
Abstract
Purpose of Review
Obesity is a trigger for multiple diseases such as diabetes mellitus, hypertension, and cardiovascular diseases. Epidemiological studies have shown that obesity may be a risk factor for periodontal disease. Recently, there have been reports of presumed mechanisms of the associations between periodontitis and lipid metabolism or thermogenesis. This review aims to discuss the link between periodontal disease and energy regulatory function based on recent findings.
Recent Findings
It has been demonstrated that activation of the C–C motif chemokine ligand/C–C chemokine receptor 7 pathway in adipose tissue induces inflammation and impairment of lipid metabolism and energy regulation in mice. Porphyromonas gingivalis administration has been shown to induce further weight gain and increased adipose tissue in diet-induced obese mice. Additionally, it has been reported that Porphyromonas gingivalis–induced endotoxemia potentially affect obesity by altering endocrine functions in brown adipose tissue in mice. Several cohort studies have shown that obesity is associated with tooth loss 5 years later, and periodontal conditions of obese individuals are significantly worse 2 and 6 months after the treatment compared with those of non-obese individuals. It has also been reported that body mass index is positively associated with the periodontal inflamed surface area index, a measure of periodontal inflammation. These results suggest that not only the enhancement of inflammation due to obesity but also the activation of inflammatory signaling may affect energy regulation.
Summary
Loss of adipose tissue homeostasis induces increase and activation of immune cells in adipose tissue, leading to impaired immune function in obesity. Various cytokines and chemokines are secreted from obese adipose tissue and promote inflammatory signaling. Some of these signaling pathways have been suggested to affect energy regulation. The combination of obesity and periodontitis amplifies inflammation to levels that affect the whole body through the adipose tissue. Obesity, in turn, accelerates the exacerbation of periodontitis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Periodontal disease is a chronic inflammatory disease that is accompanied by periodontal tissue destruction. Low-grade inflammation, such as periodontal disease, affects glucose metabolism [1]. In a new classification of periodontal disease published by the American Academy of Periodontology (AAP) and the European Federation of Periodontology (EFP) in 2018, diabetes mellitus (DM) was added as a risk factor for the progression of periodontal disease [2, 3]. Obesity is an important risk factor for type 2 (T2) DM, which accounts for 90% of DM cases, and the number of obese individuals is also increasing worldwide. In 2016, 39% of adults were overweight (25 kg/m2 ≤ body mass index [BMI] < 30 kg/m2) and 13% were obese (BMI ≥ 30 kg/m2) [4]. Animals, including humans, can sustain life by storing excess energy in the form of fat and using it during starvation. This is an ingenious biological mechanism acquired during the course of a long evolution. However, excessive energy intake and lack of exercise induce obesity, leading to health threats. Epidemiological studies have shown that obesity may be a risk factor for periodontal disease [5]. Recently, associations between periodontitis and lipid metabolism or thermogenesis have been reported [6•]. This review aims to discuss the link between periodontal disease and energy regulatory function based on recent findings.
Energy Metabolism and Thermogenesis
Fat is not only taken directly from the diet, but also newly synthesized mainly in the liver and adipose tissue using excess energy. The fat obtained is stored in adipocytes. In obesity, adipocytes become hypertrophied, but when they can no longer be stored, fat is released as free fatty acid (FFA). Elevated blood FFA is taken up by skeletal muscle and liver and accumulates mainly as triglyceride, causing lipotoxicity [7]. Adipose tissue is roughly classified into white adipose tissue (WAT) and brown adipose tissue (BAT). White adipocytes store fat, while brown adipocytes are thermogenic adipocytes that play a role in maintaining body temperature and increasing basal metabolism in cold environments. In humans, brown adipocytes are abundant in newborns, but decrease after infancy. BAT activity in human adults is characterized by inducibility and reversibility [8]. In recent years, beige adipocytes that emerge in response to certain environmental cues, such as chronic cold exposure, have been found in WAT [9]; it has been shown that the tissue called BAT is composed primarily of beige adipocytes [10]. Thermogenesis in brown adipocytes and beige adipocytes occurs through the uncoupling of oxidative phosphorylation of mitochondrial uncoupling protein 1 (UCP1).
Biogenesis of beige adipocytes is induced in response to external stimuli, such as cold exposure, exercise, and treatment with β3-adrenoreceptor (β3-AR) agonists [11]. Adenosine monophosphate-activated protein kinase (AMPK) deletion in adipocytes results in cold intolerance and reduced thermogenesis in response to β3-AR stimulation [12]. AMPK activity in adipose tissue is diminished in humans and rodents with obesity and insulin resistance [13,14,15,16]. Activation of adipocyte AMPK improves obesity-induced insulin resistance [17]. AMPK also regulates mitochondrial biogenesis by activating peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) [18]. PGC1α is a critical transcriptional activator that regulates the number of genes involved in various metabolic pathways, such as fatty acid synthesis, oxidation, and gluconeogenesis. PGC1α stimulates UCP1 expression and increases the number of mitochondria and mitochondrial oxidative capacity [19, 20]. Suppression of adipose PGC1α results in systemic insulin resistance and obesity-associated inflammation [21]. Recently, ceramides and their metabolites, sphingolipids, have attracted attention as lipotoxic substances. Systemic and adipose tissue-specific inhibition of ceramide synthesis induces browning of subcutaneous WAT in obese mice [22]. M2 macrophages, known to enhance cold-induced thermogenesis [23], were increased in adipose tissue due to the inhibition of ceramide synthesis [22].
