
Abstract
The genetic diversity and relationship was studied in a collection of 65 Brassica accessions, which included 54 B. rapa and 11 of five other Brassica species used as references. These 54 B. rapa accessions included 42 Chinese accessions and 12 exotic accessions. All accessions were analyzed by using 36 simple sequence repeats (SSR) and 43 sequence-related amplified polymorphism (SRAP) primers, and 401 polymorphic fragments were detected by SSR and SRAP markers. The average number of polymorphic fragments detected by SRAP markers was 6.23 ranging from 2 to 11 and that revealed by SSR was 3.69 ranging from 2 to 7. The unweighted pair-group method with arithmetic mean cluster analysis indicated that all accessions could be divided into five major clusters except three accessions as outliers. The exotic B. rapa accessions appeared in Cluster I except one yellow sarson accession from India, Chinese B. rapa revealed in Cluster II, and 11 accessions of other Brassica expressed them Cluster III, Cluster IV, and Cluster V. The results of principal component analysis and population structure analysis were in accordance with the cluster analysis. Molecular variance analysis revealed that the genetic variation was 26.10% among populations and 73.90% within Brassica species, which indicated existence of considerable genetic variation among exotic and Chinese B. rapa species, and exotic B. rapa can be used for broadening the genetic background of Chinese B. napus, and vice versa.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genus Brassica includes three basic diploid species Brassica rapa L. (AA genome, n = 10), B. oleracea L. (CC genome, n = 9), B. nigra L. (BB genome, n = 8), and three compound species B. napus L. (AACC genome, n = 19), B. juncea (L.) Czern & Coss (AABB genome, n = 18), and B. carinata A. Braun (BBCC genome, n = 17) (U N 1935). B. rapa is one of the most economically important oilseed crops worldwide, and it is also grown as a major vegetable and fodder crop (Prakash and Hinata 1980; Liu 1984). The oleiferous form of B. rapa, namely B. rapa subsp. oleifera (turnip rape) is the third most important Brassica oilseed crop, and it is grown in China, Canada, India, and Northern Europe (Ramchiary and Lim 2011). A large number of subspecies in B. rapa are recognized as oilseed type which are distributed in Europe, Central Asia, and India, and leaf vegetable type is generally spread in East Asia (Song et al. 1988; Zhao et al. 2005). All the subspecies in B. rapa have a wide range of variation and genetic diversity (McGrath and Quiros 1992; Zhao et al. 2005; Takuno et al. 2007; Warwick et al. 2008; Fu and Gugel 2009; Annisa and Cowling 2013). B. rapa has broad geographical distribution than B. napus and is widely distributed throughout Europe and East to South Asia. The classical Old World is one of center of origin of B. rapa, with several centers of diversity in East Asia owing to ancient trade routes in Asia and recent migration to the New World (Song et al. 1990; Guo et al. 2014).
Due to extensive era of domestication, natural selection and breeding, B. rapa possesses more desirable characters for instance, drought resistance, cold resistance, heat tolerance, early maturity, and many other characteristics (He et al. 2002; Chen et al. 2010). B. napus is an allopolyploid species that resulted from interspecific hybridization between B. rapa (A genome) and B. oleracea (C genome) (Jesske et al. 2013). Nowadays, B. napus is grown worldwide for its high yield and disease resistance. The availability of plentiful germplasm and great genetic diversity makes B. rapa the ideal prospect for improvement of B. napus (Zhao et al. 2005; Liu and Meng 2006). Therefore, new alleles can be easily transferred into B. napus genome for broadening the gene pool and increasing genetic diversity of B. napus for future breeding programs. Rapeseed breeders in China have used indigenous B. rapa and exotic B. napus introductions in breeding programs and have developed semi-winter B. napus varieties adapted to local conditions (Liu 1984; Qian et al. 2006; Li et al. 2013). However, there are no reports about using the foreign B. rapa accessions to widening the genetic background of Chinese B. napus.
