
Abstract
Selection and use of genetically diverse genotypes are key factors in any crop breeding program to develop cultivars with a broad genetic base. Molecular markers play a major role in selecting diverse genotypes. In the present study, a reference set representing a wide range of sorghum genetic diversity was screened with 40 EST-SSR markers to validate both the use of these markers for genetic structure analyses and the population structure of this set. Grouping of accessions is identical in distance-based and model-based clustering methods. Genotypes were grouped primarily based on race within the geographic origins. Accessions derived from the African continent contributed 88.6 % of alleles confirming the African origin of sorghum. In total, 360 alleles were detected in the reference set with an average of 9 alleles per marker. The average PIC value was 0.5230 with a range of 0.1379–0.9483. Sub-race, guinea margaritiferum (Gma) from West Africa formed a separate cluster in close proximity to wild accessions suggesting that the Gma group represents an independent domestication event. Guineas from India and Western Africa formed two distinct clusters. Accessions belongs to the kafir race formed the most homogeneous group as observed in earlier studies. This analysis suggests that the EST-SSR markers used in the present study have greater discriminating power than the genomic SSRs. Genetic variance within the subpopulations was very high (71.7 %) suggesting that the germplasm lines included in the set are more diverse. Thus, this reference set representing the global germplasm is an ideal material for the breeding community, serving as a community resource for trait-specific allele mining as well as genome-wide association mapping.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sorghum [Sorghum bicolor (L.) Moench, 2n = 2x = 20] is the emerging model crop species for the tropical grasses with C4 photosynthesis. Sorghum has a genome size of 740 Mb (Paterson et al. 2009). The complete genome sequence of sorghum was made available for public use in early 2008 (http://genome.jgi-psf.org/Sorbi1/Sorbi1.info.html, Paterson et al. 2009), which enabled the sorghum geneticist to understand the complex traits at sequence level. Sorghum is the fifth most important cereal crop and occupies the second position among the staple food grains in semi-arid tropics. It is well adapted to withstand harsh conditions, the characteristic feature of tropical regions.
During the initial stages of molecular markers’ evolution, RFLP markers played a significant role in identifying the genomic regions responsible for agronomically important traits, genetic diversity analyses and comparative genome mapping. In the later stages, SSR markers proved highly useful in applied breeding programs because of their reproducibility and co-dominant nature. In sorghum, SSR markers have been developed by different research groups (Brown et al. 1996; Taramino et al. 1997; Bhattramakki et al. 2000; Kong et al. 2000; Schloss et al. 2002; Ramu et al. 2009; Srinivas et al. 2008, 2009; Li et al. 2009). Genetic diversity studies in sorghum were initiated early in the 1990s using molecular markers, especially RFLP probes (Deu et al. 1994, 1995, 2006). During the initial stages, no separate clustering was observed according to race or with their geographic origin. This may be because of using less number of markers for their differentiation and also inclusion of less number of samples. Later, differentiation according to race and origin was first established by Deu et al. (1994) using RFLP probes. Later on, diversity analyses were carried out using different marker systems, AFLP (Menz et al. 2004), RAPD (Agrama and Tuinstra 2003), SSRs (Folkertsma et al. 2005; Barnaud et al. 2007; Caniato et al., 2007; Deu et al. 2008; Ng’uni et al. 2011; Bouchet et al. 2012) and next-generation sequencing technologies (NGS) between sweet and grain sorghums (Zheng et al. 2011). SSRs were used widely for linkage mapping and diversity analyses of selected germplasm subsets, ranging from a core collection (Deu et al. 2006) to more specific sets of locally adapted lines (Deu et al. 2008, 2010; Barro-Kondombo et al. 2010; Ng’uni et al. 2011; Ji et al. 2011; Ngugi and Onyango 2012) and trait-specific germplasm sets for aluminum tolerance (Caniato et al. 2007) and sweet sorghum lines (Ali et al. 2008; Murray et al. 2009; Wang et al. 2009).
The major disadvantage associated with the anonymous SSR is the high cost of development and the time-consuming process. Alternatively, molecular markers/tools in a particular crop can be developed rapidly and in an inexpensive way through data mining (Kantety et al. 2002; Varshney et al. 2002; Jayashree et al. 2006; Ellis and Burke 2007). Nowadays, because of NGS technologies and genome sequencing projects, a vast amount of nucleotide sequences are flooding the public databases. This includes both genomic and genic sequences. Genic sequences (expressed sequence tags or ESTs) are derived from either ends of cDNA of plant genome-expressed regions. ESTs or cDNA sequences have gained much attention in this functional genomics era. SSRs have their usefulness even in this functional genomics era because of their reproducibility, and multi-allelic and high-throughput nature. Identification of SSRs from ESTs (genic microsatellites) is the preferred alternative for rapid and inexpensive marker development (Gupta et al. 2003; Kantety et al. 2002; Varshney et al. 2002; Jayashree et al. 2006; Ellis and Burke 2007). ESTs, if found with SSRs, can be linked directly to the genes, which may have some agronomic importance.
