
Abstract
Pongamia is a commercially important tree; its seed oil has the potential to be used as a biofuel worldwide. The seed traits have huge genetic diversity, which suggests its scope for genetic improvement. However, lack of improved genetic stock is a major bottleneck to establishing plantations for biodiesel production. Assessment of genetic diversity, population genetic structure, and biodiesel properties in 18 Pongamia accessions were studied using eight morphological traits and 11 SSR markers. The accessions exhibited a high degree of genetic variability at both levels, indicating the potential scope of this population for further tree breeding. Certain morphological traits are governed by additive genes and may provide indirect selection to yield improvement. Based on the dissimilarity of the coefficient, NRCP10 and NRCP20 were identified as the most distinct accessions, and, interestingly, these two accessions also have a higher cetane index for better biofuel properties. The dendrogram divided 18 accessions into two major clusters. The principal component analysis revealed a total of 70.92% variability in this population. On the other hand, molecular characterization using eleven SSR markers exhibited a significant level of polymorphism across the accessions. A dendrogram based on SSR markers clearly separated the 18 accessions into five groups, which resulted in NRCP11, NRCP14 and NRCP21 being the most distinct accessions. Also, the dendrogram conforms to the first and second coordinates by principal component analysis. The population structure of these 18 accessions also revealed the presence of two gene pools. In addition, analysis of molecular variance found 97% of the variability within the population, while only 3% occurred between populations. The genotypes identified in this study using morphological and molecular markers could be selected as donor parents in Pongamia tree breeding programs to improve economic trait values.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pongamia pinnata (L.) Pierre is a diploid (2n = 22) multipurpose tree species (Fabaceae) native to India and Southeast Asia (Al Muqarrabun et al., 2013; Karmee & Chadha, 2005; Kesari et al., 2009). Subsequently, it was introduced in to China, New Zealand, Australia, and the United States (Scott et al., 2008). In agroforestry programs, this species has been widely considered due to its ease of propagation in marginal lands, nitrogen-fixing capacity, used as green manure, fast-growth attributes, and, most importantly, high seed oil yield (Pavithra et al., 2014; Scott et al., 2008). This tree has also gained momentum across the world due to its biodiesel properties (Karmee & Chadha, 2005). Additionally, it has several important applications in medicine as an antimicrobial and in agriculture as a bio-insecticide and nematicide (Shivanna & Rajakumar, 2010; Yadav et al., 2004).
Increasing fuel product prices will probably have negative consequences for the global automobile industry in the future, which drives a search for alternative renewable energy sources. In this search, Pongamia is one of the promising sources of non-edible oil yielding trees that could be considered for biodiesel production (Azam et al., 2005; Karmee & Chadha, 2005; Kesari & Rangan, 2010). The seed oil content (30–40%) is one of the important traits responsible for its commercial usage, since it can be translated into biodiesel by trans-esterification (Karmee & Chadha, 2005; Naik et al., 2008). The major impediment to efficiently exploiting the biodiesel potential of this species is a lack of improved genetic stocks (Sharma et al., 2016). Genetic diversity estimation is an imperative step for successful tree breeding (Kesari et al., 2009). It allows tree breeders to have efficient parental selection for hybridization programs (Badu-Apraku et al., 2021). Phenotypic characterization of bigger populations allows germplasm assemblage and grouping, which aids in the selection of promising genotypes for tree breeding. Despite its importance, it has certain constraints in plant breeding since it is strongly influenced by the environment and is labor intensive. Considering morphological traits has limited value, DNA markers may be a better choice for genotyping (Uchoi et al., 2016). Likewise, analysing a population structure is more important in the context of genetic diversity and association mapping studies (Kumar et al., 2020). It also provides information on associations between markers and traits of interest. In this study, the population structure was assessed using SSR markers. Unlike other molecular markers, SSR has a high rate of polymorphism, co-dominant, multi-allelic, and wide genome coverage (Adjebeng-Danquah et al., 2020; Rajarajan & Ganesamurthy, 2011; Rajarajan et al., 2021; Rohini et al., 2020; Uchoi et al., 2016). Despite the advancement of molecular markers, there have been few studies on the genetic variation in Pongamia genotypes for seed and oil yield traits (Kesari & Rangan, 2011; Kesari et al., 2010; Sharma et al., 2016). There has been no significant research combining morphological and SSR markers to understand the genetic diversity, population structure, and biodiesel properties of Pongamia. However, characterizing genetic diversity of Pongamia pinnata gained significant concern due to socioeconomic prominence.
