management against root-rot of mungbean [Vigna radiata (L.) Wilczek] incited by Macrophomina phaseolina. Journal of Plant Pathology and Microbiology, 3(5), 136. https://doi.org/10.4172/2157-7471.1000136
Kumari, S., Yadav, B. L., Verma, H. P., Meena, J. S., & Pancholi, P. (2015). Effect of sodic water, biofertilizer and phosphorus on physical properties of soil, yield attributes and yield of mungbean. Annals of Agricultural Research, 36(4), 394–399.
Kundu, A., Paul, S., Dey, A., & Pal, A. (2017). High throughput sequencing reveals modulation of microRNAs in Vigna mungo upon Mungbean Yellow Mosaic India Virus inoculation highlighting stress regulation. Plant Science, 257, 96–105. https://doi.org/10.1016/j.plantsci.2017.01.016
Kunkel, B. N., & Johnson, J. M. (2021). Auxin plays multiple roles during plant–pathogen interactions. Cold Spring Harbor Perspectives in Biology, 13(9), a040022. https://doi.org/10.1101/cshperspect.a040022
Langner, T., Kamoun, S., & Belhaj, K. (2018). CRISPR crops: Plant genome editing toward disease resistance. Annual Review of Phytopathology, 56, 479–512. https://doi.org/10.1146/annurev-phyto-080417-050158
Liu, H. H., Huang, C. C., Lin, Y. H., Tseng, M. N., & Chang, H. X. (2022). Superoxide initiates the hyphal differentiation to microsclerotia formation of Macrophomina phaseolina. Microbiology Spectrum, 10(1), e02084-e2121. https://doi.org/10.1128/spectrum.02084-21
Lonardi, S., Muñoz-Amatriaín, M., Liang, Q., Shu, S., Wanamaker, S. I., Lo, S., Tanskanen, J., Schulman, A. H., Zhu, T., Luo, M. C., Alhakami, H., Ounit, R., Hasan, A. M., Verdier, J., Roberts, P. A., Santos, J. R. P., Ndeve, A., Doležel, J., Vrána, J., … Close, T. J. (2019). The genome of cowpea (Vigna unguiculata [L.] Walp.). The Plant Journal, 98(5), 767–782. https://doi.org/10.1111/tpj.14349
Lu, C., Song, B., Zhang, H., Wang, Y., & Zheng, X. (2015). Rapid diagnosis of soybean seedling blight caused by Rhizoctonia solani and soybean charcoal rot caused by Macrophomina phaseolina using LAMP assays. Phytopathology, 105(12), 1612–1617. https://doi.org/10.1094/PHYTO-01-15-0023-R
Mah, K. M., Uppalapati, S. R., Tang, Y., Allen, S., & Shuai, B. (2012). Gene expression profiling of Macrophomina phaseolina infected Medicago truncatula roots reveals a role for auxin in plant tolerance against the charcoal rot pathogen. Physiological and Molecular Plant Pathology, 79, 21–30. https://doi.org/10.1016/j.pmpp.2012.03.004
Mahdizadeh, V., Safaie, N., & Aghajani, M. A. (2011). New hosts of Macrophomina phaseolina in Iran. Journal of Plant Pathology, 93(S4), 70.
Mahmoud, A. F., Abou-Elwafa, S. F., & Shehzad, T. (2018). Identification of charcoal rot resistance QTLs in sorghum using association and in silico analyses. Journal of Applied Genetics, 59, 243–251. https://doi.org/10.1007/s13353-018-0446-5
Mallaiah, B., & Rao, V. K. (2016). Integrated management of dry root rot of greengram [Vigna radiata (L.) Wilczek] incited by Macrophomina phaseolina (Tassi.) Goid. International Journal of Tropical Agriculture, 34(3), 607–614.
Mansoor, F., Sultana, V., & Ehteshamul-Haque, S. (2007). Enhancement of biocontrol potential of Pseudomonas aeruginosa and Paecilomyces lilacinus against root rot of mungbean by a medicinal plant Launaea nudicaulis L. Pakistan Journal of Botany, 39(6), 2113–2119.