Unlike anonymous SSRs, EST-SSRs are derived from the expressed portion of the genome and, thus, have better chances of transferability across the species/taxa. EST-SSRs were successfully developed in different crops through data mining, e.g., in rice (Cho et al. 2000), sorghum (Ramu et al. 2009; Srinivas et al. 2008, 2009), pearl millet (Senthilvel et al. 2008), barley (Eujayl et al. 2002; Thiel et al. 2003), wheat (Gupta et al. 2003; Yu et al. 2004), tall-fescue grass (Saha et al. 2004), sugarcane (da Silva and Solis-Gracia 2006), rye grass (Studer et al. 2008), etc., for different purposes including linkage mapping (Ramu et al. 2009; Srinivas et al. 2008, 2009 in sorghum, Yu et al. 2004 in wheat, Thiel et al. 2003; Varshney et al. 2006 in barley), diversity analysis (Cho et al. 2000 in rice, Leigh et al. 2003; Gupta et al. 2003 in wheat, Eujayl et al. 2002; Varshney et al. 2008 in barley) and transferability testing (Saha et al. 2004; Balyan et al. 2005; Mian et al. 2005; Zhang et al. 2006).
In sorghum, EST-SSR markers were limited to transferability study across the other cereals (Wang et al. 2005) and have not yet been used for assessing diversity of germplasm subsets. Therefore, the present study aimed to assess the pattern of genetic diversity using EST-SSR markers and test their discriminative power in comparison with the genomic SSRs in a reference set of sorghum.
Materials and methods
Plant material
A sorghum reference set was defined based on a genetic diversity analysis of global composite germplasm collection (GCGC, 3,367 accessions) using 41 genomic/neutral SSR markers in the frame of a project sponsored by the Generation Challenge Programme (GCP) of the CGIAR. The reference set captured 78 % of the sorghum GCGC genetic diversity (Billot et al. 2013). A complete list of 384 accessions along with passport information is provided in Supplemental table S1. This set was built to represent along with wild relatives, all five basic races and their intermediate races from sorghum worldwide distribution (47 different countries). Trait-specific and most commonly used genotypes in breeding programs were also included.
DNA extraction and quantification
Seeds for the reference set were collected from ICRISAT, Patancheru, India and CIRAD, Montpellier, France. These seeds were grown in pots under natural conditions. DNA was isolated from 10-day-old seedlings using the protocol developed by Mace et al. (2003). After extraction, 1 μl of DNA sample for all accessions was loaded in 0.8 % agarose gels. Uncut λ DNA was loaded as a control to assess the quality and the quantity of DNA. Based on uncut λ DNA standards, DNA samples were normalized to a uniform concentration (2.5 ng/μl) and used for SSR genotyping.
Genotyping with EST-SSR markers
Forty-five EST-SSR markers were selected from the set of 55 SSRs developed and mapped at ICRISAT (Ramu et al. 2009). These markers had reasonable coverage on sorghum genome representing all ten linkage groups of sorghum. EST-SSR markers were grouped into different multiplex sets based on their amplicon sizes in internal standard genotype, BTx623 (Table 1). In addition, three pooled control panel DNAs were used as standards (Billot et al. 2012; http://sorghum.cirad.fr/SSR_kit), to check the consistency of each run in ABI 3130xl Genetic Analyzer.
The forward primers were synthesized adding the M13-forward primer sequence (5′CACGACGTTGTAAAACGAC3′) at the 5′end. A fluorescently labeled M13-forward primer sequence was also added in the PCR mix to amplify the fluorescently labeled PCR product, which can help further screening in the ABI 3130xl Genetic Analyzer. PCR was performed in a 5 μl reaction volume with final concentrations of 2.5 ng DNA, 2 mM MgCl2, 0.1 mM of dNTPs, 1× PCR buffer, 0.08 pM of M13-tailed forward primer, 0.16 pM of M13-labeled and reverse primers and 0.1 U of Taq DNA polymerase (AmpliTaq Gold®, Applied Biosystems, USA) in a GeneAmp® PCR System 9700 thermal cycler (Applied Biosystems, USA) with the following cyclic conditions: initial denaturation at 94 °C for 15 min (to activate Taq DNA polymerase), then ten cycles of denaturation at 94 °C for 15 s, annealing at 61 °C for 20 s (temperature reduced by 1 °C for each cycle) and extension at 72 °C for 30 s. This was followed by 40 cycles of denaturation at 94 °C for 10 s, annealing at 54 °C for 20 s and extension at 72 °C for 30 s with a final extension of 20 min at 72 °C. Amplified PCR products according to their multiplexes with internal ROX-400 size standard were run through the ABI 3130xl Genetic Analyzer.