Therefore, this study aimed to investigate the genetic diversity, population structure, and biodiesel properties in P. pinnata using morphological and molecular markers for its genetic improvement.
Materials and methods
Plant material
Eighteen candidate plus trees (accessions) of Pongamia pinnata were collected from four states in India viz. Madhya Pradesh, Uttar Pradesh, Haryana, and Rajasthan (Table 1). The progeny trial was established in 2005 at Central Agroforestry Research Institute, Jhansi (Ahlawat et al., 2016). The plus trees were selected on the basis of high fruiting, seed yield, clean bole, and close canopy. These progenies were raised in a randomized block design (RBD) for three replications with five plants of each progeny per replication. Spacing of 5 m × 5 m was maintained in the field.
Oil and biodiesel properties
The seeds of each plus tree were combined to represent an entry, which was then analysed using a Soxhlet apparatus (Ambassador (BPI-32) Soxhlet Extraction Unit, New Delhi) using petroleum ether as the solvent. The 3 g of seed powder and 150 mL of petroleum ether were used for each set. Extraction was done at 80 °C for 5 h, followed by a rapid solvent recovery at 120 °C for 60 min. The mean of three replicates of each accession was calculated. Each accession’s oil content was determined and given as a percentage (w/w).
Saponification number (SN) and iodine value (IV) were estimated using FAMEs composition of oil as described by Kalayasiri et al., (1996).
where Ai is the percentage, D is the number of double bonds MWi is the molecular weight of each component.
According to Krisnangkura (1986), the Cetane Number (CN) of FAMEs was computed for estimating the biodiesel properties of each individual tree accession’s oil.
Morphological characterization
The morphological traits, viz. pod, seed, and oil characters, were studied in the present study for 18 accessions. Each morphological traits were calculated from three replications.
Pod traits
The pod length (PDL), breadth (PDB) and thickness (PDT) were calculated using calipers. The length of the pod was measured from the tip to the base; the width of the pod was measured from left to right; the thickness of the pod was determined on the part that contains seeds. The pot traits are expressed in mm.
Seed and oil traits
The seed traits such as seed length (SDL), breadth (SDB) and thickness (SDT) was calculated using caliper. The length of the seed was measured from the tip to the base; the width of the seed was measured from left to right; the thickness of the seed was observed from the front to the back and expressed in mm. While, 100-seed weight was measured by counted 100 seeds and weighted and expressed in grams. The oil content for each accession was calculated and expressed as percentage (w/w) of dry seed using following equation
where, Wfb is final beaker weight, Wib is initial beaker weight and Ws is the sample weight.
Statistical analysis
Simple statistical measures, such as mean and standard error, were considered in the quantitative data analyses. INDOSTAT (Indostat Services, Hyderabad, India) was used to analyze genetic variability measures: genotypic coefficient of variation (GCV), phenotypic coefficient of variation (PCV), heritability (h2) and genetic advance as percent mean (GAM). The dendrogram was constructed based on the group average clustering method, and principal component analysis was performed to find relationships among the variables and accessions using XLSTAT (Addinsoft, Paris), based on the morphological attributes.
