
Abstract
Express sequence tag (EST)-simple sequence repeat (SSR) was employed to assess the genetic diversity in elite germplasm collections of purslane (Portulaca oleracea L.) which is a saline and drought tolerant annual herb containing high amount of vitamins, minerals, and antioxidant properties. The 10 primers gave a total of ninety five bands among which 95 were polymorphic (100 %). The genetic diversity as estimated by Shannon’s information index was 1.85, revealing a high level of genetic diversity in the germplasm. The average numbers of observed allele, effective allele, expected heterozygosity, polymorphic information content (PIC), and Nei’s information index were 9.5, 5.14, 0.81, 0.33, 0.77, and 0.79, respectively. The UPGMA dendrogram based on Nei’s genetic distance grouped the whole germplasm into eight distinct clusters. The analysis of molecular variance (AMOVA) revealed that 71.87 % of total variation occurred within population while 28.13 % from among populations. Considering all these criteria and results from marker-assisted diversity analysis, accessions that are far apart based on their genetic coefficient (like Ac11 and Ac17; Ac4 and Ac42; Ac16 and Ac31; Ac38 and Ac17; Ac21 and Ac31; Ac30 and Ac10) could be selected as parents for further breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Purslane (Portulaca oleracea L.) is listed in the World Health Organization as one of the most used medicinal plants and it has been given the term “Global Panacea” (Dweck 2001; Samy et al. 2004). Purslane has recently been identified as the richest vegetable source of α-linolenic acid, an essential omega-3 and six fatty acids (Simopoulos et al. 2005), which is five times higher than spinach (Uddin et al. 2014). The lack of dietary sources of omega-3 and six fatty acids has resulted in a growing level of interest to introduce P. oleracea as a new cultivated vegetable (Yazici et al. 2007). Scientifically, purslane provides a rich plant source of nutritional benefits with high antioxidant properties (Alam et al. 2014b). P. oleracea is not only high in nutritional value but is also highly adaptable to many different environments (Alam et al. 2014c, d). Uddin et al. (2012) showed that P. oleracea flourishes in numerous biogeographical locations worldwide and is highly adaptable to many drought, saline, and nutrient-deficient conditions. These characteristics give P. oleracea a competitive advantage over many other cultivated crops and have led some to consider P. oleracea as the ‘power food for the future’ (Simopoulos et al. 1995).
Many research findings have been published worldwide regarding nutritional quality, medicinal properties, drought, and salinity effect on morpho-physiological attributes. but research on genetic diversity among diverse cultivars and collected germplasms are scanty. Availability of depth information on the genetic diversity and relationship among available germplasms are vital before initiating any plant breeding program. Genetic diversity in crop species can be determined using morphological (phenotypic), biochemical, and molecular (DNA) markers (Rao 2004). However, the morphological markers are strongly affected by environmental factors compared to biochemical markers and therefore are dependent on environmental conditions during cultivation. The limitations associated with morphological and biochemical markers are overcome by molecular markers (Rao 2004). Molecular diversity assessed using molecular markers is independent of the influence of environment and estimated using DNA from any growth stage (Tatikonda et al. 2009). Among the various molecular markers, polymerase chain reaction (PCR)-based markers, such as random amplified polymorphic DNA (RAPD; Williams et al. 1990), inter-simple-sequence-repeat (ISSR; Zietkiewiez et al. 1994), amplified fragment-length polymorphism (AFLP; Vos et al. 1995), and sequence-related amplified polymorphism (SRAP; Li and Quiros 2001) have become popular for genetic diversity analyses; their application does not require any prior sequence information. However, DNA microsatellites, or SSR markers often used for genetic analysis, are excellent genetic markers because they are hyper variable, co-dominant, and therefore highly informative (Du et al. 2012; Blair et al. 2010). On the other hand, express sequence tag (EST)-derived microsatellites are such type of markers which are easily obtained by electronic search of EST databases. EST-microsatellite (eSSR) polymorphism is associated with transcribed regions of the genome and reflects the genetic diversity inside or adjacent to the genes (Varshney et al. 2005), which may be functionally more informative than genomic SSR (gSSR) that are more widely used (Hu et al. 2011).