Data analysis
Raw data produced from the ABI 3130xl Genetic Analyzer were first scanned through GeneScan 3.7 (Applied Biosystems, USA) and further fragment analysis was carried out using Genotyper 3.7 software (Applied Biosystems, USA). Based on relative migration with internal size standard (ROX-400, Applied Biosystems), PCR amplicon sizes were scored in base pairs (bp). These raw data were further processed through the AlleloBin program (http://www.icrisat.org/bt-software-downloads.htm) to get perfect allele calls.
Five EST-SSR markers (Xisep0209, Xisep0621, Xisep0728, Xisep1128 and Xisep1150) were deleted from the data analysis due to poor data quality (many missing data points). Allele numbers, gene diversity, heterozygosity and polymorphic information content (PIC) were calculated with PowerMarker v3.25 software (Liu and Muse 2005). Between samples, genetic distances were assessed through simple matching index as implemented in DARwin v5 software (Perrier et al. 2003; Perrier and Jacquemoud-Collet 2006). A dendrogram was constructed using the neighbor-joining (NJ) method as implemented in the same software.
Analysis of population structure and AMOVA
Bayesian cluster analysis, a model-based approach, as implemented in STRUCTURE 2.2 (Pritchard et al. 2000; Falush et al. 2003) was employed to detect the population structure and to assign the individuals in the reference set of sorghum into subpopulations (K) based on the 40 EST-SSR markers data. An admixture model with correlated allele frequencies was used with burn-in length of 100,000 and 200,000 Monte Carlo Markov chain (MCMC) replicates with five independent runs for each K ranging from 5 to 15. After obtaining the optimum number of subpopulations, an analysis of molecular variance (AMOVA) was performed using Arlequin ver. 3.1 (Excoffier et al. 2005) to estimate the genetic variance components within subpopulations and between subpopulations.
Results
All EST-SSR markers used in this study were polymorphic across accessions of the sorghum reference set. Availability of marker data ranged from 91.4 (Xisep0108) to 100 % (Xisep0101) with a total of 1.83 % missing data. These markers yielded a total of 360 alleles with an average of 9 alleles per marker (Table 1). Out of 360 alleles observed, cultivated sorghum accessions alone contributed 320 alleles (88.9 %), whereas the wild genotypes alone contributed 257 alleles (71.4 %). The number of alleles per locus ranged from 3 (Xisep1038) to 39 (Xisep1012) (Table 1). Complete marker data produced across 384 reference set accessions with 40 EST-SSR markers are included in Supplemental table S2.
Unique and rare alleles
Cultivated sorghum genotypes accounted for 103 (28.6 %) unique alleles, while wild genotypes showed 40 unique alleles (11.1 %). Among the 40 unique alleles in wild genotypes, 8 were contributed by only one accession, IS 18868, which belongs to the verticilliflorum subspecies that purportedly originates from the USA. A total of 217 alleles were detected in common between cultivated and wild genotypes. A large number of alleles (134 alleles, 37.2 %) were found as ‘rare alleles’ (frequency <1 %). Rare alleles were not observed for two markers viz., Xisep0523 and Xisep1029, whereas the maximum numbers of rare alleles were noted for Xisep1012 (13 alleles) followed by Xisep0829 (11 alleles).
The PIC values for these markers in the reference set of sorghum varied from 0.1379 (Xisep0120) to 0.9483 (Xisep1012) with an average of 0.523. Xisep1012 was the most informative, with the most alleles and highest PIC value. The observed heterozygosity varied from 0.0 (Xisep1038) to 0.1076 (Xisep1103) with a mean of 0.039. Gene diversity varied from 0.1429 (Xisep0120) to 0.9505 (Xisep1012) with an average of 0.5690 (Table 1).