Inflammation and Energy Metabolism in Adipose Tissue
Adipocytes secrete multiple biologically active molecules, such as hormones and cytokines (known as adipokines). Normal adipocytes secrete anti-inflammatory adipokines to attenuate inflammation and play essential roles in the control of whole-body metabolism [24, 25]. The pathophysiological features of obesity include adipose tissue inflammation and infiltration of activated immune cells, such as macrophages [24]. Abnormal adipokine secretion is observed in hypertrophied adipocytes [24, 25]. Infiltration of immune cells such as macrophages is enhanced in obese adipose tissue, and the interaction with adipocytes promotes abnormal secretion of adipokines [24]. The coronavirus disease 2019 (COVID-19) pandemic has provided an opportunity for widespread public awareness that infectious diseases are more likely to become severe in metabolic disorders such as obesity. In obesity, the lack of adipose tissue homeostasis increases and activates immune cells in adipose tissue and promotes the production of inflammatory cytokines and chemokines [26,27,28,29]. It has also been suggested that such changes in immune cell response impair cell-mediated immune function, leading to enhanced activation of the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome and increased susceptibility to infection [30].
Intake of a high-fat diet (HFD) changes the gut microbiota, which induces endotoxemia followed by chronic inflammation in various tissues, including adipose tissue [31,32,33]. Changes in the composition and number of gut microbiota lead to an increase in toll-like receptor 4 (TLR4) ligands that flow into the liver via the portal vein [34], which has been suggested to be relevant to multiple diseases such as obesity, cardiovascular disease, inflammatory disease, and diabetes [32]. We hypothesized that lipopolysaccharide (LPS) from the gut microbiota activates monocytes in the portal vein and accumulates in the adipose tissue. A comprehensive analysis of the gene expression in adipocytes co-cultured with macrophages stimulated with low concentrations of LPS was performed to examine the factors involved in chronic inflammation of adipose tissue. As a result, it was observed that co-cultured adipocytes promoted the expression of genes such as LPS-binding protein (LBP) and CD14, which are required for the action of LPS [35]. In the culture medium of the co-culture system, a marked increase in the secretion of proteins such as LBP, soluble CD14, C–C motif chemokine ligand 5 (CCL5), and serum amyloid A (SAA) was observed compared with those of adipocytes and macrophages individually cultured or without LPS stimulation. In addition, the amount of interleukin 6 (IL6) produced from adipocytes increased significantly via macrophage-derived tumor necrosis factorα (TNFα) [36, 37], suggesting the induction of acute phase proteins such as C-reactive protein (CRP) and SAA [38]. SAA induces IL6 production in dendritic cells and promotes T helper 17 (Th17) cell differentiation [39]. Th17 cells secrete inflammatory cytokines, such as IL17, and have been suggested to be involved in periodontal disease [40, 41].
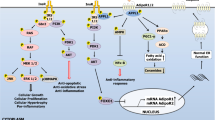
Based on the results of the adipocyte-macrophage co-culture system, it has been suggested that IL17 promotes Th17 cell infiltration through increased CCL20 production in adipose tissue in a chronically inflamed state, thereby exacerbating inflammation [42]. A comprehensive analysis of the gene expression in adipocytes co-cultured with macrophages showed a marked increase in gene expression of many CC chemokines, such as monocyte chemoattractant protein (MCP1), CCL5, and CXC chemokines, in adipocytes. Among them, CCL19 contributes to the attraction of dendritic cells and T cells. Serum CCL19 levels were markedly increased in both hereditary (ob/ob) and diet-induced obese mice. Therefore, we investigated the effects on inflammation and metabolism in mice lacking the CCL19 receptor, CC chemokine receptor 7 (CCR7) gene. Compared with wild-type mice, Ccr7−/− mice showed an increase in UCP1 expression in BAT, high rectal temperature during cold stimulation, and a significant increase in thermogenesis. In addition, they were resistant to HFD-induced obesity, adipose tissue and liver inflammation, fatty liver, and dyslipidemia, and insulin response and glucose tolerance were maintained normally [43]. Subsequently, for further investigation of the effects of CCL19/CCR7 axis, adipocyte-specific Ccl19 knock-in (KI) mice were generated. Ccl19 KI mice demonstrated that activation of the CCL19/CCR7 pathway in adipose tissue induced inflammation and that adipocyte CCL19 inhibited AMPKα by activating extracellular signal-regulated kinase 1/2 (ERK1/2), resulting in impaired lipid metabolism and energy regulation (Fig. 1). In addition, a 40% HFD enhanced these pathological changes and induced insulin resistance and weight gain [44•].