In purslane, AFLP-based drought tolerance and diversity analysis of ten collected accessions from six countries are the first and only available reports (Ren et al. 2011). Whereas, Lokhande et al. (2009) described the genetic diversity analysis of 14 collected clones of sea purslane (Sesuvium portulacastrum L.) using RAPD markers. On the other hand, development of EST-SSR primers to assess genetic diversity has been reported in many crops, e.g., in celery (Fu et al. 2014), sorghum (Ramu et al. 2013), in soybean (Zhang et al. 2013), in cotton (Jena et al. 2012), cucumber (Hu et al. 2011), pepper (Kong et al. 2012), and in jatropha (Wen et al. 2010a, b).
However, studies about the comparison between the collected purslane germplasms and the genetic relationships obtained using molecular markers like EST-SSRs are yet to be reported. The present study surveys the molecular diversity in forty-five accessions of purslane collected from seven states of Western Peninsular Malaysia, using ten EST-SSR primer combinations. The genotyping data were used to understand the relationships among forty-five germplasms and identify genetically diverse lines for using them in genetic improvement of purslane.
Materials and methods
Plant materials
A total of 45 purslane accessions were used in this study collected from seven states of Western Peninsular Malaysia (Fig. 1). Among 45 samples, 11 samples were collected from Selangor, 5 samples from Melaka, 5 samples from Negeri Sembilan, 9 samples from Kedah, 5 samples from Perak, 5 samples from Penang, and rest 5 samples from Perlis. The collected purslane samples were transplanted in glass house, field-2, Faculty of Agriculture, Universiti Putra Malaysia, and all the molecular study was done in Plant Breeding and Molecular Biology Laboratory, AgroBio Complex, Faculty of Agriculture, Universiti Putra Malaysia. Brief morphological descriptions and collection details of purslane accessions have already been published in our previous article (Alam et al. 2014a).

Map showing the seven states (white arrow) of Western Peninsular Malaysia from where purslane samples were collected
Genomic DNA extraction
To extract genomic DNA, young and fresh leaves were collected from all 45 purslane accessions. Genomic DNA from each accession was isolated from fresh leaf tissue of 20-day-old plants using the CTAB (cetyltrimethylammonium bromide) method modified from Doyle and Doyle (1990). About 0.5 g of fresh leaf tissue was ground in liquid nitrogen and taken into a 2-ml micro centrifuge tube and then combined with 1,000 µl of CTAB buffer [100 mM Tris–HCl pH 8.0, 20 mM EDTA (ethylenediaminetetraacetate) pH 8.0, 1.4 mM NaCl, 2 % (w/v) CTAB, 2 % (w/v) PVP (polyvinylepyrrolidone) and ß-mercaptaphenol] and 3 µl mercapethanol. The solution was then allowed to centrifuge at 500 rpm at 65 °C for one hour (60 min) by gently shaking the tubes at 10-minutes interval to mix appropriately. Finally, the whole samples were centrifuged at 13,000 rpm for 5 min to precipitate solid parts of the cell. Afterwards, the total amount of supernatant was transferred into a new 2-ml eppendorf tube. Then equal volume of chloroform:isoamyl alcohol (24:1, v/v) was added in each sample, and the tube was gently inverted to homogenize the mixture and centrifuged at 13,000 rpm for 5 min to precipitate polysaccharides. The upper phase was then transferred into a new 2-ml eppendorf tube excluding green and white lower phase and again incorporated with 600 µl cold isopropanol and mixed gently by inverting the tube at least 50 times. All samples were then incubated at −20 °C for 30 min. After that, it was centrifuged for 10 min at 13,000 rpm to get DNA pellet. Carefully pipetted off the supernatant without losing the pellet and let the pellet air-dried. The DNA pellet was then washed and dry out 1–2 times adding 600 µl of 4 °C 75 % ethanol until white DNA fiber appeared. Finally, 50 µl of TE buffer (10 mM tris–HCl pH 8.0, 1 mM EDTA pH 8.0) and 1 µl of RNase (DNase free) were added and mixed by finger vortexing to dissolve DNA completely and incubated at 37 °C for 1 h to break down RNA if any was present in the isolated DNA.
DNA identification and quantification
One µl of each DNA sample was put on NanoDrop spectrophotometry (ND-1000, NanoDrop Technologies Inc., Wilmington, DE, USA), and relative purity with concentration of the extracted DNA was estimated from the computer-displayed value. The final concentration of each DNA sample was diluted with 1× TE buffer (10 mM Tris–HCl, pH 8.0, 1 mM EDTA, pH 8.0) to get required concentration and kept in a refrigerator of −20 °C for PCR analysis.