Allelic distribution according to the race
Maximum numbers of alleles were present in the bicolor race (212 alleles, 58.89 %), followed by the caudatum race (208 alleles, 57.78 %). Durra and guinea (excluding guinea margaritiferum (Gma)) race accessions contributed 177 (49.17 %) and 180 alleles (50 %), respectively. Kafir race’s contribution was limited to only 107 alleles (29.72 %). Among the guinea races, guinea margaritiferum (Gma) contributed 91 alleles (25.28 %). All EST-SSR markers screened were found polymorphic in bicolor and guinea races. Five markers (Xisep0114, Xisep0422, Xisep0829, Xisep1038 and Xisep1231) were found monomorphic across the kafir race accessions. Xisep0120 and Xisep0422 markers were found monomorphic in the caudatum and durra races, respectively. Within the guinea race, gma race accessions had nine monomorphic markers, namely, Xisep0242, Xisep0422, Xisep0444, Xisep0607, Xisep0639, Xisep0824, Xisep0829, Xisep0948 and Xisep1035.
Allelic distribution according to geographic origin
All the markers were polymorphic across all the genotypes originating from the African continent. Genotypes originating from the African continent had 319 alleles (88.61 %), whereas those from other parts of world contributed 306 alleles (85.0 %). Among the African origin genotypes, those from Central Africa contributed 183 alleles (50.83 %), Eastern Africa 251 alleles (69.72 %), Southern Africa 212 alleles (58.89 %) and Western Africa 225 alleles (62.50 %).
Reference set accessions from the Asian continent were grouped into three, namely, Eastern Asia, Middle East and India. All the markers were polymorphic in the genotypes of Indian origin contributed 174 alleles (48.33 %). Genotypes from Eastern Asia contributed 119 alleles (33.05 %), whereas those from Middle East contributed 139 alleles (38.61 %). Five markers, Xisep0203, Xisep0422, Xisep0805, Xisep1038 and Xisep1231, were found monomorphic in Eastern Asia accessions, while Xisep0422, Xisep0449, Xisep0504 and Xisep1231 were monomorphic in Middle East accessions. In both cases, Xisep0422 and Xisep1231 markers were found monomorphic.
Cluster analysis
Reference set accessions were clustered according to geographic origin and by race within each origin. Factorial analysis (FA) (Fig. 1a, b) and NJ analysis (Fig. 2) have clearly identified the different clusters according to their race and geographic origin. Guinea margaritiferum race genotypes from Western Africa were clustered on top of the second quadrant (II) (Fig. 1b) of FA, when axes 2 and 3 were considered. Wild genotypes were clustered in the middle of the intersection point between quadrant I and II. Genotypes belonging to the kafir race were clustered in III quadrant, while genotypes belonging to races caudatum and guinea were grouped in IV and I quadrants. The durra race grouped together between quadrant I and IV when viewed on axis 1–2 of the FA. On these axes, caudatum accessions grouped in quadrant II, while bicolor race accessions were grouped between durra and caudatums.

Factorial analysis with 40 EST-SSR markers screened against a reference set of sorghum. Basic races have been represented with different colors [B-bicolor in blue, C-caudatum in violet, D-durra in pink, G-guinea (excluding Gma) in green, Gma-guinea margaritiferum in light green, K-kafir in orange, wild in red

NJ dendrogram for sorghum reference set based on the allelic data of 40 EST-SSRs. Cluster numbers and color codes are the same as those used in the STRUCTURE analysis presented in Fig. 3
For a better understanding, the dendrogram produced by NJ analysis was divided into eight major clusters. Accessions originating from Western Africa (AfricaW) were grouped into two separate clusters. One cluster was exclusively made up of accessions belonging to guinea margaritiferum (Gma) in the VIIa cluster which formed a group along with wild genotypes (Fig. 2). Another cluster consisted of mostly guinea (G) and guinea gambicum (Gga) and their intermediate races (cluster II). Cluster III was formed mainly with the accessions belonging to the kafir (K) race, most of which originated from Southern Africa (AfricaS). This cluster also included other accessions from Southern Africa belonging to race bicolor (B) and from Eastern Africa belonging to intermediate race durra-caudatum (DC). Accessions that originated from India were mainly grouped into two clusters. One group of accessions belonging to the guinea race was grouped in cluster IV along with some other intermediate races derived from Southern Africa. Another group of accessions was grouped in Cluster Va which belonged to the durra race. Durras from the Middle East were clustered in Cluster Vb. Cluster VI was divided into two subgroups, a and b. Cluster VIa mainly consisted of bicolor and intermediate races from Eastern Asia, whereas VIb consisted of durra and intermediate accessions with durra from Africa being grouped together. Accessions belonging to the caudatum race derived from Eastern Africa (AfricaE), Central Africa and Western Africa were largely grouped in cluster I. This cluster also hosted the accessions having the intermediate races between caudatum and guinea.