Possible mechanisms of influences of the CCL19/CCR7 pathway in obese adipose tissue. Activation of the CCL19/CCR7 pathway induces inflammation and that adipocyte CCL19 inhibits AMPKα by activating ERK1/2, resulting in impaired lipid metabolism and energy regulation. The enhancement of these pathological changes induces insulin resistance
López-Cotarelo et al. reported that MEK1/2/ERK1/2 signaling induced by CCR7 ligands had an inhibitory effect on AMPK activity in human mature dendritic cells. In their experiments, both CCL19 and CCL21 were used as CCR7 ligands [45]. Our results of a comprehensive analysis of the gene expression in adipocytes co-cultured with macrophages showed a marked increase in Ccl19 gene, but not in Ccl21 gene. The expression level of Ccl19 was one of the highest of the expression of all chemokines produced from adipocytes. Epidemiological studies have shown that serum CCL19 levels are elevated in individuals with obesity [46]. Furthermore, it has also been reported that adipose tissue CCL19 expression correlates with BMI, HbA1c, CRP, homeostasis model assessment of insulin resistance (HOMA-IR), IL12, and CCL5 [47]. A previous report exists regarding the downregulation of AMPKα through the activation of ERK1/2 in muscle cells. Hwang et al. demonstrated that endoplasmic reticulum stress induced by thapsigargin and tunicamycin increased the phosphorylation of ERK1/2 and suppressed AMPKα activation in muscle cells [48]. Several publications have also shown that ERK1/2 activation is promoted in adipose tissues of humans and mouse models with obesity and diabetes and in 3T3-L1 adipocytes treated with TNFα or insulin [49,50,51]. However, the relationship between ERK and AMPK was not examined in these studies.
As shown in these results, various cytokines and chemokines are secreted from obese adipose tissue, promoting inflammatory signaling. Some of these signaling pathways have been suggested to affect energy regulation.
Association of Periodontal inflammation and Obesity
In periodontal disease, fimbriae, endotoxin, and proteases, which are the main virulence factors of periodontal pathogens, act on monocytes, macrophages, and gingival fibroblasts in the periodontal tissue. These cells induce an inflammatory response in the periodontal tissue via inflammatory mediators, such as IL1β, TNFα, prostaglandin E2 (PGE2), and matrix metalloproteinases (MMPs), and tissue destruction progresses. This chronic periodontal inflammatory reaction leads to leakage of host- and microbial-derived factors into the bloodstream. Once in the systemic circulation, these factors could contribute to the pathophysiology of systemic diseases, either directly or indirectly [52]. Multiple studies have shown the presence of periodontal bacteria in atherosclerotic coronary arteries [53,54,55]. Non-hematogenous dissemination of periodontal bacteria has also been suggested. Swallowed periodontal bacteria may translocate via the oro-digestive route and colonize ectopically in the gut. Periodontal bacteria have been detected in the gut of patients with inflammatory bowel disease [56, 57]. Although the presence of periodontal bacteria in extra-oral tissues may be transient, the release of virulence factors like endotoxin and toxic proteases [58, 59], or inflammatory mediators, can be important, given the chronicity of periodontitis and the frequency of bacteremia. In fact, serum levels of TNFα are elevated in patients with periodontitis [60, 61].
Our previous study demonstrated that administration of low-concentration LPS (1 ng/mL/g body weight) significantly upregulated the serum levels of LBP, CCL5, and SAA in hereditary obese (ob/ob) mice and diet-induced obese mice compared with those in the control group (ob/ + mice and normal diet-fed mice) [38]. This result suggests that local inflammation, such as periodontitis, is more likely to have a systemic effect in obese patients than in non-obese patients. As mentioned earlier, immune cells are activated in the obese adipose tissue. Also, some of the immune cells activated by bacterial products or host inflammatory factors may migrate to the adipose tissue via the circulation. In the combination of obesity and periodontitis, local inflammation is inferred to be amplified to levels that affect the whole body through adipose tissue. Thus, obesity may exacerbate periodontal disease.
Several recent animal model studies have demonstrated the relationship between the exacerbation of periodontitis and obesity. Saturated fatty acids induce inflammation and promote alveolar bone resorption in mice [62, 63]. In obese rats, it has been shown that protein kinase C (PKC) activation and oxidative stress may induce insulin resistance in gingiva leading to periodontal disease progression [64]. Furthermore, caloric restriction has been reported to prevent alveolar bone loss in experimental periodontitis in obese rats [65]. Sato et al. reported that HFD-induced changes in the gut microbiota exacerbated alveolar bone resorption via uric acid elevation [66]. Other researchers have reported that Porphyromonas gingivalis (P. gingivalis) administration induced further weight gain and increased adipose tissue in diet-induced obese mice [68]. Furthermore, it is also indicated the possibility that P. gingivalis–induced endotoxemia potentially affects obesity by altering endocrine functions in BAT [6•]. These results suggest that not only exacerbation of inflammation due to obesity but also activation of inflammatory signaling may affect energy regulation. It should be noted that administration of a single periodontal pathogen or endotoxin to animals, as in these animal studies, does not exactly reproduce human periodontitis associated with multiple pathogens [62, 63]. In addition, care must be taken in interpreting the results depending on the method and period of administration, strains, and bacterial species [6•, 66, 69]. Considering that the microbiota changes with oral and intravenous administration of P. gingivalis [66, 70, 67•], the indirect effect of P. gingivalis can also be said to be large. Further clarifying the mechanism of the relation between periodontal disease and obesity may reveal new and effective treatment strategies that will contribute to the improvement of periodontitis and obesity-related diseases in the future.