Molecular marker analysis
The genomic DNA was isolated from young leaves using the modified CTAB (Cetyl trimethyl ammonium bromide) method (Doyle & Doyle, 1990). DNA purity was assessed by UV-Spectrophotometer (Eppendorf, Germany). A total of 20 SSR repeats were randomly selected from the assembled Pongamia transcriptome sequences in NCBI (SRA046342.1) (Huang et al., 2012), using MIcroSAtellite (MISA, http://pgrc.ipk-gatersleben.de/misa). The minimum number of repeats used to select the SSRs was 7 for di-nucleotide repeats, 4 for tri-nucleotide repeats, and 3 for tetra, penta and hexanucleotide repeats. Primer pairs for each SSR were designed using the standalone Primer3 (http://primer3.sourceforge.net/). Out of these markers, only 11 primers were found to be polymorphic (Table 2). Amplification was carried out in 20 µl volumes containing 2 µl genomic DNA (20 ng), 2 µl of 10 × Taq Buffer with MgCl2, 1 µl of dNTP (10 mM), Taq DNA polymerase (3.0 U/µl), 1 µl of each forward and reverse primer (0.2 mM), and 12.7 µl of nuclease-free water. The thermal cycler (Eppendorf Master-Cycler, Germany) with thermal cycling program follows: initial denaturation for 3 min at 93 °C, followed by 35 cycles of denaturation for 30 s at 93 °C, with an optimized annealing temperature for 30 s (54.6–56.3 °C), then extension for one minute at 72 °C, and the final extension for 8 min at 72 °C. The amplified fragments were separated on 3% agarose gel using 80 V for 2 h by gel electrophoresis (Syngene, USA).
Data analysis
The amplified fragments were used to construct a binary matrix (presence/absence of bands), to generate a dendrogram based on the unweighted neighbor-joining method, and principal coordinate analysis using DARwin software (Perrier & Flori, 2003). According to Botstein et al., (1980), each primer set was calculated for polymorphic information content (PIC). STRUCTURE software (v.2.3.3) was used to study the genetic structure using a Bayesian model (Falush et al., 2007). Also, STRUCTURE HARVESTER (Irfan et al., 2010) was used to resolve the estimated value of Ln probability of data-LnP (K). K, which ranged from 0 to 10, was calculated using three iterations of the data in this analysis. GenAlex 6.4 was used to analyze molecular variance (AMOVA) according to Peakall and Smouse, (2012).
Results and discussion
Morphological characterization of pod, seed and oil traits
The mean values of the eight morphological traits in the 18 accessions exhibited significant variation (Table 3). In NRCP13, the maximum pod length and pod breadth were observed, while the minimum pod length was found in NRCP17. Similarly, the minimum pod breadth was recorded in NRCP21. The thickness of the pods also varied significantly across the 18 accessions. The maximum seed length, seed breadth, and 100-seed weight were observed in NRCP13. Seed thickness varied significantly across the accessions. Based on the mean performance for pod and seed traits NRCP13 has superior performed than other accessions. Similarly, Kaushik et al., (2007) has reported Pongamia CPT-33 had maximum values for most of the pod and seed traits. In addition, the oil content value in this study ranged from 30.93 percent (NRCP7) to 38.15% (NRCP13) in Table 3. The oil content showed high variation among the genotypes as it was evident from the range of mean values.
Biofuel properties
Across the 18 Pongamia accessions, there were substantial variations observed in the biofuel traits, such as saponification number (SN), iodine value (IV), and cetane index (CI) (Table 4). The maximum saponification number and Iodine value were found in NRCP11 and NRCP23, while the minimum SN and IV were found in NRCP20. Nevertheless, NRCP20 has the highest cetane index. In this study, the saponification number and the cetane index obtained are similar to the previous report (Sharma et al., 2016). According to the Indian biodiesel standard (IS15607: 2005), the minimum cetane number should be 51; however, in this study the average cetane number was 53.49 showing that these accessions exhibit high biodiesel potential (Mukta et al., 2009; Sharma et al., 2016).
Morphological trait based genetic variability and diversity
In this study, the GCV was less than the PCV for pod, seed, and oil content traits indicating that non-additive gene action predominated (Table 5). Estimation of broad-sense heritability showed maximum heritability for the 100-seed weight followed by oil content. Table 5 also shows maximum genetic advance for the 100-seed weight followed by pod breadth. Based on these results, the 100-seed weight, seed thickness, pod length, pod breadth, and pod thickness had high heritability and genetic advance over other traits, indicating these traits are influenced by additive genes. In addition, oil yield had high heritability coupled with medium genetic advance indicating the additive gene effects on oil trait and as a result, the selection of these traits would be effective for oil and other yield associated trait improvement. Furthermore, the results in this study correspond with Ahlawat et al., (2016) and Kaushik et al., (2007) for Pongamia pod and seed traits.