Most of the wild genotypes were grouped in cluster VIIb. There was no specific cluster differentiation according to their geographic origin (Fig. 2). But there was clear demarcation in grouping pattern according to their subspecies. Most of the arundinaceum and aethiopicuum subspecies accessions were grouped together, while verticilliflorum and drummodii subspecies accessions formed another group. Some of the landraces were also grouped into the same cluster along with wild genotypes in cluster VIIc. Cluster VIII consisted of accessions from different geographic origins. A dendrogram was also constructed without wild accessions (Supplemental figure S3), which does not affect much of the grouping pattern of other germplasm.
STRUCTURE and AMOVA
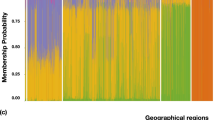
This reference set representing global sorghum germplasm collection representing all basic races and intermediate races from all geographic origins. Hence, cluster analysis (distance based) in combination with STRUCTURE (model-based) analysis was used to identify the possible number of subpopulations. Cluster analysis clearly showed that Gma accessions were separated from guinea race and clubbed with wild accessions. STRUCTURE analysis also clearly assigned the individuals into subpopulations according to their geographic origin followed by race. Gma race accessions were separated from guinea and wild accessions when K = 6. This K was taken as the prime source of a number of subpopulations for further investigation. Southern Africa-derived accessions (kafir and guinea races) were grouped as a single subpopulation. When K = 7, Southern Africa-derived accessions [kafir (cluster III) and guinea races (cluster IV)] were separated into two subgroups. Further at K = 8, clusters V and VI were grouped into three subpopulations, separating Middle East- and East Asia-derived accessions as a single population from Indian durras and intermediate races from the African continent. When K increased to 10, accessions from Middle East and East Asia were further separated into two subpopulations. Hence, K = 10 was taken as the possible number of subpopulations present in the reference set of sorghum (Fig. 3). AMOVA was performed with these ten subpopulations and more genetic variation within the subpopulations (71.7 %) than between the subpopulations (28.3 %) was observed.

STRUCTURE analysis identified ten subpopulations among reference set accessions of sorghum. Labels below refer to the cluster number in NJ analysis
Discussion
Any crop improvement program starts with identification of variability among the genotypes. Diversity analysis at the molecular level using PCR-based markers is the cheapest and rapid method of identifying the relationships among the genotypes. The user-friendly nature of SSR markers was successfully exploited in crop species for better understanding of the genetic diversity, domestication process and geographic divergence and distribution.
During the early stages, RFLP markers were used for diversity analysis (Aldrich et al. 1992; Tao et al. 1993). Because of very few numbers of genotypes included in those studies, the prevailing structure could not be deciphered well using RFLP and RAPD markers. However, an assessment of the population structure mainly depends on the type and number of markers used and representativeness of the samples analyzed. In the present study, samples were selected from a global genetic diversity analysis using SSR markers. A reference set of sorghum (Supplemental table S1) was selected for diversity analysis representing a significant portion of genetic variation with all five basic races, their intermediate races and wild genotypes. Markers selected for screening (Table 1) have complete genome coverage across the ten linkage groups of sorghum.
All of the EST-SSR markers (40) used in this study were polymorphic (100 %). Gupta et al. (2003) identified that only 55 % of the 20 EST-SSR markers used were polymorphic among 52 wheat accessions. Eujayl et al. (2002) reported a lower level of polymorphism (25 %) when 42 EST-SSR markers screened against 64 durum wheat germplasm lines. This clearly indicates that the percentage of polymorphism depends on the number and nature of the material used under analysis. In the present study, data analysis was carried out with 40 EST-SSR markers. Five markers (Xisep0209, Xisep0621, Xisep0728, Xisep1128 and Xisep1150) were removed before the analysis because of missing data. The AlleloBin program, used for converting raw allele calls to perfect allele calls, uses a step-wise mutation model (SMM; Kimura and Crow 1964). In the present study, 33 EST-SSR markers followed the SMM model, whereas 7 markers (Xisep0203, Xisep0523, Xisep0617, Xisep0630, Xisep1038, Xisep1140 and Xisep1202) did not follow the SMM model. However, these seven markers fit well in an alternative model called ‘infinite alleles model’ (IAM; Ohta and Kimura 1973), which assumes that each mutation (insertion and deletion) creates a new allele. A similar pattern of allele scoring was observed with genomic SSR markers in Folkertsma et al. (2005).