Clinical Studies on the Association of Periodontitis and Obesity
Several cohort and intervention studies related to obesity and periodontitis have been conducted. Recent cohort studies indicated that obesity is a risk factor for tooth loss [71] and that obesity is associated with worse mean probing pocket depth following periodontal therapy [72•]. Several reports have investigated the association between the degree of obesity and systemic effects of periodontal inflammation. Munenaga et al. reported that Japanese patients with T2DM accompanied by severe periodontal disease and BMI around 25 kg/m2 showed slightly increased systemic inflammation as assessed by high-sensitivity (hs)-CRP (hs-CRP levels over 500 ng/mL). In contrast, subjects with low hs-CRP levels (< 500 ng/mL) were characterized by a significantly lower BMI (approximately 23 kg/m2) than that in the group with hs-CRP levels ≥ 500 ng/mL [73]. Other researchers have suggested that periodontal disease has a significant impact on systemic inflammation in patients with normal BMI, but not in patients with a BMI of around 35 kg/m2 [74]. These studies suggest that systemic influences of local inflammation may be enhanced by mild obesity but largely masked by severe obesity. Therefore, it is considered that whether the systemic effects due to local inflammation, such as periodontal disease, manifest or not depends on the degree of obesity.
In recent years, clinical studies have been performed using the data of periodontal inflamed surface area (PISA), which is a measure of the amount of inflamed periodontal tissue. PISA is a quantified value of the surface area of the bleeding pocket epithelium [75, 76]. As the correlation between PISA and glycated hemoglobin (HbA1c) has been reported [76,77,78], it can be said to be an index for understanding the degree of systemic influence due to local periodontal inflammation. Recently, it has been reported that BMI is positively associated with PISA [79]. On the other hand, Takeda et al. reported that PISA in T2DM patients was not related to obesity parameters [77]. However, it is necessary to establish a larger control group to evaluate the relationship between PISA and obesity. Other researchers also reported that although obesity is not an independent risk factor for the progression of periodontitis, obesity and periodontitis have many common risk factors for each progression [80]. At present, there is limited clinical evidence on the association between periodontal disease and overweight/obesity. Further studies are required to clarify this association.
Conclusions
Loss of adipose tissue homeostasis induces increase and activation of immune cells in adipose tissue, leading to impairment of immune function in obesity. Various cytokines and chemokines are secreted from obese adipose tissue, promoting inflammatory signaling. Some of these signaling pathways may affect energy regulation. The combination of obesity and periodontitis amplifies inflammation to levels that affect the whole body through the adipose tissue. Obesity, in turn, accelerates the exacerbation of periodontitis.
Further understanding of the link between periodontal disease and energy metabolism disorders is expected to lead to new and more effective therapeutic strategies for both diseases. Obesity is a risk factor for many diseases, including DM, hypertension, and cardiovascular disease. Therefore, improving periodontitis and controlling body weight can contribute significantly to the maintenance of health and future life.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Borgnakke WS, Ylostalo PV, Taylor GW, Genco RJ. Effect of periodontal disease on diabetes: systematic review of epidemiologic observational evidence. J Clin Periodontol. 2013;40(Suppl 14):135–52. https://doi.org/10.1902/jop.2013.1340013.
Papapanou PN, Sanz M, Buduneli N, Dietrich T, Feres M, Fine DH, et al. Periodontitis: consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Periodontol. 2018;89:S173–82. https://doi.org/10.1002/JPER.17-0721.
Tonetti MS, Greenwell H, Kornman KS. Staging and grading of periodontitis: framework and proposal of a new classification and case definition. J Clin Periodontol. 2018;45:S149–61. https://doi.org/10.1111/jcpe.12945.
World Health Organization. Obesity and overweight 2018. Available from: https://www.who.int/en/news-room/fact-sheets/detail/obesity-and-overweight.
Jepsen S, Suvan J, Deschner J. The association of periodontal diseases with metabolic syndrome and obesity. Periodontol. 2000;2020(83):125–53. https://doi.org/10.1111/prd.12326.
• Hatasa M, Ohsugi Y, Katagiri S, Yoshida S, Niimi H, Morita K, et al. Endotoxemia by Porphyromonas gingivalis alters endocrine functions in brown adipose tissue. Front Cell Infect Microbiol. 2021;10: 580577. https://doi.org/10.3389/fcimb.2020.580577. (It has been shown that P. gingivalis-induced endotoxemia potentially affects obesity by altering the endocrine functions of brown adipose tissue in mice.)