EST-SSR primer selection and prescreening
A total of sixty EST-SSR primers were screened with P. oleracea DNA out of these; only ten primers with clear and reproducible polymorphic bands were chosen and used for analysis of genetic diversity. Primers sequence with annealing temperature of those ten EST-SSR markers has been presented in Table 1.
PCR amplification for EST-SSR markers
Polymerase chain reaction for amplification of EST-SSR fragments was performed in 15 μl of reaction volume containing 1 μl of 70 ng template DNA, 1 μl of each forward and reverse EST-SSR primers, 7.4 μl master mix (Dream Taq green PCR master mix-2× containing green buffer, dNTPs and 4 mM MgCl2, Supplied by Fermentas-Fisher Scientific, UK), and 4.6 μl nuclease free water. PCR amplification was carried out in a thermocycler (T100TM, Bio-Rad) following three phases Touchdown PCR protocol (Don et al. 1991) with slight modifications. In the first phase, initial temperature was adjusted to 95 °C for 3 min and the initial denaturation temperature was set at 95 °C for 30 s, annealing at Tm 62 °C for 60 s, and the elongation temperature was set at 72 °C for 30 s. In the second phase, denaturation temperature was set at 95 °C for 30 s, annealing at Tm 52 °C for 60 s and the elongation at 72 °C for 2 min, and finally, the elongation temperature was set at 72 °C for 10 min in the third phase. The 1st and 2nd phases were repeated 9 and 30 times, respectively, followed by cooling and infinitive hold at 4 °C. For electrophoresis, 3.0 % MetaPhorTM agarose (Lonza, USA) gel was prepared containing 1 μl Midori green in 1× TBE buffer (0.05 M Tris, 0.05 M boric acid, 1 mM EDTA, pH 8.0). The gel was run at constant voltage of 80 V for 80 min and visualized Molecular Imager® (GelDocTM XR, Bio-Rad). However, most of the patterns with extremely good polymorphism and useful information were often accompanied with a background smear. To reduce this smear, 2 % formamide was used in the reaction. All the patterns generated were repeated at least three times in order to obtain reproducible data.
Data scoring
Molecular weight for each band was measured using Alfa Imager software version 5.5. Each amplification product position was considered a particular locus. The amplified products for EST-SSR marker alleles at a specific position in a gel were scored visually as “1” for present and “0” for absent of a band to generate a binary data matrix. A 50 bp DNA ladder was added with the first load of the gel to score of the bands in each gel. The only bands that were scored are those that are >50 bp in length and reproducible. The scoring of the visual bands was done according to the size of the DNA ladder bands defined by the producers (www.genedirex.com).
Data analysis
Amplified bands were visually scored as present (1) or absent (0) for each EST-SSR marker. With the marker data, binary matrices were established and then used to calculate Jaccard’s similarity coefficients (Jaccard 1908). Cluster analysis was performed to construct dendrogram based on the similarity matrix data using the unweighted pair group method using arithmetic averages (UPGMA) and the SHAN clustering program. All analyses were performed with the NTSYS-pc 2.10 software (Rohlf 2002). Polymorphism information content (PIC) values were calculated to evaluate diverse level of EST-SSR marker according to Anderson et al. (1993) using the formula: PIC = 1−ΣP 2 ij , where P ij is the frequency of the jth allele (marker) for the ith EST-SSR locus. The binary data were also subjected to PCA (principal component analysis) to investigate the structure of our collection. The PCA of the 45 purslane accessions were calculated by EIGEN module of NTSYS-pc 2.10 (Rohlf 2002) software. The genetic diversity parameters of each geographic group, which included the percentage of polymorphic loci (PLP), observed number of alleles per locus (na), effective number of alleles per locus (ne), Nei’s gene diversity (h), and Shannon’s information index (I) were calculated by POPGENE software (version 3.2) (Yeh et al. 2000).
Analysis of molecular variance (AMOVA)
After obtaining the optimum number of subpopulations, an analysis of molecular variance (AMOVA) was performed using Arlequin software version 3.1 (Excoffier et al. 2005) to estimate the genetic variance components within populations and between populations.
Results
Allelic variation among the accessions using EST-SSRs
Ten EST-SSRs were used to estimate the genetic diversity of 45 accessions of P. oleracea. The sizes of the amplicons ranged from 100–650 bp. The 10 EST-SSRs produced 95 alleles of which all 95 (100 %) were polymorphic (Fig. 2). The number of alleles for each EST-SSR ranged from six (EST-SSR15) to twelve (EST-SSR9, EST-SSR41 and EST-SSR46) with a mean of 9.5. The PIC values for the evaluated 10 EST-SSR primers were relatively high (0.65–0.83) with a mean value of 0.77, which indicate the effectiveness, polymorphism, and validity of the selected primers (Table 2).