Evaluation of genetic diversity and relationship among Chinese and exotic B. rapa has significant implications for genetic improvement and incorporation of novel alleles from the B. rapa gene pool into B. napus (Liu and Meng 2006; Allender and King 2010). Various molecular marker’s approaches have been employed to investigate the genetic diversity in B. rapa accessions worldwide. Zhao and Becker (1998) used isozyme markers to analyze genetic variation among 12 Chinese and 24 European oilseed B. rapa accessions. Their results revealed that existence of two clear-cut groups of Chinese and European origin in B. rapa. Zhao et al. (2005, 2007) used amplified fragment length polymorphism (AFLP) markers to discover genetic diversity of 161 B. rapa including traditional and modern cultivars, and breeding materials from different geographical locations of the world (10 of 27 B. rapa subsp. oleifera used from China), and 96 accessions of leafy vegetable types cultivated in China. Their results revealed that different morphotypes (oilseed, root, or leafy types) are often more related to other morphotypes from the same region (East Asia vs. Europe) than similar morphotypes from different regions, suggesting either an independent origin in both regions and/or a long and separate domestication and breeding history in both regions. Tanhuanpää et al. (2016) analyzed genetic diversity in 61 accessions of B. rapa using single nucleotide polymorphism (SNP) markers and reported great variation in the diversity indices between accessions and divided them into three groups that correspond to morphotypes and flowering habit but not to geography. Pino Del Carpio et al. (2011) used phenotypic, metabolic and molecular markers to assess 168 accessions of B. rapa and concluded that metabolic data provided a similar classification as the genotypic AFLP data (Zhao et al. 2005, 2007). Warwick et al. (2008) evaluated patterns and levels of genetic diversity in 93 Canadian spring turnip rape accessions by AFLP markers and revealed three main clusters corresponding to European (I), Indian (III), and a mixed Asian/European/Indian (II) proposed origins of the taxa. Fu and Gugel (2009) investigated genetic variability of Canadian elite cultivars of summer turnip rape by simple sequence repeat (SSR) markers. They discovered two distinct groups of nine cultivars separated by high vs. low glucosinates content. Annisa and Cowling (2013) studied genetic diversity in a global collection of 164 oilseed B. rapa accessions (including only one Chinese B. rapa accession) using SSR markers, in context with the cytological and morphological methods. On the basis of SSR data, their results indicated three distinct groups belonging to South Asia, Southern Europe, and Northern Europe. Guo et al. (2014) studied the genetic diversity in a global collection of 173 B. rapa accessions by using SSR markers, and reported three molecular genetic groups, one from the classical Old World (Europe and West Asia–North Africa), second from East Asia, and third from East, Central, South, and Southeast Asia. Takahashi et al. (2016) proposed that Central Asia is the sole geographical origin of turnips and most of Japanese turnips were derived from Central Asia.
Brassica rapa also showed cytoplasmic diversity based on PCR products using primers specific to mitochondrial genomes or plasmids (Zhang et al. 2013) and chloroplast SSR markers (Zamani-Nour et al. 2013). All above studies suggested that B. rapa is a genetically diverse species in both genomic and cytoplasmic DNA. However, detailed research on comparison of the genetic diversity and genetic relationship between Chinese and exotic B. rapa has not been carried out by combining SSR and sequence-related amplified polymorphism (SRAP) technology. Therefore, the present study was designed for investigation of genetic diversity among Chinese and some exotic oilseed B. rapa accessions for providing significant insights for oilseed B. rapa and B. napus breeding.
The aims of the present study were (1) to characterize the levels and patterns of genetic diversity among a set of Chinese and some exotic B. rapa accessions and (2) to compare the diversity between Chinese and the exotic B. rapa accessions.
Materials and methods
Plant samples
– A collection of 65 accessions including 42 Chinese and 12 exotic B. rapa along with three B. napus, two each of B. juncea, B. carinata, B. nigra, and B. oleracea were analyzed in this study. They contained 41 accessions of B. rapa subsp. oleifera (turnip rape), eight B. rapa subsp. chinensis, three B. rapa subsp. pekinensis, one each of B. rapa subsp. chinensis var. tai-tsai Lin and ssp. trilocularis (Table 1). Eleven other Brassica accessions of B. napus, B. juncea, B. carinata, B. nigra, and B. oleracea were included as reference. All these accessions were planted in the experimental field of Northwest A&F University at Yangling, Shaanxi, PR China, on September 20, 2013.
Genomic DNA extraction
– Young leaves of 15 plants of each accession were randomly collected from plants grown in the field and ground in liquid nitrogen. Total genomic DNA was isolated using the cetyltrimethylammonium bromide (CTAB) method as described previously (Murray and Thompson 1980). The DNA pellet was dissolved in 100 μL TE buffer and detected for integrity and quality on 0.8% agarose gel. The concentration of the DNA samples was calculated by using Epoch Microplate Spectrophotometer (Epoch, USA), and the final dilution was done before use at 100 ng µL−1.