The Euclidean distance-based cluster analysis used in this study grouped 18 accessions into two major clusters (Fig. 1a). There were ten accessions in Cluster I and eight in Cluster II. Cluster I was further subdivided into 1A and 1B, with six and four accessions, respectively. Cluster II was separated into groups 2A and 2B having three and five accessions, respectively. Furthermore, among the accessions, NRCP20 and NRCP10, which had the highest dissimilarity index, were shown to be the most diversified. Ahlawat et al., (2016) found a similar pattern of clustering in Pongamia based on pod and seed traits. Consequently, variations between these accessions are due to genotype-environment interactions and/or spontaneous mutations (Rajarajan et al., 2021). The clustering results suggest that these diverse genotypes are important in parental selection for tree improvement programs because they will offer unique alleles to have transgressive segregants for yield improvement (Rajarajan et al., 2021; Uchoi et al., 2016). Also, the biplot analysis in this study agreed with the clustering pattern based on morphological traits, clearly categorizing the accessions into two distinct coordinates as PC1 and PC2, with a total variability of 70.92% (Fig. 1b). This type of multivariate analysis for grouping accessions could provide tree breeders with an idea about accessions’ variabilities.

a Dendrogram based on morphological traits constructed using Squared Euclidean distance and group average clustering method. b Biplot analysis based of PCA by morphological traits
SSR marker analysis for genetic diversity
In this study, out of fifteen SSR markers, eleven were found to be polymorphic, which revealed a high polymorphism across the eighteen accessions (Table 6). A total of 313 bands were produced by 11 primers; the alleles generated ranged from 2 to 6 with a mean of 3.6 per locus (Table 6). The primer P011 produced the maximum alleles (6) per locus (Fig. 2). The polymorphism percentage ranged from 50.0% to 100%, with a mean of 88.8%. Also, the PIC value estimated for 11 primers ranged from 0.14 to 0.77 with an average of 0.53. P007 had the highest PIC value. The polymorphic information content of these SSR primers are higher than the earlier reports by different markers RAPD (Ahlawat et al., 2016; Kesari et al., 2010), ISSR (Kesari et al., 2010) AFLP (Kesari et al., 2010; Pavithra et al., 2013; Sharma et al., 2016, 2017) in Pongamia, indicating the discrimination potential of these SSR markers in genetic diversity studies (Table 7).

SSR banding profile (P007) of 18 Pongamia accessions. Where M is 100 bp Ladder and 1–18 Pongamia accessions
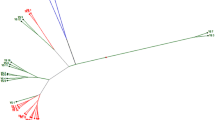
The similarity coefficient based on SSR markers ranged from 0.36 to 0.87, with an average of 0.55 indicating the existence of greater genetic diversity across the accessions. Cluster analysis based on the neighbor-joining method grouped all 18 accessions into five clusters according to their similarities (Fig. 3a). Cluster I was composed of six accessions, Cluster II and III each had four, cluster IV had three and cluster V had a single accession. According to the dissimilarity coefficient, the accessions NRCP11, NRCP14, and NRCP21 were identified as the most distinct and would provide divergent alleles for breeding programs (Rohini et al., 2020). Also, the principal coordinate analysis of the 18 accessions clearly divided the accessions into four different coordinates, which once again confirms the dendrogram pattern (Fig. 3b).