Polymorphic banding patterns of EST-SSR13 and EST-SSR26 among all 45 genotypes. Lane M 50 bp DNA ladder (Genedirex). 1 Ac1; 2 Ac2; 3 Ac3; 4 Ac4; 5 Ac5; 6 Ac6; 7 Ac7; 8 Ac8; 9 Ac9; 10 Ac10; 11 Ac11; 12 Ac12; 13 Ac13; 14 Ac14; 15 Ac15; 16 Ac16; 17 Ac17; 18 Ac8, 19 Ac19; 20 Ac20; 21 Ac21; 22 Ac22; 23 Ac23; 24 Ac24; 25 Ac25; 26 Ac26; 27 Ac27; 28 Ac28; 29 Ac29; 30 Ac30; 31 Ac31; 32 Ac32; 33 Ac33; 34 Ac34; 35 Ac35; 36 Ac36; 37 Ac37; 38 Ac38; 39 Ac39; 40 Ac40; 41 Ac41; 42 Ac42; 43 Ac43; 44 Ac44 and 45 Ac45)
The maximum numbers of effective alleles (6.683) were found in EST-SSR46 and the lowest (3.248) were seen in EST-SSR15 (Table 2). Furthermore, the highest expected heterozygosity was estimated in EST-SSR46 (0.859), and the lowest was recorded in EST-SSR15 (0.699), respectively, with the mean value of 0.81 (Table 2). The Nei’s varied from 0.6921 to 0.8504 with the highest in EST-SSR46 and lowest in EST-SSR15 with the mean value of 0.7963 and the Shannon’s information index varied from 1.4219 to 2.1089, respectively, with the highest in EST-SSR46 and the lowest in EST-SSR15 and their mean value was 1.8496 (Table 2).
Cluster analysis
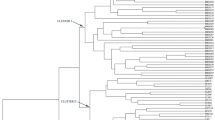
The UPGMA and neighbor-joining dendrogram from EST-SSR data were constructed using Power marker software. The tree from cluster analysis showed the genetic relationships among 45 purslane accessions, and 8 distinct groups were generated from the analysis (Fig. 3; Table 3). Here, cluster IV constructed with the highest number (10) of accessions followed by cluster II and VII, both were composed of 9 accessions, cluster I had 7 accessions, clusters V and VIII were comprised with 3 accessions, and both the clusters III and VI contained 2 accessions, respectively (Table 3). The genetic differences among 45 P. oleracea accessions ranges from 0.90 to 0.27 (Supplementary table) where the highest distance observed in several accessions and the least distances was found among both in Ac27 and Ac28, and Ac28 and Ac29.

Dendrogram based on UPGMA, depicting the genetic relationship among the P. oleracea accessions using EST-SSR markers among 45 P. oleracea accessions
Another dendrogram constructed based on the source of sample collection from seven states in Western Peninsular Malaysia where broadly divided into two major groups: with Melaka (MLK) and Selangor (SLG) in one cluster while other states belongs to the second cluster (Fig. 4). Individually, Melaka and Selangor comprised cluster I, Kedah and Negeri Sembilan formed cluster II, Perak separately consisted cluster III, and finally cluster IV was composed with Perlis and Penang (Fig. 4). The pairwise population matrix of Nei’s genetic distance has been presented in Table 4.

The figure describes the pattern of clustering which divided the seven states into four groups based on molecular data analysis using EST-SSR markers (MLK Melaka, SLG Selangor, KDH Kedah, NSB Nigeri Sembilan, PRK Perak, PLS Perlis and PNG Penang)
Principal component analysis
The patterns of cluster analysis were also confirmed by the Principal Component Analysis (Figs. 5, 6). The principal component analysis (PCA) results derived from EST-SSR markers indicated 81.9 % of total variation came from the first three principal components with PC1, PC2, and PC3 having 63.4, 11.9, and 6.6 %, respectively (Table 5). From PC1, the highest value was 0.259 followed by 0.219 and 0.193, while the least were −0.439, −0.396, and −0.359, respectively. The highest (0.259) was found in Ac44, while the least was found in Ac40 (Table 5).