Sequence-related amplified polymorphism (SRAP) analysis
– Forty-three pairs of SRAP primers (Table S1) were chosen to analyze the tested accessions because of their superior polymorphism shown in previous work of our laboratory. These 43 selected SRAP primer combinations were synthesized by BioAsia (Shanghai, China). PCR reactions were performed in a 10 μL volume containing 2 μL DNA template, 0.6 μL (10 μM) of each primer, 5 μL PCR MIX containing 1 × PCR buffer, 500 µM dNTP each, 20 mM Tris-HCL (pH 8.3), 100 mM KCl, 3 mM MgCl2; Golden DNA Polymerase; 2.5 U µl−1 (Tiangen Biotech Co., Ltd, Beijing, China), and sterile distilled water. The amplification program was performed in C1000 thermal cycler (Bio-Rad Co. Ltd. America) following these steps: 5 min at 94 °C; 30 s at 94 °C, 60 s at 56 °C, 45 s at 72 °C, 40 cycles, and a final extension at 72 °C for 5 min. Polymorphism at each locus was assessed by electrophoresis of PCR products on 8% polyacrylamide (w/v) gel in 1 × Tris–borate–ethylenediaminetetraacetic acid (EDTA) (TBE) and visualized by silver staining.
Simple sequence repeats (SSR) analysis
– SSR analysis was performed by PCR with 36 pairs of SSR primers (Table S2). The primers were selected for high polymorphism and easy to score quality. The SSR reaction system is same as described for SRAP experiment. The PCR reaction was performed as follows: 1 min at 95 °C; 1 min at 94 °C, 1 min at 35 °C, and 1 min at 72 °C for 5 cycles; 1 min at 94 °C, 1 min at 50 °C, and 1 min at 72 °C for 34 cycles, and 7 min incubation at 72 °C.
All the experiments for SRAP and SSR were carried out for twice, and only the reproducible bands for each sample were recorded in both replications.
Data collection and analysis
– In the SRAP molecular analysis only strong, reproducible and clearly distinguished polymorphic fragments among all accessions were collected and used in the data analysis. The profile data produced by SRAP markers were scored manually for each fragment in every accession for each primer pair and recorded as 1 representing the presence of a fragment, 0 representing the absence of a fragment, and 9 as the missing of a fragment. SSR results were analyzed by loci, and alleles were detected for each locus. For jointly analyzing the data of SRAP and SSR, we also used the same scores as 1, 0, and 9 to record the SSR results as to record SRAP data (scoring of a representative gel of SSR is shown in Table S3). The following formula was used to calculate the polymorphic information content (PIC) per marker: \( {\text{PIC}} = 1 - \sum\limits_{j = 1}^{n} {P_{ij}^{2} } \), where P ij is the frequency of the jth allele of the ith marker locus and n is the total number of alleles.
The data were analyzed using the qualitative routine to generate simple matching coefficients (SMC), calculated as SMC = a −1(n − d), where a is the number of fragments in common between two accessions, n is the number of fragments in the matrix, and d is the number of fragments absent in both accessions (Sokal and Michener 1958). SMC was used to construct a dendrogram by the unweighted pair-group method with arithmetic average (UPGMA) and the sequential, hierarchical, and nested clustering (SHAN) routine in the NTSYS-pc2.10 program (Rohlf 1998). The principal component analysis (PCA) was done by first calculating a correlation matrix among markers. Eigenvalues and eigenvectors were then obtained from the correlation matrix and were used to calculate the coordinates of each accession. The 0, 1 matrix of SRAP and SSR markers was also used for population structure analysis by Structure version 2.3.4 (Pritchard et al. 2000). All markers were treated as dominant markers, so the recessive allele model was used. The admixture model was used in the analysis. Length of burn-in period before the start of data collection and number of Markov Chain Monte Carlo analysis repeats after burn-in were both set at 10,000. The number of groups (K) ranged from 1 to 10. For the analysis of molecular variance (AMOVA), all accessions were classified into three groups, Chinese B. rapa, exotic B. rapa, and other Brassica. The components of variance attributable to different varieties and breeding lines were estimated from the genetic distance matrix, as specified in the AMOVA procedure in ARLEQUIN version 2.000. A nonparametric permutation procedure with 3000 permutations was used to test the significance of variance components associated with the different possible levels of genetic structure in this study (Excoffier et al. 1992). The pairwise F st values, a value of F statistic analogs computed from AMOVA, were used to compare genetic distances between any two groups.