In total, 40 EST-SSR markers produced 360 alleles with an average of 9 alleles per marker (Table 1). This is the maximum number of alleles per marker reported using EST-SSR markers in any cereal to date. In case of tall-fescue grass, an average of 2.78 alleles/marker were reported (Saha et al. 2004), while 1.8 alleles/marker in bread wheat (Gupta et al. 2003) with 20 EST-SSRs, 4.5 alleles/markers in durum wheat with 42 EST-SSRs (Eujayl et al. 2002), 3 alleles/markers in 54 barley accessions using 38 EST-SSR markers (Thiel et al. 2003) and 4.6 alleles/marker in Crotalaria species (Wang et al. 2006) were reported. Compared to the results obtained with neutral genomic SSRs, the average number of alleles per marker detected in this study is comparable to that found in a limited size core collection (Caniato et al. 2007), or on geographically limited studies (e.g., Barnaud et al. 2007, Deu et al. 2008), but is lower than that found on the same material (Billot et al. 2013). This illustrates that although EST-SSR markers present a high discriminating power, they are under selective pressure.
The PIC values of markers can provide an estimate of the discrimination power in a set of accessions by taking not only the number of alleles, but also the relative frequencies of each allele (Smith et al. 2000). The average PIC value of EST-SSR markers (0.5230) was a bit higher in this reference set of sorghum in comparison with previous studies using EST-SSR markers for genetic diversity analysis in other crops, e.g., 0.443 in bread wheat (Gupta et al. 2003) and 0.45 in barley (Thiel et al. 2003). However, the average PIC value was lower compared to the PIC values of genomic SSR markers in sorghum [0.62 in both the studies of Agrama and Tuinstra (2003) and Caniato et al. (2007)]. However, this is higher than the PIC value reported by Folkertsma et al. (2005) using 100 guinea race accessions and 21 genomic SSR markers and by Ali et al. (2008) using 72 sorghum accessions with 41 SSR markers. This is on par with PIC (0.54) reported by Wang et al. (2009) in a study involving 96 sweet sorghum lines and 95 SSRs. High PIC values and large number of alleles per markers can also be attributed to the nature of the materials studied. As the reference set was derived from global germplasm collection covering worldwide origins and racial characterizations, and including wild samples, it is expected to produce a large number of alleles. SSR markers containing di-nucleotide repeats produced more alleles and hence greater PIC values (Table 1). These results were in harmony with previous studies by Smith et al. (2000), Agrama and Tuinstra (2003), Casa et al. (2005) and Deu et al. (2008).
Allelic distribution
The maximum number of alleles was reported in the bicolor race (58.89 %) followed by the caudatum race (57.78 %). Five markers were found to be monomorphic across the kafir accessions, while nine markers were found to be monomorphic among the Gma accessions. A large number of monomorphic markers and low percentages of allele contribution from Gma and kafir (25.28 and 29.72 %, respectively) to the total alleles are the major reasons for their separate clusters as compared to other races.
Genotypes from the African continent reported the maximum number of alleles (88.61 %) suggesting that the maximum diversity was found in that continent and also greatly supporting the idea that sorghum originated from Africa. In Africa, Eastern African accessions contributed the maximum number of alleles (69.72 %) supporting greatly the idea that sorghum originated from Eastern Africa. No marker was found to be monomorphic in African-originated accessions, whereas five markers were found to be monomorphic among the accessions derived from Eastern Asia and four markers were monomorphic among the accessions derived from the Middle East. In both these cases, Xisep0422 and Xisp1231 were found to be monomorphic.
Wild species are the most divergent (capturing 71.39 % of total alleles) in the present study as reported by Deu et al. (1994, 2006). Cultivated and wild genotypes shared 217 alleles in common. Among 40 unique alleles reported by wild accessions, 8 were contributed only from one wild accession, IS 18868, belonging to S. bicolor subspecies verticilliflorum. This accession has passport data indicating its origin as the USA, but this likely means that it entered the global germplasm collection via the USA rather than as a direct field collection from Africa. These rare alleles may be a target of plant breeders if these alleles have any significant role in trait-specific characters. A large number of rare alleles in the present study (37.2 %) were detected due to the presence of many unique alleles in the wild and cultivated germplasm. Two markers did not detect any rare alleles and they had only few alleles.