Three-dimensional plots of PCA indicating relationships among 45 purslane accessions based on EST-SSR markers

Two-dimensional plots of PCA indicating relationships among 45 purslane accessions based on EST-SSR markers
Analysis of molecular variance (AMOVA)
The analysis of molecular variance unveiled that variation among the population was found to be low (28.13 %), while variation among the population accounted for 71.87 % of the total variation of P. oleracea accessions (Table 6).
Discussion
Any crop improvement program starts with identification of variability among the genotypes. Study of genetic diversity is a critical component of applied plant breeding for optimizing the choice of parents in a crop-breeding program. An effective germplasm assessment provides the scientific basis for selection of parents/donors for recombination breeding or hybrid breeding, and to breed for specific agro-ecological conditions and situations (Kumar et al. 2012). Diversity analysis at the molecular level using PCR-based markers is the cheapest and rapid method of identifying the relationships and/or differences among the genotypes. Formerly, genetic diversity was mostly measured based on the morphological and physiological variations of quantitative and economically significant traits, but these methods have several weaknesses, such as time consuming and laborious, and this method cannot define the exact level of genetic diversity among germplasm because the trait appears by interaction between genes and environment (Schulman 2007). The genetic uniqueness of each accession can be determined and quantified by the use of DNA profiling which employing various molecular markers such as RFLPs, DAFs, RAPDs, and microsatellites (Brown and Kresovich 1996); however, PCR-based markers are more suitable for large-scale analysis. Among the PCR-based markers, microsatellites are becoming more popular, both for genetic diversity and breeding research (Joshi et al. 2000). Proficient and consistent use of molecular markers such as EST-SSR for study of genetic diversity in any food crop or tree crop requires selection and application of primers which will give clear, distinct, reliable, and sufficient information required to study the divergence that occur within the crop (Arolu et al. 2012). In our research, the number of polymorphic loci detected per primer combination varies according to the primer. The EST-SSR markers amplified distinct band patterns among the 45 P. oleracea accessions, and each marker revealed polymorphism.
The 10 EST-SSR markers detected 95 alleles (100 %) with a mean of 9.5, showing a higher discrimination power. Also, PIC values presented a similar result (Table 2). The PIC values that ranged from 0.65 to 0.83 with a mean of 0.77. Our result is in agreement with the findings of Hu et al. (2011) where they estimated genetic diversity among cucumber germplasm collections using SSR and EST-SSR markers. Furthermore, in our study, the number of alleles for each EST-SSR ranged from 6 to 12 with a mean of 9.5 (Table 2) which is very similar with the outcomes of watermelon diversity analysis using EST-SSR by Mujaju et al. (2013) and Hwang et al. (2011).
The EST-SSR markers also demonstrated different levels of genetic diversity among P. oleracea accessions under present study. The Shannon’s information index varied from 1.42 to 2.11 with the mean value of 1.85, which is about 10-fold higher that the reported mean value (0.16) by Mujaju et al. (2013) in watermelon. Greater value of Shannon’s information index in the present study was another indication of presence of higher genetic diversity in the genotypes under this study. The Nei’s gene diversity varied from 0.69 to 0.85 with the mean value of 0.79. This average value was higher than the value recorded by Wen et al. (2010a, b) in Jatropha curcas L. who found 0.38 which also indicated presence of adequate genetic diversity (Table 2). This average value was higher than the value recorded in rice by Zai-quan et al. (2012) who found 0.33.
Expected heterozygosity from this study found to vary from 0.69 to 0.86 with the mean value of 0.81. This finding is also 8-fold and 20-fold higher than the expected heterozygosity mean output (0.10 and 0.04) described in watermelon accessions (Mujaju et al. 2013) and in Sorghum (Ramu et al. 2013), respectively, using EST-SSR markers.
The genetic diversity parameters such as expected heterozygosity, Shannon’s information index, Nei’s gene diversity, and PIC have positive correlation to the number of allele. It was observed from the current study, that EST-SSR marker had the highest values of expected heterozygosity (0.81), Shannon’s information index (1.85), Nei’s gene diversity (0.79), and PIC (0.77; Table 2). Since, higher value of genetic diversity parameter implies greater diversity. So, EST-SSR maker was more effective and suitable for genetic diversity analysis in P. oleracea germplasm collections.