Marquez, N., Giachero, M. L., Declerck, S., & Ducasse, D. A. (2021). Macrophomina phaseolina: General characteristics of pathogenicity and methods of control. Frontiers in Plant Science, 12, 634397. https://doi.org/10.3389/fpls.2021.634397
Marquez, N., Giachero, M. L., Gallou, A., Debat, H. J., Cranenbrouck, S., Di Rienzo, J. A., Pozo, M. J., Ducasse, D. A., & Declerck, S. (2018). Transcriptional changes in mycorrhizal and nonmycorrhizal soybean plants upon infection with the fungal pathogen Macrophomina phaseolina. Molecular Plant-Microbe Interactions, 31(8), 842–855. https://doi.org/10.1094/MPMI-11-17-0282-R
Mayek-PÉrez, N., GarcÍa-Espinosa, R., López-CastaÑeda, C., Acosta-Gallegos, J. A., & Simpson, J. (2002). Water relations, histopathology and growth of common bean (Phaseolus vulgaris L.) during pathogenesis of Macrophomina phaseolina under drought stress. Physiological and Molecular Plant Pathology, 60(4), 185–195. https://doi.org/10.1006/pmpp.2001.0388
Méndez-Aguilar, R., Reyes-Valdés, M. H., Hernández-Delgado, S., López-Salinas, E., Cumpián-Gutiérrez, J., Cantú-Almaguer, M. A., & Mayek-Pérez, N. (2017). Identification and mapping of QTLs associated with resistance to Macrophomina phaseolina and drought stress in common beans. Annual Report of the Bean Improvement Cooperative, 60. Published by USDA.
Mengistu, A., Ray, J. D., Smith, J. R., Arelli, P. R., Bellaloui, N., Chen, P., Shannon, G., & Boykin, D. (2018). Effect of charcoal rot on selected putative drought tolerant soybean genotypes and yield. Crop Protection, 105, 90–101. https://doi.org/10.1016/j.cropro.2017.11.012
Mishra, G. P., Aski, M. S., Bosamia, T., Chaurasia, S., Mishra, D. C., Bhati, J., Kumar, A., Javeria, S., Tripathi, K., Kohli, M., Kumar, R. R., Singh, A. K., Devi, J., Kumar, S., & Dikshit, H. K. (2021). Insights into the host-pathogen interaction pathways through RNA-Seq analysis of Lens culinaris Medik. in response to Rhizoctonia bataticola infection. Genes, 13(1), 90.
Mohanapriya, R., Naveenkumar, R., & Balabaskar, P. (2017). Survey, virulence and pathogenicity of root rot incidence of cowpea in selected districts of Tamilnadu caused by Macrophomina phaseolina (Tassi) Goid. Internacional Journal of Current Microbiology and Applied Science, 6, 694–705. https://doi.org/10.20546/ijcmas.2017.603.080.
Muchero, W., Ehlers, J. D., Close, T. J., & Roberts, P. A. (2011). Genic SNP markers and legume synteny reveal candidate genes underlying QTL for Macrophomina phaseolina resistance and maturity in cowpea [Vigna unguiculata (L) Walp.]. BMC Genomics. https://doi.org/10.1186/1471-2164-12-8
Murthy, K., Niranjana, S. R., & Shetty, H. S. (2003). Effects of chemical fungicides and biological agent on seed quality improvement in pulses. Seed Research, 31(1), 121–124.
Nair, R., & Schreinemachers, P. (2020). Global status and economic importance of mungbean. In R. M. Nair, R. Schafleitner & Lee, S-H (Eds.) (pp. 1–8) The Mungbean Genome. Springer. https://doi.org/10.1007/978-3-030-20008-4_1.