Results
Marker polymorphism
– A total of 401 polymorphic bands were detected by SSR and SRAP primer pairs in the all 65 accessions presented in Table 2. The average number of polymorphic fragments detected by SRAP markers was 6.23 ranging from 2 to 11, whereas that for SSR was 3.69 ranging from 2 to 7 (Table S1 and Table S2). The maximum number of 11 polymorphic bands was detected by SRAP primer pair Em14Me32, while in case of SSR molecular marker maximum number of 7 bands was revealed by primer pair BrGMS635. The average PIC value 0.61 detected by SRAP markers was higher than that of SSR markers 0.42. Genetic distance varied from 0.0261 to 0.5112 and 0.0538 to 0.5600 for SRAP and SSR analysis, respectively (Table 2).
Cluster and principal component analysis
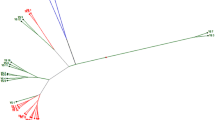
– The molecular marker data of 401 SRAP and SSR polymorphic bands were jointly used to calculate genetic similarity coefficient for all accessions, and a dendogram was constructed from these similarity coefficient data with the UPGMA method (Fig. 1). Genetic similarity estimates varied from 0.65 to 0.97 for all 65 accessions. The 65 accessions were divided into five major clusters at the coefficient value of 0.733, and three accessions (No. 30, 44, and 42) were revealed as outliers. Cluster I contained all exotic accessions of B. rapa subsp. oleifera except accession No. 42, B. rapa subsp. trilocularis (yellow sarson) from India. Cluster II contained 38 B. rapa accessions from China. The Chinese accessions in the cluster II could be subdivided into some small groups. Three B. rapa subsp. pekinensis accessions (No. 4, 26, and 33) were clustered together into a small group. Eight B. rapa subsp. chinensis (No. 17, 18, 22, 27, 28, 29, 30, and 36) was scattered relative widely in some small groups of the cluster II, indicating that this subspecies has a relatively larger variation. Cluster III included two each of B. juncea and B. nigra accessions. Cluster IV consisted five accessions, three (No. 55, 56, and 57) B. napus, and two (No. 37, 41) B. rapa accessions. Cluster V contained four (No. 60, 61, 64, and 65) accessions, two each of B. carinata and B. oleracea, respectively. Most Brassica species inclined to cluster within their own varietal groups in the cluster tree. However, two B. rapa accessions from China “YongshouhuaipingYoucai” (No. 37) and “LinyoucuimubanpoYoucai” (No. 41) unexpectedly clustered together with B. napus accessions in cluster IV.

Clustering of 65 Brassica accessions by unweighted pair-group method with arithmetic mean method with SMC index. Brief results of population structure analysis also showed in the right part in comparison with the cluster analysis
The principal component analysis (PCA) result was similar to the cluster analysis (Fig. 2). The first two principal components accounted for 15.80 and 6.79% of the total variation, respectively. All 11 other Brassica accessions scattered widely from the B. rapa accessions. PCA results divided all B. rapa accessions into two major groups, Chinese origin and exotic B. rapa, except two accessions No. 37 and 41.

Biplot of the first two major principal components extracted from SRAP and SSR data
Population structure analysis
– The break point of the estimated Ln probability of data [Ln P (D)] was obtained when K = 5, suggesting the presence of five groups, GI-GV (Fig. 3). Group I contained four accessions, two of each B. carinata, B. oleracea, Group II included five accessions, three of B. napus together with two B. rapa accessions, Group III included four accessions, two of each B. juncea and B. nigra, respectively, Group IV formed by 41 accessions of B. rapa from China. Group V contained 11 exotic B. rapa accessions. In general, the population structure analysis demonstrated highly accordance with cluster analysis and PCA results.