A better resolution of the relationship among the 45 purslane accessions was provided by the UPGMA cluster analysis using EST-SSR markers. EST-SSR marker produced eight major clusters among 45 P. oleracea accessions (Fig. 3). It is notable that EST-SSRs produced a total of eight clusters and the reason could be explained in this way that EST-SSR markers might target larger number of repeated sequences specifically in the centromeric region that might heavily influence the classification pattern (Parsons et al. 1997). These clustering patterns prove the acceptability and adaptability of EST-SSR markers for the genetic diversity analysis among purslane germplasm collections. Additionally another dendrogram was constructed based on the germplasm collections from seven states of Western Peninsular Malaysia where Selangor and Melaka were grouped in same cluster may be due to the agro-geographical relationship and becoming neighbor states. But very interestingly, Negeri Sembilan was clustered with Kedah though they are situated far away from each other; may be any how, the primary plant materials were transferred to Kedah from N. Sembilan or vice versa. Same grouping was also observed among Penang and Perlis as grouping pattern of Selangor and Melaka. Whereas, Perak was clustered separately but broadly with the same group of Penang, Perlis and Perak leaving their neighbor state Kedah (Fig. 4).
Pairwise population matrix of Nei’s genetic distance showed the maximum distance among Penang and Melaka (0.47). Both states are situated to the opposite corner from each other and the minimum (0.13) was found among Melaka and Selangor (Table 4). Yan et al. (2009) conducted AFLF marker-based genetic diversity analysis among P. oleracea accessions sampled worldwide regarding α-linolenic fatty acid (ALA) composition analysis. Whereas, the first based on taxonomy Danin et al. (1978) noted the high level of diversity in Australian P. oleracea accessions which differed to those in all other parts of the world.
It was found that cluster analysis was profoundly supported by the PCA. The genetic diversity results which were observed under this study supported by the consonance between cluster and PC analyses (Figs. 3, 5; Table 5). Principal components analysis (PCA) indicated 81.9 % of total variation among all the accessions, and the first three principal components (PC1, PC2, and PC3) from the Eigen vectors analysis were 63.4, 11.9, and 6.6 %, respectively (Table 5). Fu et al. (2014) also stated that the cluster analysis and PCA results were almost same who determined the genetic diversity among celery (Apium graveolens L.) cultivars using EST-SSR markers. The similar findings have also been reported by Zhang et al. (2013) using EST-SSR markers in vegetable soybean genetic diversity analysis.
The AMOVA revealed that higher percentage of variation was attributed to variation within populations within populations (71.87 %) and among populations (28.13 %) of P. oleracea accessions (Table 6). Li et al. (2011) opined that, assessment of genetic diversity within a population is essential to characterize germplasm and provides insights into evolutionary aspects, conservation, utilization, and establishment of breeding programs.
Conclusion
There was a high level of genetic diversity among accessions of P. oleracea in this study suggesting that EST-SSR markers were effective in detection of polymorphism in this species, and this is the first report on using EST-SSR markers to study the genetic diversity on P. oleracea germplasm collections. To broaden the genetic base and improvement of the P. oleracea, populations having the lowest genetic similarities could be selected as parents. Therefore, hybridization should be made between two distant populations. Results from marker-assisted diversity analysis accessions that are far apart based on their genetic coefficient (like Ac11 and Ac17; Ac4 and Ac42; Ac16 and Ac31; Ac38 and Ac17; Ac21 and Ac31; Ac30 and Ac10 from EST-SSRs respectively) could be selected as parents for further breeding programs. This will bring about greater diversity which will lead to high productive index in terms of increase in yield and overall quality.