Nelson, J. R., Verma, S., Bassil, N. V., Finn, C. E., Hancock, J. F., Cole, G. S., Knapp, S.J., & Whitaker, V. M. (2021). Discovery of three loci increasing resistance to charcoal rot caused by Macrophomina phaseolina in octoploid strawberry. G3: Genes, Genomes, Genetics, 11(3), jkab037. https://doi.org/10.1093/g3journal/jkab037.
Nene, Y. L., Haware, M. P., & Reddy, M. V. (1981). Chickpea diseases: resistance-screening techniques. Information Bulletin No. 10 (pp. 10), International Crops Research Institute for the Semi-Arid Tropics, Patancheru, India.
Noreen, R., Ali, S. A., Hasan, K. A., Habiba, F. U., Tariq, A., Ara, J., & Ehteshamul-Haque, S. (2019). Role of fluorescent Pseudomonas associated with root nodules of mungbean in the induction of nodulation by the rhizobia in mungbean. Pakistan Journal of Botany, 51(3), 1161–1168.
Notomi, T., Mori, Y., Tomita, N., & Kanda, H. (2015). Loop-mediated isothermal amplification (LAMP): Principle, features, and future prospects. Journal of Microbiology, 53(1), 1–5. https://doi.org/10.1007/s12275-015-4656-9
Olaya, G., & Abawi, G. S. (1996). Effect of water potential on mycelial growth and on production and germination of sclerotia of Macrophomina phaseolina. Plant Disease, 80(12), 1347–1350. https://doi.org/10.14601/Phytopathol_Mediterr-2613.
Ortiz, V., Chang, H. X., Sang, H., Jacobs, J., Malvick, D. K., Baird, R., Mathew, F. M., de Jensen, C. E., Wise, K. A., Mosquera, G. M., & Chilvers, M. I. (2023). Population genomic analysis reveals geographic structure and climatic diversification for Macrophomina phaseolina isolated from soybean and dry bean across the United States, Puerto Rico, and Colombia. Frontiers in Genetics, 14, 1103969. https://doi.org/10.3389/fgene.2023.1103969
Pandey, A. K., Burlakoti, R. R., Rathore, A., & Nair, R. M. (2020). Morphological and molecular characterization of Macrophomina phaseolina isolated from three legume crops and evaluation of mungbean genotypes for resistance to dry root rot. Crop Protection, 127, 104962. https://doi.org/10.1016/j.cropro.2019.104962
Pandey, A. K., Yee, M., Win, M. M., Lwin, H. M. M., Adapala, G., Rathore, A., Sheu, Z.-M., & Nair, R. M. (2021). Identification of new sources of resistance to dry root rot caused by Macrophomina phaseolina isolates from India and Myanmar in a mungbean mini-core collection. Crop Protection, 143, 105569. https://doi.org/10.1016/j.cropro.2021.105569
Pineda-Fretez, A., Orrego, A., Iehisa, J. C. M., Flores-Giubi, M. E., Barúa, J. E., Sánchez-Lucas, R., Jorrín-Novo, J., & Romero-Rodríguez, M. C. (2023). Secretome analysis of the phytopathogen Macrophomina phaseolina cultivated in liquid medium supplemented with and without soybean leaf infusion. Fungal Biology, 127(5), 1043–1052. https://doi.org/10.1016/j.funbio.2023.04.001
Polakala, S. R., Rayalu, K. S. R., Behara, M., Kala, A. S., Prasad, N. H., & Chandra, K. J. (2023). Management of charcoal rot by soil solarization and biosolarization. In Macrophomina phaseolina (pp. 301–312). Academic Press.
Pootakham, W., Nawae, W., Naktang, C., Sonthirod, C., Yoocha, T., Kongkachana, W., Sangsrakru, D., Jomchai, N., U-Thoomporn, S., Somta, P., Laosatit, K., & Tangphatsornruang, S. (2021). A chromosome-scale assembly of the black gram (Vigna mungo) genome. Molecular Ecology Resources, 21(1), 238–250. https://doi.org/10.1111/1755-0998.13243
Prameela Devi, T., & Singh, R. H. (1998). Studies on virulence of Macrophomina phaseolina isolates from blackgram and greengram. Journal of Mycology and Plant Pathology, 22(2), 196–198.