Structure of genetic diversity
Based on floral and grain morphology, sorghum cultivars were grouped into five basic races and ten intermediate races. Reference set accessions were grouped primarily according to race within their geographic origin (Fig. 2) and in agreement with previous studies using RFLP markers (Deu et al. 1994, 2006), SSRs and RFLPs (Menz et al. 2004), SSRs (Deu et al. 2008) and recently by using DArT markers (Mace et al. 2008; Bouchet et al. 2012). Cluster analysis of accessions based on EST-SSR allelic variation divided them into eight groups (Fig. 2). On a finer discrimination level, a racial pattern was found within eight clusters on the dendrogram. Racial discrimination by markers was first observed by Deu et al. (1994) in sorghum.
Average gene diversity (H e, also known as expected heterozygosity) among the reference set is 0.5690 with 40 EST-SSR markers (Table 1). As expected with EST-SSR markers, this is lower in comparison with previously published results in a small set of highly polymorphic SSRs in materials from Morocco (H e = 0.84) by Dje` et al. (1999), in Eritrea (H e = 0.78) by Ghebru et al. (2002), Niger accessions (H e = 0.61), in South Africa (H e = 0.60) by Uptmoor et al. (2003) and in Kenyan sorghums (0.6627) by Ngugi and Onyango (2012). The observed heterozygosity reported in the present study is much less (0.039), which confirms the use of single locus/single copy markers and is expected of autogamous accessions maintained by several generations of enforced self-pollination. Besides, the large differences between the expected and observed heterozygosity is reinforced by the population structure.
In previous studies, accessions belong to bicolor race were found scattered across all clusters and considered as the most heterogeneous and ancient race with wider geographical distribution and diverse uses (forage, broom-corn and sweet stalks) (Doggett 1988; Dje` et al. 2000; Deu et al. 2006; Mace et al. 2008). However, in the present study, EST-SSR markers showed their ability to differentiate the bicolor race mainly into two major groups (Fig. 2). African bicolors was grouped in cluster III along with the homogeneous kafir race derived from Southern Africa. Some of the bicolor accessions derived from Africa were grouped closely with wild accessions in cluster VIIc. A couple of bicolor accessions derived from India were grouped in clusters Va and VIa. Grouping of bicolor race with wild accessions confirms their resemblance to spontaneous weedy sorghum and the most primitive grain sorghum (Casa et al. 2005) and also their sharing rare alleles with wild sorghum. This suggests that EST-SSR markers have good discriminating power in relation to the bicolor race. Similarly, Mace et al. (2008) also found the grouping of one bicolor race (IS 12179C) with wild genotypes.
Caudatum is the race of greatest interest because it provides the genes responsible for higher grain yields with excellent seed quality (Mace et al. 2008), whereas the guinea race is of interest to breeders due to its great genetic diversity (Deu et al. 1994, 1995). The caudatum race was grouped mainly into three different clusters (Fig. 2), which were largely geographic origin specific. Most of the African caudatums were grouped in cluster I, whereas a group of accessions from Eastern Africa (AfricaE) were grouped in cluster IV. Eastern Asian and North American origin caudatums were grouped in cluster VIa. A couple of caudatum race accessions from Eastern Africa (AfricaE) were grouped with wild accessions (VIIb).
Durra race accessions were grouped primarily in cluster Va (Fig. 2), largely comprising durra genotypes originating from India. Most of the durras were considered as drought-tolerant genotypes based on their adaptations to very high temperatures and/or receding moisture conditions (Deu et al. 2006). Durra accessions and intermediate races with durra, durra–caudatum (DC) genotypes from Middle East were grouped in cluster Vb. A group of DC was grouped in cluster VIII. This is contrary to the conclusions drawn by Deu et al. (2008) where all intermediate races with durra were clustered in the same cluster of durra.
Kafir race is grouped in only cluster III primarily derived from Southern Africa (AfricaS) (Fig. 2). Interestingly, an intermediate race with caudatum (KC) also grouped in the same cluster. This suggests that the kafir race is the least divergent and expected to be the most homogeneous group among all the races. The same conclusions were drawn in previous studies (Deu et al. 1994, 1995, 2006; Cui et al. 1995; Menkir et al. 1997; Dje` et al. 2000). The homogenous nature in the kafir race was supported by the presence of a lower number of alleles (29.72 %) and five monomorphic markers. These results are in agreement with the recent origin and restricted geographic distribution of the kafir race (Doggett 1988; Deu et al. 2006).