References
Alam MA, Juraimi AS, Rafii MY, Hamid AA, Aslani F, Hasan MM, Zainudin MAM, Uddin MK (2014a) Evaluation of antioxidant compounds, antioxidant activities and mineral composition of 13 collected purslane (Portulaca oleracea L.) accessions. Biomed Res Int 2014:1–10
Alam MA, Juraimi AS, Rafii MY, Hamid AA, Aslani F (2014b) Screening of Purslane (Portulaca oleracea L.) accessions for high salt tolerance. Sci World J 2014:1–12
Alam MA, Juraimi AS, Rafii MY, Hamid AA, Aslani F, Alam MZ (2014c) Effects of salinity on phenolic compounds and antioxidant activities of 13 collected purslane (Portulaca oleracea L.) germplasms. Food Chem 169:439–447
Alam MA, Juraimi AS, Rafii MY, Hamid AA, Aslani F (2014d) Collection and identification of different Purslane (Portulaca oleracea L.) accessions available in Western Peninsular Malaysia. Life Sci J 11:431–437
Anderson JA, Churchill GA, Autrique JE, Tanksley SD, Sorrells ME (1993) Optimizing parental selection for genetic linkage maps. Genome 36:181–186
Arolu IW, Rafii MY, Hanafi MM, Mahmud TMM, Latif MA (2012) Molecular characterization of Jatropha curcas germplasm using inter simple sequence repeat (ISSR) markers in Peninsular Malaysia. Aust J Crop Sci 6:1666–1673
Blair MW, González LF, Kimani PM, Butare L (2010) Genetic diversity, inter-gene pool introgression and nutritional quality of common beans (Phaseolus vulgaris L.) from Central Africa. Theor App Genet 121:237–248
Brown SM, Kresovich S (1996) The use of molecular markers in plant germplasm conservation. In: Paterson AH (ed) Genome mapping in plants, Clandes, New York, pp 85–93
Danin A, Baker I, Baker HG (1978) Cytogeography and taxonomy of the Portulaca oleracea L. polyploid complex. Israel J Bot 27:177–211
Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS (1991) ‘Touchdown’ PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res 19:4008
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Du QZ, Zhang DQ, Li BL (2012) Development of 15 novel microsatellite markers from cellulose synthase genes in Populus tomentosa (Salicaceae). Am J Bot 99:46–48
Dweck AC (2001) Purslane (Portulaca oleracea): the global panacea. Pers Care Mag 2:7–15
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Fu N, Wang P, Liu X, Shen H (2014) Use of EST-SSR markers for evaluating genetic diversity and fingerprinting Celery (Apium graveolens L.) cultivars. Molecules 19:1939–1955
Hu J, Wang L, Li J (2011) Comparison of genomic SSR and EST-SSR markers for estimating genetic diversity in cucumber. Biol Plant 55:577–580
Hwang J, Kang J, Son B, Kim K, Park Y (2011) Genetic diversity in watermelon cultivars and related species based on AFLPs and EST-SSRs. Notulae Botanicae Horti Agrobotanici Cluj-Napoca 39:285–292
Jaccard P (1908) Nouvelles recherches sur la distribution florale. Bulletin de la Société Vaudoise des Sciences Naturelles 44:223–270
Jena SN, Srivastava A, Rai KM, Ranjan A, Singh SK, Nisar T, Srivastava M, Bag SK, Mantri S, Asif MH (2012) Development and characterization of genomic and expressed SSRs for levant cotton (Gossypium herbaceum L.). Theor Appl Genet 124:565–576
Joshi SP, Gupta VS, Aggarwal RK, Ranjekar PK, Brar DS (2000) Genetic diversity and phylogenetic relationship as revealed by inter simple sequence repeat (ISSR) polymorphism in the genus Oryza. Theor Appl Genet 100:1311–1320
Kong Q, Zhang G, Chen W, Zhang Z, Zou X (2012) Identification and development of polymorphic EST-SSR markers by sequence alignment in pepper, Capsicum annuum (Solanaceae). Am J Bot 2012:59–61
Kumar H, Kaur G, Banga S (2012) Molecular characterization and assessment of genetic diversity in sesame (Sesamum indicum L.) germplasm collection using ISSR markers. J Crop Improv 26:540–557
Li G, Quiros CF (2001) Sequence-related amplified polymorphism (SRAP), a new marker system based on a simple PCR reaction: its application to mapping and gene tagging in Brassica. Theor Appl Genet 103:455–461
Li G, Ra WH, Park JW, Kwon SW, Lee JH, Park CB, Park YJ (2011) Developing EST-SSR markers to study molecular diversity in Liriope and Ophiopogon. Biochem Systemat Ecol 39:241–252
Lokhande VH, Nikam TD, Patade VY, Surasanna P (2009) Morphological and molecular diversity analysis among the Indian clones of Sesuvium portulacastrum L. Genet Res Crop Evol 56:705–717
Mujaju C, Sehic J, Nybom H (2013) Assessment of EST-SSR markers for evaluating genetic diversity in Watermelon accessions from Zimbabwe. Am J Plant Sci 4:1448–1456