a Radial neighbor-joining tree based on 74 alleles from 11 SSR loci among 18 Pongamia germplasm. b Principal coordinates analysis (PCoA) of 18 Pongamia accessions using SSR markers
Population structure analysis
Information on genetic differentiation among and within populations is important for determining the relationship between accessions based on allele frequency distribution (Kumar et al., 2020; Rohini et al., 2020). In this study, we attempted to investigate the population structure and genetic relationships among Pongamia accessions using SSR markers (Fig. 4a). The two subpopulations (SUP1 & SUP2) were estimated based on the highest delta K value (K = 2) (Fig. 4b). The SUP1 and SUP2 had 11 and 7 accessions, respectively. Similarly, Pavithra et al., (2014) reported Pongamia population structure using AFLP markers. In addition, the structure analysis chart (red and green colors) using STRUCTURE HARVESTER clearly defined two gene pools present in this population, which also shows intermixing between the accessions based on the 20% genetic background of other accessions (Fig. 4b). Accordingly, NRCP11, NRCP12, NRCP16, and NRCP26 were found to be intermixed accessions. Similarly, Rohini et al., (2020) reported intermixed accessions in Citrus jambhiri population. Thus, aid in identifying pure genotypes from intermixed ones in a genetic resource repository for efficient utilization and characterization (Rajarajan et al., 2021; Rohini et al., 2020). Also, in this study, the results of structure analysis correspond with principal component analysis (PCoA) and neighbor-joining tree.

Population structure analysis of Pongamia pinnata using 11 SSR markers: a Delta K analysis. b Structure analysis of two sub-populations of 18 Pongamia accessions and c analysis of molecular variance of 18 accessions of Pongamia pinnata based on SSR markers
Furthermore, the AMOVA analysis instigated in this study found a high degree of genetic diversity within subpopulations and a low level among the population at 97% and 3%, respectively (Fig. 4c). The reason for high variation within the population might be due to outcrossing or spontaneous mutations. Furthermore, one could expect a high gene flow in the population by the low level of variation among the population (Kumar et al., 2020). Likewise, in this study, a high level of gene flow justified only 3% differentiation among the population. This kind of study will help tree breeders to monitor and maintain genetic variation in the population for tree breeding programs.
Evaluation of morphological traits is a crucial step in determining genetic diversity and identifying superior genotypes (Rajarajan et al., 2021). Yet, the environment, which is considered a critical limitation, significantly impacts morphological trait expression. As a result, molecular markers should be used in concert with morphological traits to precisely measure the level of genetic diversity in the target species. Similarly, we assessed genetic diversity at both the morphological and molecular levels in this study. However, there was no association between the two assessments, which could be due to different phenotypes resulting from genotype and environment interaction. Overall, this study highlights the potential of combining markers and morphological traits to assess genetic differentiation and population structure in Pongamia pinnata for genetic improvement.
Conclusion
In this study, genetic variability for 100-seed weight, pod length, and the pod and seed thickness, had high heritability with high genetic advance, indicating that these traits are governed by additive genes and selection based on these traits would be very useful in trait improvement. Furthermore, multivariate analysis revealed that NRCP10 and NRCP20 are more diversified than other accessions. Also, these accessions have a high cetane index, and useful in biodiesel production. In SSR marker analysis, accessions NRCP11, NRCP14, and NRCP21 found to be highly diversified. In addition, the population genetic structure analysis of Pongamia provided an idea about the population structure and its variability among and within the population for utilization in tree breeding.
References
Adjebeng-Danquah, J., Manu-Aduening, J., Asante, I. K., Agyare, R. Y., Gracen, V., & Offei, S. K. (2020). Genetic diversity and population structure analysis of Ghanaian and exotic cassava accessions using simple sequence repeat (SSR) markers. Heliyon, 6(1), e03154.
Ahlawat, S. P., Kumar, R. V., Ranjan, R., Pandey, S. K., Joshi, D. C., & Dhyani, S. K. (2016). Morphological and molecular level of genetic diversity among Pongamia [Pongamia pinnata (L.) Pierre] accessions. Indian Journal of Biotechnology, 15(1), 85–94.
Al Muqarrabun, L. M. R., Ahmat, N., Ruzaina, S. A. S., Ismail, N. H., & Sahidin, I. (2013). Medicinal uses, phytochemistry and pharmacology of Pongamia pinnata (L.) Pierre: A review. Journal of Ethnopharmacology, 150(2), 395–420.
Azam, M. M., Waris, A., & Nahar, N. M. (2005). Prospects and potential of fatty acid methyl esters of some non-traditional seed oils for use as biodiesel in India. Biomass and Bioenergy, 29(4), 293–302.