Population structure of the tested Brassica accessions suggested by structure analysis (K = 5). The dominant five colors represent five inferred groups (GI-GV). Each bar represents each accession. The estimated genetic fraction of each accession of each inferred group was indicated in different colors. The numbers under each bar is the same accession numbers in Table 1. (Color figure online)
Analysis of molecular variance
– On the basis of Brassica species and their geographical origin, all the 65 accessions used in this study were classified into three groups, exotic B. rapa, Chinese B. rapa, and other Brassicas, for analysis of molecular variance (AMOVA). The AMOVA results indicated that 26.10% of the total variation was due to differences among populations and 73.90% variation was due to difference within populations (Table 3). The pairwise F st values of three groups were all significant (Table 4).
Discussion
B. rapa is both an economically and agriculturally important crop with wide geographical ranges (Prakash and Hinata 1980; Liu 1984). Various studies have been conducted to explore the genetic diversity in B. rapa (McGrath and Quiros 1992; Zhao et al. 2005; Takuno et al. 2007; Warwick et al. 2008; Fu and Gugel 2009; Annisa and Cowling 2013). The previous studies suggested that the classical Old World is the center of origin of B. rapa, with centers of diversity in East Asia along with ancient trade routes in Asia, and recent migration to the New World (Song et al. 1990; Zhao et al. 2005; Guo et al. 2014). In the present investigation, with the combination of SSR and SRAP data, the 65 Brassica accessions tested were divided into five major clusters based on the results of cluster, PCA, and structure analysis. Eleven other Brassica accessions were separated from B. rapa accessions and formed three clusters (Cluster III, Cluster IV, and Cluster V). The majority of the tested B. rapa accessions appeared into two main clusters (Cluster I and Cluster II). Cluster I contained 11 of 12 exotic accessions, with only one exotic accession No. 42, yellow sarson with Indian origin not revealed in this cluster. Cluster II contained 38 B. rapa accessions from China. According to our expectations, exotic and indigenous Chinese B. rapa used in this study were clearly distinguished from each other. It can be concluded that genetic relationships among accessions had the tendency to associate with their geographical origins. Our results are in accordance with previous studies on that the cluster of B. rapa was aligned with geographical origin (Zhao et al. 2005, 2007; Takuno et al. 2007; Warwick et al. 2008; Annisa and Cowling 2013). We supported that Europe and East Asia are two centers of diversity of B. rapa (Song et al. 1990; Zhao et al. 2005; Guo et al. 2014). Brassica accessions also gathered based on their own species. However, two B. rapa accessions (No. 37, 41) from China appeared out of its own group and joined B. napus group, unexpectedly. These two accessions were collected in the border area between B. napus and B. rapa cultivated region in Shaanxi, China, and B. rapa is readily crossed with B. napus to form viable progenies, perhaps these two accessions were the natural hybrid of B. napus and B. rapa, and their genetic makeup were more close to B. napus.
To measure the source and percentage of variation in B. rapa, we divided the tested Brassica accessions into three groups: domestic B. rapa, exotic B. rapa, and other Brassica in AMOVA analysis. The results of AMOVA showed that 73.90% of the total genetic variation was found within the groups, and 26.10% among groups, which is in accordance with the previous results (Takuno et al. 2007; Ofori et al. 2008; Zhao et al. 2009; Pino del Caprio et al. 2011; Annisa and Cowling 2013) and is probably due to high rates of cross-pollination of the species. Although the variance among groups is relative low compared with genetic variance within groups, it confirmed that certain genetic variation exists between exotic and Chinese B. rapa tested in the present study. Therefore, exchange of the genes between exotic and Chinese B. rapa by hybridization will be more beneficial for Brassica species in future breeding programs.
The Chinese B. rapa accessions displayed in the cluster II could be divided into some subgroups. Three B. rapa subsp. pekinensis and eight B. rapa subsp. chinensis accessions were appeared separately within Cluster II; this was not in agreement with the previous results (He et al. 2002; Zhao et al. 2005). They reported separation of leafy B. rapa from oilseed B. rapa. Our results supported the view that the oilseed B. rapa was selected from root or leafy domesticated types in the Old World and East Asia (Reiner et al. 1995; Gómez-Campo and Prakash 1999). Since the number of exotic accession is limited, we cannot reach a conclusive remark about the relationship of the exotic B. rapa in the present study. However, there existed considerable genetic difference between Chinese and exotic B. rapa tested in the present study as shown by 26.10% genetic variation between them revealed by AMOVA analysis. In the present study, three accessions (No. 30, 42, and 44) of B. rapa were found to be genetically distinct from other B. rapa, and among them, No. 42 was yellow sarson from India. Annisa and Cowling (2013) reported that the 74 Indian oilseed B. rapa in their experiment formed a unique cluster, which is distinguished from the European oilseed B. rapa. Hence, it would be desirable to put these genotypes in hybridization program to broaden the genetic diversity of the B. napus and to further improve the breeding work of the latter.