Parsons JB, Newbury HT, Jackson MT, Ford-Lloyd BV (1997) Contrasting genetic diversity relationships are revealed in rice (Oryza sativa L.) using different marker types. Mole Breed 3:115–125
Ramu P, Billot C, Rami JF, Senthilvel S, Upadhyaya HD, Reddy LA, Hash CT (2013) Assessment of genetic diversity in the sorghum reference set using EST-SSR markers. Theor Appl Genet 126:2051–2064
Rao NK (2004) Plant genetic resources: advancing conservation and use through biotechnology. Afr J Biotechnol 3:136–145
Ren S, Weeda S, Akande O, Guo Y, Rutto L, Mebrahtu T (2011) Drought tolerance and AFLP-based genetic diversity in purslane (Portulaca oleracea L.). J Biotech Res 3:51–61
Rohlf FJ (2002) NTSYS-pc: numerical taxonomy system ver.2.1. Exeter Publishing Ltd., New York
Samy J, Sugumaran M, Lee KLW (2004) Herbs of Malaysia: an introduction to the medicinal, culinary, aromatic and cosmetic use of herbs. Times, Kuala Lumpur
Schulman AH (2007) Molecular markers to assess genetic diversity. Euphytica 158:313–321
Simopoulos AP, Norman HA, Gillaspy JE (1995) Purslane in human nutrition and its potential for world agriculture. World Rev Nutr Diet 77:47–74
Simopoulos AP, Tan DX, Mancheste LC, Reiter RJ (2005) Purslane: a plant source of omega-3 fatty acids and melatonin. J Pineal Res 39:331–332
Tatikonda L, Wani SP, Kannan S, Beerelli N, Sreedevi TK, Hoisington DA, Devi D, Varshney RK (2009) AFLP-based molecular characterization of an elite germplasm collection of Jatropha curcas L., a biofuel plant. Plant Sci 176:505–513
Uddin MK, Juraimi AS, Anwar F, Hossain MA, Alam MA (2012) Effect of salinity on proximate mineral composition of purslane (Portulca oleracea L.). Aust J Crop Sci 6:1732–1736
Uddin MK, Juraimi AS, Hossain MS, Nahar MAU, Ali ME, Rahman MM (2014) Purslane weed (Portulaca oleracea): A prospective plant source of nutrition, omega-3 fatty acid, and antioxidant attributes. Sci World J 2014:1–6
Varshney RK, Graner A, Sorrells ME (2005) Genetic microsatellite markers in plants: features and applications. Trends in Biotechnol 23:48–55
Vos P, Hogers R, Bleeker M, Reijans T, Van de Lee T et al (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Wen M, Wang H, Xia Z, Zou M, Cheng LuC, Wang W (2010a) Development of EST-SSR and genomic-SSR markers to assess genetic diversity in Jatropha curcas L. BMC Res Notes 42:1–8
Wen M, Wang H, Xia Z, Zou M, Lu C, Wang W (2010b) Development of EST-SSR and genomic-SSR markers to assess genetic diversity in Jatropha Curcas L. BMC Res Notes 3:42–50
Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18:6531–6535
Yan G, Aryamanesh N, Wang S (2009) Purslane – A potential vegetable crop. Rural Industries Research and Development Corporation, Australia, pp 1–26 RIRDC Publication No. 09/088
Yazici I, Turkan I, Sekmen AH, Demiral T (2007) Salinity tolerance of purslane (Portulaca oleracea L.) is achieved by enhanced antioxidative system, lower level of lipid peroxidation and proline accumulation. Environ Exp Bot 61:49–57
Yeh FC, Yang R, Boyle TJ, Ye Z, Xiyan JM (2000) PopGene32, Microsoft Windowsbased Freeware for Population Genetic Analysis, Version 1.32. Molecular Biology and Biotechnology Centre, University of Alberta, Edmonton
Zai-quan C, Fu-you Y, Ding-qing L, Teng-qiong Y, Jian F, Hui-jun Y, Qiao-fang Z, Dun-yu Z, Wei-jiao L, Xing-qi H (2012) Genetic diversity of wild rice species in Yunnan province of China. Rice Sci 19:21–28
Zhang G, Xu S, Mao W, Hu Q, Gong Y (2013) Determination of the genetic diversity of vegetable soybean [Glycine max (L.) Merr.] using EST-SSR markers. J Zhejiang Univ Sci B (Biomed & Biotechnol) 14:279–288
Zietkiewiez E, Rafalski A, Labuda D (1994) Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomics 20:176–183
Acknowledgments
The authors sincerely acknowledge UPM Research University Grant (01-02-12-1695RU) for financial support of the project and IGRF (International Graduate Research Fellowship, UPM) for PhD Fellowship.
Conflict of interests
The authors declare that there is no conflict of interests regarding the publication of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Alam, M.A., Juraimi, A.S., Rafii, M.Y. et al. Application of EST-SSR marker in detection of genetic variation among purslane (Portulaca oleracea L.) accessions. Braz. J. Bot 38, 119–129 (2015). https://doi.org/10.1007/s40415-014-0103-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40415-014-0103-0