Badu-Apraku, B., Garcia-Oliveira, A. L., Petroli, C. D., Hearne, S., Adewale, S. A., & Gedil, M. (2021). Genetic diversity and population structure of early and extra-early maturing maize germplasm adapted to sub-Saharan Africa. BMC Plant Biology, 21(1), 1–15.
Botstein, D., White, R. L., Skolnick, M., & Davis, R. W. (1980). Construction of a genetic linkage map in man using restriction fragment length polymorphisms. American Journal of Human Genetics, 32(3), 314.
Doyle, J. J., & Doyle, J. L. (1990). Isolation ofplant DNA from fresh tissue. Focus, 12(13), 39–40.
Falush, D., Stephens, M., & Pritchard, J. K. (2007). Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Molecular Ecology Notes, 7(4), 574–578.
Huang, J., Lu, X., Yan, H., Chen, S., Zhang, W., Huang, R., & Zheng, Y. (2012). Transcriptome characterization and sequencing-based identification of salt-responsive genes in Millettia pinnata, a semi-mangrove plant. DNA Research, 19(2), 195–207.
Irfan, M., Hayat, S., Hayat, Q., Afroz, S., & Ahmad, A. (2010). Physiological and biochemical changes in plants under waterlogging. Protoplasma, 241(1), 3–17.
Kalayasiri, P., Jeyashoke, N., & Krisnangkura, K. (1996). Survey of seed oils for use as diesel fuels. Journal of the American Oil Chemists’ Society, 73(4), 471–474.
Karmee, S. K., & Chadha, A. (2005). Preparation of biodiesel from crude oil of Pongamia pinnata. Bioresource Technology, 96(13), 1425–1429.
Kaushik, N., Kumar, S., Kumar, K., Beniwal, R. S., Kaushik, N., & Roy, S. (2007). Genetic variability and association studies in pod and seed traits of Pongamia pinnata (L.) Pierre in Haryana, India. Genetic Resources and Crop Evolution, 54(8), 1827–1832.
Kesari, V., & Rangan, L. (2010). Development of Pongamia pinnata as an alternative biofuel crop—current status and scope of plantations in India. Journal of Crop Science and Biotechnology, 13(3), 127–137.
Kesari, V., & Rangan, L. (2011). Genetic diversity analysis by RAPD markers in candidate plus trees of Pongamia pinnata, a promising source of bioenergy. Biomass and Bioenergy, 35(7), 3123–3128.
Kesari, V., Madurai Sathyanarayana, V., Parida, A., & Rangan, L. (2010). Molecular marker-based characterization in candidate plus trees of Pongamia pinnata, a potential biodiesel legume. AoB Plants, 2010, 1–12.
Kesari, V., Krishnamachari, A., & Rangan, L. (2009). Effect of auxins on adventitious rooting from stem cuttings of candidate plus tree Pongamia pinnata (L.), a potential biodiesel plant. Trees, 23(3), 597–604.
Krisnangkura, K. (1986). A simple method for estimation of cetane index of vegetable oil methyl esters. Journal of the American Oil Chemists’ Society, 63(4), 552–553.
Kumar, D., Chhokar, V., Sheoran, S., Singh, R., Sharma, P., Jaiswal, S., et al. (2020). Characterization of genetic diversity and population structure in wheat using array based SNP markers. Molecular Biology Reports, 47(1), 293–306.
Mukta, N., Murthy, I., & Sripal, P. (2009). Variability assessment in Pongamia pinnata (L.) Pierre germplasm for biodiesel traits. Industrial Crops and Products, 29(2–3), 536–540.
Naik, M., Meher, L., Naik, S., & Das, L. (2008). Production of biodiesel from high free fatty acid Karanja (Pongamia pinnata) oil. Biomass and Bioenergy, 32(4), 354–357.
Pavithra, H. R., Gowda, B., Prasanna, K. T., & Shivanna, M. B. (2013). Pod and seed traits in candidate plus trees of Pongamia pinnata (L.) Pierre from southern peninsular India in relation to provenance variation and genetic variability. Journal of Crop Science and Biotechnology, 16(2), 131–142.