Chouchani ET, Kajimura S. Metabolic adaptation and maladaptation in adipose tissue. Nat Metab. 2019;1:189–200. https://doi.org/10.1038/s42255-018-0021-8.
Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans. Diabetes. 2009;58:1526–31. https://doi.org/10.2337/db09-0530.
Sidossis L, Kajimura S. Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. J Clin Invest. 2015;125:478–86. https://doi.org/10.1172/JCI78362.
Shinoda K, Ohyama K, Hasegawa Y, Chang HY, Ogura M, Sato A, et al. Phosphoproteomics identifies CK2 as a negative regulator of beige adipocyte thermogenesis and energy expenditure. Cell Metab. 2015;22:997–1008. https://doi.org/10.1016/j.cmet.2015.09.029.
Ikeda K, Maretich P, Kajimura S. The common and distinct features of brown and beige adipocytes. Trend Endocrinol Metab. 2018;29:191–200. https://doi.org/10.1016/j.tem.2018.01.001.
Mottillo EP, Desjardins EM, Crane JD, Smith BK, Green AE, Ducommun S, et al. Lack of adipocyte AMPK exacerbates insulin resistance and hepatic steatosis through brown and beige adipose tissue function. Cell Metab. 2016;24:118–29. https://doi.org/10.1016/j.cmet.2016.06.006.
Xu XJ, Gauthier MS, Hess DT, Apovian CM, Cacicedo JM, Gokce N, et al. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res. 2012;53:792–801. https://doi.org/10.1194/jlr.P022905.
Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, et al. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab. 2010;298:E751–60. https://doi.org/10.1152/ajpendo.00745.2009.
Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123:2764–72. https://doi.org/10.1172/JCI67227.
Lindholm CR, Ertel RL, Bauwens JD, Schmuck EG, Mulligan JD, Saupe KW. A high-fat diet decreases AMPK activity in multiple tissues in the absence of hyperglycemia or systemic inflammation in rats. J Physiol Biochem. 2013;69:165–75. https://doi.org/10.1007/s13105-012-0199-2.
Luo T, Nocon A, Fry J, Sherban A, Rui X, Jiang B, et al. AMPK activation by metformin suppresses abnormal extracellular matrix remodeling in adipose tissue and ameliorates insulin resistance in obesity. Diabetes. 2016;65:2295–310. https://doi.org/10.2337/db15-1122.
Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–60. https://doi.org/10.1038/nature07813.
Betz MJ, Enerback S. Human brown adipose tissue: what we have learned so far. Diabetes. 2015;64:2352–60. https://doi.org/10.2337/db15-0146.
Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr. 2011;93:884S-S890. https://doi.org/10.3945/ajcn.110.001917.
Kleiner S, Mepani RJ, Laznik D, Ye L, Jurczak MJ, Jornayvaz FR, et al. Development of insulin resistance in mice lacking PGC-1α in adipose tissues. Proc Natl Acad Sci U S A. 2012;109:9635–40. https://doi.org/10.1073/pnas.1207287109.
Chaurasia B, Kaddai VA, Lancaster GI, Henstridge DC, Sriram S, Galam DLA, et al. Adipocyte ceramides regulate subcutaneous adipose browning, inflammation, and metabolism. Cell Metab. 2016;24:820–34. https://doi.org/10.1016/j.cmet.2016.10.002.
Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011;480:104–8. https://doi.org/10.1038/nature10653.
Chait A, den Hartigh LJ. Adipose tissue distribution, inflammation and its metabolic consequences, including diabetes and cardiovascular disease. Front Cardiovasc Med. 2020;7:22. https://doi.org/10.3389/fcvm.2020.00022.
Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–77. https://doi.org/10.1038/nrm2391.
Louwen F, Ritter A, Kreis NN, Yuan J. Insight into the development of obesity: functional alterations of adipose-derived mesenchymal stem cells. Obes Rev. 2018;19:888–904. https://doi.org/10.1111/obr.12679.
Ghilotti F, Bellocco R, Ye W, Adami HO, Trolle LY. Obesity and risk of infections: results from men and women in the Swedish National March Cohort. Int J Epidemiol. 2019;48:1783–94. https://doi.org/10.1093/ije/dyz129.
Honce R, Schultz-Cherry S. Impact of obesity on influenza A virus pathogenesis, immune response, and evolution. Front Immunol. 2019;10:1071. https://doi.org/10.3389/fimmu.2019.01071.
Donath MY, Dinarello CA, Mandrup-Poulsen T. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat Rev Immunol. 2019;19:734–46. https://doi.org/10.1038/s41577-019-0213-9.
Rodrigues TS, de Sá KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med. 2021;218: e20201707. https://doi.org/10.1084/jem.20201707.
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–72. https://doi.org/10.2337/db06-1491.
Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–9. https://doi.org/10.1038/nature11552.
Delzenne NM, Neyrinck AM, Bäckhed F, Cani PD. Targeting gut microbiota in obesity: effects of prebiotics and probiotics. Nat Rev Endocrinol. 2011;7:639–46. https://doi.org/10.1038/nrendo.2011.126.
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–85. https://doi.org/10.1038/nature10809.
Yamashita A, Soga Y, Iwamoto Y, Asano T, Li Y, Abiko Y, et al. DNA microarray analyses of genes expressed differentially in 3T3-L1 adipocytes co-cultured with murine macrophage cell line RAW 264.7 in the presence of the toll-like receptor 4 ligand bacterial endotoxin. Int J Obese. 2008;32:1725–9. https://doi.org/10.1038/ijo.2008.153.
Yamashita A, Soga Y, Iwamoto Y, Yoshizawa S, Iwata H, Kokeguchi S, et al. Macrophage-adipocyte interaction: marked interleukin-6 production by lipopolysaccharide. Obesity. 2007;15:2549–52. https://doi.org/10.1038/oby.2007.305.
Iwashita M, Sakoda H, Kushiyama A, Fujishiro M, Ohno H, Nakatsu Y, et al. Valsartan, independently of AT1 receptor or PPARγ, suppresses LPS-induced macrophage activation and improves insulin resistance in cocultured adipocytes. Am J Physiol Endocrinol Metab. 2012;302:E286–96. https://doi.org/10.1152/ajpendo.00324.2011.
Nakarai H, Akiko Y, Nagayasu S, Iwashita M, Kumamoto S, Ohyama H, et al. Adipocyte-macrophage interaction may mediate LPS-induced low-grade inflammation: potential link with metabolic complications. Innate Immun. 2012;18:164–70. https://doi.org/10.1177/1753425910393370.
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaozet U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–98. https://doi.org/10.1016/j.cell.2009.09.033.
Zenobia C, Hajishengallis G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol 2000. 2015;69:142–59. https://doi.org/10.1111/prd.12083.
Wang W, Wang X, Lu S, Lv H, Zhao T, Xie G, et al. Metabolic disturbance and Th17/Treg imbalance are associated with progression of gingivitis. Front Immunol. 2021;12: 670178. https://doi.org/10.3389/fimmu.2021.670178.
Shinjo T, Iwashita M, Yamashita A, Sano T, Tsuruta M, Matsunaga H, et al. IL-17A synergistically enhances TNFα-induced IL-6 and CCL20 production in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2016;477:241–6. https://doi.org/10.1016/j.bbrc.2016.06.049.
Sano T, Iwashita M, Nagayasu S, Yamashita A, Shinjo T, Hashikata A, et al. Protection from diet-induced obesity and insulin resistance in mice lacking CCL19-CCR7 signaling. Obesity. 2015;23:1460–71. https://doi.org/10.1002/oby.21127.
• Hayashi M, Iwashita M, Nishimura Y, Shinjo T, Sano T, Yamashita A, et al. Adipose-specific C-C motif chemokine ligand (CCL) 19 overexpression drives the mice to both insulin resistance and weight gain. BMJ Open Diabetes Res Care. 2021;1: e001871. https://doi.org/10.1136/bmjdrc-2020-001871. (It has been demonstrated that activation of the CCL19/CCR7 pathway in adipose tissue induced inflammation and impairment of lipid metabolism and energy regulation in mice.)
López-Cotarelo P, Escribano-Díaz C, González-Bethencourt IL, Gómez-Moreira C, Deguiz ML, Torres-Bacete J, et al. A novel MEK-ERK-AMPK signaling axis controls chemokine receptor CCR7-dependent survival in human mature dendritic cells. J Biol Chem. 2015;290:827–40. https://doi.org/10.1074/jbc.M114.596551.
Kitahara CM, Trabert B, Katki HA, Chaturvedi AK, Kemp TJ, Pinto LA, et al. Body mass index, physical activity, and serum markers of inflammation, immunity, and insulin resistance. Cancer Epidemiol Biomarkers Prev. 2014;23:2840–9. https://doi.org/10.1158/1055-9965.EPI-14-0699-T.
Kochumon S, Al-Rashed F, Abu-Farha M, Devarajan S, Tuomilehto J, Ahmad R, et al. Adipose tissue expression of CCL19 chemokine is positively associated with insulin resistance. Diabetes Metab Res Rev. 2019;35: e3087. https://doi.org/10.1002/dmrr.3087.
Hwang SL, Jeong YT, Li X, Kim YD, Lu Y, Chang YC, et al. Inhibitory cross-talk between the AMPK and ERK pathways mediates endoplasmic reticulum stress-induced insulin resistance in skeletal muscle. Br J Pharmacol. 2013;169:69–81. https://doi.org/10.1111/bph.12124.
Hong S, Song W, Zushin P-JH, Liu B, Jedrychowski MP, Mina AI, et al. Phosphorylation of Beta3 adrenergic receptor at serine 247 by ERK MAP kinase drives lipolysis in obese adipocytes. Mol Metab. 2018;12:25–38. https://doi.org/10.1016/j.molmet.2018.03.012.