Radadiya, N., Mangukia, N., Antala, V., et al. (2021). Transcriptome analysis of sesame-Macrophomina phaseolina interactions revealing the distinct genetic components for early defense responses. Physiology and Molecular Biology of Plants, 27, 1675–1693.
Rai, A., Irulappan, V., & Senthil-Kumar, M. (2022). Dry root rot of chickpea: A disease favored by drought. Plant Disease, 106(2), 346–356. https://doi.org/10.1094/PDIS-07-21-1410-FE
Rashid, Z., Kaur, H., Babu, V., Singh, P. K., Harlapur, S. I., & Nair, S. K. (2021). Identification and validation of genomic regions associated with charcoal rot resistance in tropical maize by genome-wide association and linkage mapping. Frontiers in Plant Science, 12, 726767. https://doi.org/10.3389/fpls.2021.726767
Reddy, P. S., Fakrudin, B., Rajkumar, Punnuri, S. M., Arun, S. S., Kuruvinashetti, M. S., & Seetharama, N. (2008). Molecular mapping of genomic regions harboring QTLs for stalk rot resistance in sorghum. Euphytica, 159, 191–198. https://doi.org/10.1007/s10681-007-9472-9.
Rettinassababady, C., & Ramadoss, N. (1999). Occurrence of root rot in rice fallow blackgram (Macrophomina phaseolina). Legume Research, 22(2), 139–140.
Rocha, D. C., Oliveira, M. B., de Freitas, M. A., & Petrofeza, S. (2017). Rapid detection of Macrophomina phaseolina in common bean seeds using a visual loop-mediated isothermal amplification assay. Australasian Plant Pathology, 46, 205–212. https://doi.org/10.1007/s13313-017-0477-0
Sanchez, S., Grez, J., Contreras, E., Gil, P. M., & Gambardella, M. (2019). Physiological response and susceptibility of strawberry cultivars to the charcoal rot caused by Macrophomina phaseolina under drought stress conditions. Journal of Berry Research, 9(2), 165–177. https://doi.org/10.3233/JBR-180329
Sangeeta, M., Gangopadhyay, S., Jagdish, P., & Panwar, P. (2018). Screening of mung bean varieties for resistant against Macrophomina phaseolina causing dry root rot. International Journal of Tropical Agriculture, 36(1), 147–149.
Satya, V., Vijayasamundeeswari, A., Paranidharan, V., & Velazhahan, R. (2011). Burkholderia sp. strain TNAU-1 for biological control of root rot in mung bean (Vigna radiata L.) caused by Macrophomina phaseolina. Journal of Plant Protection Research, 51(3), 273–278. https://doi.org/10.2478/v10045-0011-0045-5
Schroeder, M. M., Lai, Y., Shirai, M., Alsalek, N., Tsuchiya, T., Roberts, P., & Eulgem, T. (2019). A novel Arabidopsis pathosystem reveals cooperation of multiple hormonal response-pathways in host resistance against the global crop destroyer Macrophomina phaseolina. Scientific Reports, 9(1), 20083. https://doi.org/10.1038/s41598-019-56401-2
Seethapathy, P., Kurusamy, R., & Kuppusamy, P. (2017). Soil borne diseases of major pulses and their biological management. Innovative Farming, 2(1), 01–11.
Shahid, S., & Khan, M. R. (2016a). Biological control of root-rot on mungbean plants incited by Macrophomina phaseolina through microbial antagonists. Plant Pathology Journal, 15(2), 27. https://doi.org/10.3923/ppj.2016.27.39
Shahid, S., & Khan, M. R. (2016b). Management of root-rot of mungbean caused by Macrophomina phaseolina through seed treatment with fungicides. Indian Phytopathology, 69(2), 128–136.