References
Allender CJ, King GJ (2010) Origins of the amphidiploid species Brassica napus L. investigated by chloroplast and nuclear molecular markers. BMC Plant Biol 10:54
Annisa CS, Cowling WA (2013) Global genetic diversity in oilseed Brassica rapa. Crop Pasture Sci 64:993–1007
Chen S, Zou J, Cowling WA, Meng J (2010) Allelic diversity in a novel gene pool of canola-quality Brassica napus enriched with alleles from B. rapa and B carinata. Crop Pasture Sci 61:483–492
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Fu YB, Gugel RK (2009) Genetic variability of Canadian elite cultivars of summer turnip rape Brassica rapa revealed by simple sequence repeat markers. Can J Plant Sci 89:865–874
Gómez-Campo C, Prakash S (1999) Origin and domestication. In: Gómez-Campo C (ed) Developments in plant genetics and breeding: biology of Brassica coeno species. Elsevier, Burlington, pp 33–58
Guo YM, Chen S, Li ZY, Cowling WA (2014) Center of origin and centers of diversity in an ancient crop, Brassica rapa (Turnip Rape). J Hered 105:555–565
He YT, Tu JX, Fu TD, Li DR, Chen BY (2002) Genetic diversity of germplasm resources of (Brassica campestris L.) in China by RAPD markers. Acta Agron Sin 28:693–703
Jesske T, Olberg B, Schierholt A, Becker HC (2013) Resynthesized lines from domesticated and wild Brassica taxa and their hybrids with B. napus L. genetic diversity and hybrid yield. Theor Appl Genet 126:1053–1065. doi:10.1007/s00122-012-2036-y
Li Q, Mei J, Zhang Y, Li J, Ge X, Li Z, Qian W (2013) A large-scale introgression of genomic components of Brassica rapa into B. napus by the bridge of hexaploid derived from hybridization between B. napus and B. oleracea. Theor Appl Genet 126:2073–2080. doi:10.1007/s00122-013-2119-4
Liu HL (1984) Origin and evolution of rapeseeds. Acta Agron Sin 10:9–18
Liu RH, Meng JL (2006) RFLP and AFLP analysis of inter and intra-specific variation of Brassica rapa and B. napus shows that B. rapa is an important genetic resource for B. napus improvement. Acta Gene Sin 33:814–823
McGrath JM, Quiros CE (1992) Genetic diversity at isozyme and RFLP loci in Brassica campestris as related to crop type and geographical origin. Theor Appl Genet 83:783–790
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucl Acids Res 8:4321–4325
Ofori A, Becker HC, Kopisch-Obuch FJ (2008) Effect of crop improvement on genetic diversity in oilseed Brassica rapa (turnip-rape) cultivars, detected by SSR markers. J Appl Genet 49:207–212
Pino Del Carpio D, Basnet R, De Vos R, Maliepaard C, Visser R, Bonnema G (2011) The patterns of population differentiation in a Brassica rapa core collection. Theor Appl Genet 122:1105–1118
Prakash S, Hinata K (1980) Taxonomy, cytogenetics and origin of crop Brassicas, a review. Opera Bot 55:1–57
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Qian W, Meng J, Li M, Frauen M, Sass O, Noack J, Jung C (2006) Introgression of genomic components from Chinese Brassica rapa contributes to widening the genetic diversity in rapeseed (B. napus L.) with emphasis on the evolution of Chinese rapeseed. Theor Appl Genet 113:49–54
Ramchiary N, Lim YP (2011) Genetics of Brassica rapa L. In: Schmidt R, Bancroft I (eds) Genetics and genomics of the Brassicaceae. Springer, New York, pp 215–260
Reiner H, Holzner W, Ebermann R (1995) The development of turnip-type and oilseed-type Brassica rapa crops from the wild type in Europe-an overview of botanical, historical and linguistic facts. In: Rapeseed today and tomorrow, 9th International Rapeseed Congress, Cambridge, UK, pp 1066–1069
Rohlf FJ (1998) Numerical taxonomy and multivariate analysis system. Version 2.0. Exeter Software, Setauket, NY