Ozaki K-I, Awazu M, Tamiya M, Iwasaki Y, Harada A, Kugisaki S, et al. Targeting the ERK signaling pathway as a potential treatment for insulin resistance and type 2 diabetes. Am J Physiol Endocrinol Metab. 2016;310:E643–51. https://doi.org/10.1152/ajpendo.00445.2015.
Bashan N, Dorfman K, Tarnovscki T, Harman-Boehm I, Liberty IF, Blüher M, et al. Mitogen-activated protein kinases, inhibitory kappaB kinase, and insulin signaling in human omental versus subcutaneous adipose tissue in obesity. Endocrinology. 2007;148:2955–62. https://doi.org/10.1210/en.2006-1369.
Konkel JE, O’Boyle C, Krishnan S. Distal consequences of oral inflammation. Front Immunol. 2019;10:1403. https://doi.org/10.3389/fimmu.2019.01403.
Pucar A, Milasin J, Lekovic V, Vukadinovic M, Ristic M, Putnik S, et al. Correlation between atherosclerosis and periodontal putative pathogenic bacterial infections in coronary and internal mammary arteries. J Periodontol. 2007;78:677–82. https://doi.org/10.1902/jop.2007.060062.
Rafferty B, Jönsson D, Kalachikov S, Demmer RT, Nowygrod R, Elkind MSV, et al. Impact of monocytic cells on recovery of uncultivable bacteria from atherosclerotic lesions. J Intern Med. 2011;270:273–80. https://doi.org/10.1111/j.1365-2796.2011.02373.x.
Kozarov EV, Dorn BR, Shelburne CE, Dunn WA Jr, Progulske-Fox A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler Thromb Vasc Biol. 2005;25:e17–8. https://doi.org/10.1161/01.ATV.0000155018.67835.1a.
Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, Narushima S, et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science. 2017;358:359–65. https://doi.org/10.1126/science.aan4526.
Schirmer M, Denson L, Vlamakis H, Franzosa EA, Thomas S, Gotman NM, et al. Compositional and temporal changes in the gut microbiome of pediatric ulcerative colitis patients are linked to disease course. Cell Host Microbe. 2018;24:600–10. https://doi.org/10.1016/j.chom.2018.09.009.
Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5:eaau3333. https://doi.org/10.1126/sciadv.aau3333.
Farrugia C, Stafford GP, Potempa J, Wilkinson RN, Chen Y, Murdoch C, et al. Mechanisms of vascular damage by systemic dissemination of the oral pathogen Porphyromonas gingivalis. FEBS J. 2021;288:1479–95. https://doi.org/10.1111/febs.15486.
Andrukhov O, Ulm C, Reischl H, Nguyen PQ, Matejka M, Rausch-Fan X. Serum cytokine levels in periodontitis patients in relation to the bacterial load. J Periodontol. 2011;82:885–92. https://doi.org/10.1902/jop.2010.100425.
Gümüş P, Nizam N, Lappin DF, Buduneli N. Saliva and serum levels of B-cell activating factors and tumor necrosis factor-α in patients with periodontitis. J Periodontol. 2014;85:270–80. https://doi.org/10.1902/jop.2013.130117.
Muluke M, Gold T, Kiefhaber K, Al-Sahli A, Celenti R, Jiang H, et al. Diet-induced obesity and its differential impact on periodontal bone loss. J Dent Res. 2016;95:223–9. https://doi.org/10.1177/0022034515609882.
Li Y, Lu Z, Zhang X, Yu H, Kirkwood KL, Lopes-Virella MF, et al. Metabolic syndrome exacerbates inflammation and bone loss in periodontitis. J Dent Res. 2015;94:362–70. https://doi.org/10.1177/0022034514561658.
Mizutani K, Park K, Mima A, Katagiri S, King GL. Obesity-associated gingival vascular inflammation and insulin resistance. J Dent Res. 2014;93:596–601. https://doi.org/10.1177/0022034514532102.
Soares EL, Dos Santos FA, Mroczek T, de Lima DC, Josefino HVB, Scorsin BCM, et al. Caloric restriction prevents alveolar bone loss in the experimental periodontitis in obese rats. Life Sci. 2021;265: 118747. https://doi.org/10.1016/j.lfs.2020.118747.
Sato K, Yamazaki K, Kato T, Nakanishi Y, Tsuzuno T, Yokoji-Takeuchi M, et al. Obesity-related gut microbiota aggravates alveolar bone destruction in experimental periodontitis through elevation of uric acid. mBio. 2021;12:e0077121. https://doi.org/10.1128/mBio.00771-21.