Sharma, I., Kumari, N., & Sharma, V. (2014). Defense gene expression in Sorghum bicolor against Macrophomina phaseolina in leaves and roots of susceptible and resistant cultivars. Journal of Plant Interactions, 9(1), 315–323. https://doi.org/10.1080/17429145.2013.832425
Sharma, K., & Singh, T. (2000). Seed and seedling infection of Rhizoctonia bataticola in Vigna radiata. Journal of Mycology and Plant Pathology, 30(1), 15–18.
Sharma, N. K., Yadav, S., Gupta, S. K., Irulappan, V., Francis, A., Senthil-Kumar, M., & Chattopadhyay, D. (2023). MicroRNA397 regulates tolerance to drought and fungal infection by regulating lignin deposition in chickpea root. Plant, Cell & Environment, 46(11), 3501–3517. https://doi.org/10.1111/pce.14666
Shirai, M., & Eulgem, T. (2023). Molecular interactions between the soilborne pathogenic fungus Macrophomina phaseolina and its host plants. Frontiers in Plant Science. https://doi.org/10.3389/fpls.2023.1264569
Siddiqui, A., Dawar, S., Zaki, M. J., & Hamid, N. (2011). Role of ultra violet (UV-C) radiation in the control of root infecting fungi on groundnut and mung bean. Pakistan Journal of Botany, 43(4), 2221–2224.
Silva, E., Belinato, J. R., Porto, C., et al. (2021). Soybean metabolomics based in mass spectrometry: Decoding the plant’s signaling and defense responses under biotic stress. Journal of Agriculture and Food Chemistry, 69, 7257–7267. https://doi.org/10.1021/acs.jafc.0c07758
Singh, J., Singh, R. K., & Pandey, S. K. (2022). Diseases of mungbean and their management. In Naimuddin, R. K. Mishra, & M. Akram (Eds.) Diseases of Pulse Crops and their Management, Astral Publishers.
Singh, G., Kumar, A., Verma, M. K., et al. (2022a). Secondary metabolites produced by Macrophomina phaseolina, a fungal root endophyte of Brugmansia aurea, using classical and epigenetic manipulation approach. Folia Microbiologica, 67, 793–799. https://doi.org/10.1007/s12223-022-00976-3
Sinha, N., Patra, S. K., & Ghosh, S. (2022). Secretome analysis of Macrophomina phaseolina identifies an array of putative virulence factors responsible for charcoal rot disease in plants. Frontiers in Microbiology, 13, 847832. https://doi.org/10.3389/fmicb.2022.847832
Soni, N., Raghuwanshi, K. S., & Kulwal, P. L. (2022). Effect of abiotic factors on the incidence of dry root rot in chickpea (Cicer arietinum L.). Journal of Agriculture Research and Technology, 47(2), 205.
Talekar, S. C., Viswanatha, K. P., & Lohithasawa, H. C. (2021). Screening chickpea genotypes for resistance to Rhizoctonia bataticola in controlled conditions. Legume Research, 44(1), 101–108. https://doi.org/10.18805/LR-4061.
Tariq, R., Hussain, A., Tariq, A., Khalid, M. H. B., Khan, I., Basim, H., & Ingvarsson, P. K. (2022). Genome-wide analyses of the mung bean NAC gene family reveals orthologs, co-expression networking and expression profiling under abiotic and biotic stresses. BMC Plant Biology, 22(1), 343. https://doi.org/10.1186/s12870-022-03716-4
Thilagavathi, R., Saravanakumar, D., Ragupathi, N., & Samiyappan, R. (2007). A combination of biocontrol agents improves the management of dry root rot (Macrophomina phaseolina) in greengram. Phytopathologia Mediterranea, 46(2), 157–167.