Sokal RR, Michener CD (1958) A statistical method for evaluating systematic relationships. Univ Kans Sci Bull 38:1409–1438
Song KM, Osborn TC, Williams PH (1988) Brassica taxonomy based on nuclear restriction fragment length polymorphisms (RFLPs): 2. preliminary analysis of subspecies within B. rapa syn. campestris and B. oleracea. Theor Appl Genet 76:593–600
Song K, Osborn TC, Williams PH (1990) Brassica taxonomy based on nuclear restriction fragment length polymorphisms (RFLPs) 3. Genome relationships in Brassica and related genera and the origin of B. oleracea and B. rapa (syn. Campestris). Theor Appl Genet 79:497–506
Takahashi Y, Yokoi S, Takahata Y (2016) Genetic divergence of turnip (Brassica rapa L. em. Metzg. subsp. rapa) inferred from simple sequence repeats in chloroplast and nuclear genomes and morphology. Genetic Resour Crop Evol 63:869–879
Takuno S, Kawakara T, Ohnishi O (2007) Phylogenetic relationships among cultivated types of Brassica rapa L. em. Metzg.as revealed by AFLP analysis. Genet Resour Crop Evol 54:279–285
Tanhuanpää P, Erkkilä M, Tenhola-Roininen T, Tanskanen J, Manninen O (2016) SNP diversity within and among Brassica rapa accessions reveals no geographic differentiation. Genome 59:11–21. doi:10.1139/gen-2015-0118
U N (1935) Genomic analysis of Brassica with special reference to the experimental formation of B. napus and its peculiar mode of fertilization. Jpn J Bot 7:389–452
Warwick SI, James T, Falk KC (2008) AFLP-based molecular characterization of Brassica rapa and diversity in Canadian spring turnip rape cultivars. Plant Gene Resour 6:11–21
Zamani-Nour S, Clemens R, Möllers C (2013) Cytoplasmic diversity of Brassica napus L., Brassica oleracea L. and Brassica rapa L. as determined by chloroplast microsatellite markers. Genet Resour Crop Evol 60:953–965
Zhang RJ, Hu SW, Yan JQ, Sun GL (2013) Cytoplasmic diversity in Brassica rapa L. investigated by mitochondrial markers. Genet Resour Crop Evol 60:967–974
Zhao JY, Becker HC (1998) Genetic variation in Chinese and European oilseed rapa (B. napus) and Turnip rapa (B. campestris) analysed with isozymes. Acta Agron Sin 24:213–220
Zhao JJ, Wang XW, Deng B, Lou P, Wu J, Sun RF, Xu ZY, Vromans J, Koornneef M, Bonnema G (2005) Genetic relationships within Brassica rapa as inferred from AFLP fingerprints. Theor Appl Genet 110:1301–1314. doi:10.1007/s00122-005-1967-y
Zhao JJ, Paulo MJ, Jamar D, Lou P, Eeuwijk FV, Bonnema G, Vreugdenhil D, Koornneef M (2007) Association mapping of leaf traits, flowering time, and phytate content in Brassica rapa. Genome 50:963–973
Zhao YG, Ofori A, Lu CM (2009) Genetic diversity of European and Chinese oilseed Brassica rapa cultivars from different breeding periods. Agric Sci China 8:31–938
Acknowledgements
This work was supported by the earmarked fund for China Agriculture Research System (CARS-13), National Key Technology R&D Program (2010BAD01B02), and a grant of Northwest A&F University for S.W. Hu.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
Information of 43 pairs of SRAP primers (DOCX 23 kb)
Table S2
Information of 36 pairs of SSR primers (DOCX 21 kb)
Table S3
Scoring of a representative gel of SSR with primer BrGMS126 and accession no. in Table S3 correspond to Table 1 (XLS 547 kb)
Rights and permissions
About this article

Cite this article
Zhang, X., Chen, H., Channa, S.A. et al. Genetic diversity in Chinese and exotic Brassica rapa L. accessions revealed by SSR and SRAP markers. Braz. J. Bot 40, 973–982 (2017). https://doi.org/10.1007/s40415-017-0392-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40415-017-0392-1