• Sasaki N, Katagiri S, Komazaki R, Watanabe K, Maekawa S, Shiba T, et al. Endotoxemia by Porphyromonas gingivalis Injection Aggravates Non-alcoholic Fatty Liver Disease, Disrupts Glucose/Lipid Metabolism, and Alters Gut Microbiota in Mice. Front Microbiol. 2018;9:2470. https://doi.org/10.3389/fmicb.2018.02470. (It has been shown that P. gingivalis administration induced further weight gain and increased adipose tissue in diet-induced obese mice.)
Rojas C, García MP, Polanco AF, González-Osuna L, Sierra-Cristancho A, Melgar-Rodríguez S, Cafferata EA, et al. Humanized mouse models for the study of periodontitis: an opportunity to elucidate unresolved aspects of its immunopathogenesis and analyze new immunotherapeutic strategies. Front Immunol. 2021;12: 663328. https://doi.org/10.3389/fimmu.2021.663328.
Arimatsu K, Yamada H, Miyazawa H, Minagawa T, Nakajima N, Ryder MI, et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci Rep. 2014;4:4828. https://doi.org/10.1038/srep04828.
Komazaki R, Katagiri S, Takahashi H, Maekawa S, Shiba T, Takeuchi Y, et al. Periodontal pathogenic bacteria, Aggregatibacter actinomycetemcomitans affect non-alcoholic fatty liver disease by altering gut microbiota and glucose metabolism. Sci Rep. 2017;7:13950. https://doi.org/10.1038/s41598-017-14260-9.
Vallim AC, Gaio EJ, Oppermann RV, Rösing CK, Albandar JM, Susin C, et al. Obesity as a risk factor for tooth loss over 5 years: a population-based cohort study. J Clin Periodontol. 2021;48:14–23. https://doi.org/10.1111/jcpe.13378.
• Suvan J, Harrington Z, Petrie A, Patel K, Darbar U, Donos N, et al. Obesity as predictive factor of periodontal therapy clinical outcomes: a cohort study. J Clin Periodontol. 2020;47:594–601. https://doi.org/10.1111/jcpe.13261. (A cohort study showed that periodontal conditions of obese individuals are significantly worse 2 and 6 months after treatment compared with those of non-obese individuals. The findings indicated that obesity is a predictive risk factor for periodontal therapy.)
Munenaga Y, Hiroshima Study Group; Yamashina T, Tanaka J, Nishimura F. Improvement of glycated hemoglobin in Japanese subjects with type 2 diabetes by resolution of periodontal inflammation using adjunct topical antibiotics: results from the Hiroshima Study. Diabetes Res Clin Pract. 2013;100:53–60. https://doi.org/10.1016/j.diabres.2013.01.028.
Slade GD, Ghezzi EM, Heiss G, Beck JD, Riche E, Offenbacher S. Relationship between periodontal disease and C-reactive protein among adults in the Atherosclerosis Risk in Communities study. Arch Intern Med. 2003;163:1172–9. https://doi.org/10.1001/archinte.163.10.1172.
Nesse W, Abbas F, van der Ploeg I, Spijkervet FK, Dijkstra PU, Vissink A. Periodontal inflamed surface area: quantifying inflammatory burden. J Clin Periodontol. 2008;35:668–73. https://doi.org/10.1111/j.1600-051X.2008.01249.x.
Nesse W, Linde A, Abbas F, Spijkervet FK, Dijkstra PU, de Brabander ED, et al. Dose-response relationship between periodontal inflamed surface area and HbA1c in type 2 diabetics. J Clin Periodontol. 2009;36:295–300. https://doi.org/10.1111/j.1600-051X.2009.01377.x.
Takeda K, Mizutani K, Minami I, Kido D, Mikami R, Konuma K, et al. PISA and PESA were not significantly associated with BMI or visceral fat area (VFA) in patients with type 2 diabetes. BMJ Open Diabetes Res Care. 2021;1: e002139. https://doi.org/10.1136/bmjdrc-2021-002139.
Romano F, Perotto S, Mohamed SEO, Bernardi S, Giraudi M, Caropreso P, et al. Bidirectional association between metabolic control in type-2 diabetes mellitus and periodontitis inflammatory burden: a cross-sectional study in an italian population. J Clin Med. 2021;10:1787. https://doi.org/10.3390/jcm10081787.
Aoyama N, Fujii T, Kida S, Nozawa I, Taniguchi K, Fujiwara M, et al. Association of periodontal status, number of teeth, and obesity: a cross-sectional study in Japan. J Clin Med. 2021;10:208. https://doi.org/10.3390/jcm10020208.
Charupinijkul A, Arunyanak S, Rattanasiri S, Vathesatogkit P, Thienpramuk L, Lertpimonchai A. The effect of obesity on periodontitis progression: the 10-year retrospective cohort study. Clin Oral Investig. 2021. https://doi.org/10.1007/s00784-021-04031-2.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Oral Disease and Nutrition
Rights and permissions
About this article

Cite this article
Iwashita, M., Hayashi, M., Nishimura, Y. et al. The Link Between Periodontal Inflammation and Obesity. Curr Oral Health Rep 8, 76–83 (2021). https://doi.org/10.1007/s40496-021-00296-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40496-021-00296-4