Thombre, B., & Kohire, O. (2018). Integrated management of Macrophomina blight of mungbean (Vigna radiata L.) caused by Macrophomina phaseolina (Tassi) Goid. Indian Phytopathology, 71, 423–429. https://doi.org/10.1007/s42360-018-0055-6
Tomar, R. S., Parakhia, M. V., Rathod, V. M., Thakkar, J. R., Padhiyar, S. M., Thummar, V. D., & Golakiya, B. A. (2017). Molecular mapping and identification of QTLs responsible for charcoal rot resistance in castor (Ricinus communis L.). Industrial Crops and Products, 95, 184–190. https://doi.org/10.1016/j.indcrop.2016.10.026.
Tyagi, R. N. S., Mathur, A. K., Gaur, V. K., Chitley, K., Bansal, R. K., & Pathak, A. K. (1988). Pathological status of pulse crops in Rajasthan. Indian Phytopathology, 41, 280.
Vidhyasekaran, P., Ramanathan, A., Rathinakumar, L., & Samiyappan, R. (2002). Induction of defense-mechanisms in mungbean suspension cultured cells by an elicitor from Macrophomina phaseolina. Acta Phytopathologica Et Rntomologica Hungarica, 37(1–3), 91–98. https://doi.org/10.1556/aphyt.37.2002.1-3.10
Vinholes, P., Rosado, R., Roberts, P., Borém, A., & Schuster, I. (2019). Single nucleotide polymorphism-based haplotypes associated with charcoal rot resistance in Brazilian soybean germplasm. Agronomy Journal, 111(1), 182–192. https://doi.org/10.2134/agronj2018.07.0429
Viteri, D. M., Linares, A. M., Miranda, Z., & Shi, A. (2022). Identification of a QTL region for ashy stem blight resistance using genome-wide association and linage analysis in common bean recombinant inbred lines derived from BAT 477 and NY6020-4. Frontiers in Plant Science, 13, 1019263. https://doi.org/10.3389/fpls.2022.1019263
Wang, L., Zhang, Y., Zhu, X., Zhu, X., Li, D., Zhang, X., Gao, Y., Xiao, G., Wei, X., & Zhang, X. (2019). Development of an SSR-based genetic map in sesame and identification of quantitative trait loci associated with charcoal rot resistance. Scientific Reports, 7, 8349. https://doi.org/10.1038/s41598-017-08858-2
Yan, W., Ni, Y., Liu, X., et al. (2021). The mechanism of sesame resistance against Macrophomina phaseolina was revealed via a comparison of transcriptomes of resistant and susceptible sesame genotypes. BMC Plant Biology, 21, 1–21.
Zatybekov, A., Abugalieva, S., Didorenko, S., Rsaliyev, A., Maulenbay, A., Fang, C., & Turuspekov, Y. (2023). Genome-wide association study for charcoal rot resistance in soybean harvested in Kazakhstan. Vavilov Journal of Genetics and Breeding, 27(6), 565. https://doi.org/10.18699/VJGB-23-68
Zhang, J. Q., Zhu, Z. D., Duan, C. X., Wang, X. M., & Li, H. J. (2011). First report of charcoal rot caused by Macrophomina phaseolina on mungbean in China. Plant Disease, 95(7), 872. https://doi.org/10.1094/PDIS-01-11-0010
Acknowledgements
This work was supported by the Tamil Nadu Agricultural University Core Funding and TARE scheme under Department of Science and Technology to S.G.; Medha 2023 fellowship by Bayer Foundation to N.H.S; National Institute of Plant Genome Research Core Funding and partly under the mission program of the Department of Biotechnology (DBT) on “Characterization of genetic resources” grant no. (BT/Ag/Network/Chickpea/2019-20) to M. S-K; the Council of Scientific and Industrial Research (CSIR) senior research fellowship [CSIR File no.–09/803(0177)/2020-EMR-I] to R.M.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sadhana, N.H., Geethanjali, S., Mirchandani, R. et al. Navigating towards dry root rot resistance in mungbean: impacts, mechanisms, and management strategies. Plant Physiol. Rep. 29, 439–460 (2024). https://doi.org/10.1007/s40502-024-00807-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40502-024-00807-2