Pavithra, H. R., Shivanna, M. B., Chandrika, K., Prasanna, K. T., & Gowda, B. (2014). Genetic analysis of Pongamia pinnata (L.) Pierre populations using AFLP markers. Tree Genetics and Genomes, 10(1), 173–188.
Peakall, R., & Smouse, P. E. (2012). GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research–an update. Bioinformatics, 28(19), 2537–2539.
Perrier, X., & Flori, A. (2003). Methods of data analysis. In Genetic diversity of cultivated tropical plants (pp. 47–80). CRC Press.
Rajarajan, K., Uthappa, A. R., Handa, A. K., Chavan, S. B., Vishnu, R., Shrivastava, A., et al. (2021). Genetic diversity and population structure of Leucaena leucocephala (Lam.) de Wit genotypes using molecular and morphological attributes. Genetic Resources and Crop Evolution, 69, 71–83.
Rajarajan, K., & Ganesamurthy, K. (2011). Genetic diversity analysis of sorghum [Sorghum bicolor (L.) Moench] genotypes for drought tolerance using SSR markers. Indian Journal of Genetics and Plant Breeding, 71(1), 17.
Rohini, M. R., Sankaran, M., Rajkumar, S., Prakash, K., Gaikwad, A., Chaudhury, R., & Malik, S. K. (2020). Morphological characterization and analysis of genetic diversity and population structure in Citrus× jambhiri Lush. using SSR markers. Genetic Resources and Crop Evolution, 67, 1259–1275.
Scott, P. T., Pregelj, L., Chen, N., Hadler, J. S., Djordjevic, M. A., & Gresshoff, P. M. (2008). Pongamia pinnata: An untapped resource for the biofuels industry of the future. Bioenergy Research, 1(1), 2–11.
Sharma, S. S., Islam, M. A., Malik, A. A., Kumar, K., Negi, M. S., & Tripathi, S. B. (2016). Seed traits, fatty acid profile and genetic diversity assessment in Pongamia pinnata (L.) Pierre germplasm. Physiology and Molecular Biology of Plants, 22(2), 193–205.
Sharma, S. S., Islam, M. A., Singh, V. K., Negi, M. S., & Tripathi, S. B. (2017). Genetic diversity, population structure and association study using TE-AFLP markers in Pongamia pinnata (L.) Pierre germplasm. Tree Genetics & Genomes, 13(1), 1–10.
Shivanna, M. B., & Rajakumar, N. (2010). Ethno-medico-botanical knowledge of rural folk in Bhadravathi taluk of Shimoga district, Karnataka. Indian Journal of Traditional Knowledge, 9(1), 158–162.
Uchoi, A., Malik, S. K., Choudhary, R., Kumar, S., Rohini, M. R., Pal, D., et al. (2016). Inferring phylogenetic relationships of Indian citron (Citrus medica L.) based on rbcL and matK sequences of chloroplast DNA. Biochemical Genetics, 54(3), 249.
Yadav, P. P., Ahmad, G., & Maurya, R. (2004). Furanoflavonoids from Pongamia pinnata fruits. Phytochemistry, 65(4), 439–443.
Acknowledgements
The authors are grateful to Field Assistant Mr. Rambabu, ICAR-Central Agroforestry Research Institute, India and Indian Council of Agricultural Research (ICAR), New Delhi, India, for facilities to complete this work.
Author information
Authors and Affiliations
Contributions
KR and AKH conceived the idea. KR wrote the main manuscript text. KR, AA, HA, MR, SS and RV prepared the manuscript. KR and AA revised the manuscript at different stages of the writing process and read and approved the revised manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All the authors declared that no conflicts of interest are associated with this publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rajarajan, K., Sakshi, S., Rana, M. et al. Understanding the genetic determinants and population structure of Pongamia pinnata (L.) Pierre for oil yield and its properties using transcriptome derived SSR markers. Plant Physiol. Rep. 27, 407–418 (2022). https://doi.org/10.1007/s40502-022-00675-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40502-022-00675-8