Because of its diverse nature, the guinea race has become the choice of breeders (Deu et al. 1994, 1995; Folkertsma et al. 2005) in crop improvement programs. Guinea race exhibits a moderate level of genetic diversity and is grouped into three major clusters according to their origin and distribution (Fig. 2). Similar grouping patterns were observed with isozymes, RFLP probes and SSRs (Deu et al. 1994, 2006, 2008; Cui et al. 1995; Folkertsma et al. 2005; Barnaud et al. 2007). Guinea accessions derived mainly from Western Africa (AfricaW) were grouped into two separate clusters, margaritiferum and non-margaritiferum. Guinea guineense (Ggu) and guinea gambicum (Gga) were grouped with some other guinea lines in cluster II, whereas the most Gma accessions clustered in a small group (cluster VIIa) closely knit with wild accessions, which mainly originated from Western Africa (AfricaW) (Fig. 2). Hence, Gma may be considered as a recently evolved “‘primitive form’ of the guinea race”. Guinea accessions originating from India were grouped separately in cluster IV and closely with Southern African guineas (including guinea roxburghii, Gro) suggesting a recent introduction of Asian forms from Southern Africa. Deu et al. (2006) also found a separate cluster for guineas derived from Asia.
The singularity of Gma race accessions is in accordance with previous studies (Deu et al. 1994, 1995, 2006, 2008; Cui et al. 1995; de Oliveira et al. 1996; Folkertsma et al. 2005; Barro-Kondombo et al. 2010). The singular nature of Gma was due to the presence of only 25.27 % of alleles found across the sorghum reference set and a maximum of nine monomorphic markers. All Gma accessions in cluster VIIa originated from Western Africa (AfricaW). The distinct nature of Gma from other guinea race subgroups is remarkable, since both are infertile and cultivated in sympatry in the same season by the same farmers (Deu et al. 2006). The Gma subgroup was distinct from other guinea races and closer knit with wild genotypes than other cultivated sorghums suggesting that the Gma group represents an independent domestication event. A Gma accession of Southern African origin (IS 19455) was not grouped with Western African margaritiferum, but with other guineas originating from Southern Africa. Grouping of this accession is in agreement with previous studies by Deu et al. (1995, 2006). This suggests that Southern African margaritiferum shares a common ancestor with Western African margaritiferum and the change in its genetic background as compared to Western African guinea margaritiferum is due to their isolation and selection pressure (Deu et al. 2006). Grouping of accessions is not much affected by removal of wild accessions from cluster analysis (Supplemental figure S3).
STRUCTURE analysis clearly identified ten subpopulations according to their race within the geographic origin in the reference set germplasm lines. This in agreement with clustering observed in distance-based method, with AMOVA results indicating that a large amount of variation (71.7 %) is present within the subpopulations. This confirms that the reference set used in this study includes diverse germplasm lines and is suitable for allele mining and association mapping.
Geneflow in sorghum
Exchange of genes is one of the major factors in evolution of domesticated plant species (Harlan 1992). Gene flow from cultivated to wild, weedy and feral relatives disturbs the size and dynamics of wild and weedy populations, resulting in the disturbance of natural gene pools and endangering the wild relatives. This in turn leads to loss of natural genetic diversity (Akimoto et al. 1999; Snow et al. 2003). Molecular analysis clearly identified the existence of gene flow in crop plants (Mariac et al. 2006; Sagnard et al. 2011). In the present study, some of the landraces were grouped with wild genotypes and most of them were derived from Africa (Fig. 2). A wild accession (IS 21691, S. drummondii from Western Africa) was grouped in cluster III along with kafir and a couple of wild accessions were grouped with the guinea race in cluster II. This can be attributed to gene flow between landraces or cultivars and wild genotypes. This may be because of wild accessions growing around the cultivated sorghums in Africa as observed by Tesso et al. (2008) and Sagnard et al. (2011). Existing cultural practices in Africa (growing side by side or in close proximity) are the major source of gene flow from cultivated to wild genotypes and vice versa. Wild accessions were found in Africa in crop margins, barren lands, hill bottom areas and in the fields where crops were abandoned due to severe drought, pest and weed infestation or extreme nutrient deficiency (Tesso et al. 2008). This suggests that wild accessions have the desirable alleles to resist these factors. This would encourage sorghum breeders to use wild alleles for transfer into locally adapted improved varieties in their crop improvement programs.
In conclusion, functional diversity analysis with proper phenotyping in multiple locations will help the breeders to mine for trait-specific alleles and facilitate an effective method of identifying the gene/QTL for different agronomic traits through association mapping (Abdurakhmonov and Abdukarimov 2008; Zhu et al. 2008; Casa et al. 2008; Upadhyaya et al. 2012). Thus, the sorghum diversity panel representing global genetic diversity can be used as a community resource for allele mining and genome-wide association mapping using SNPs generated through NGS technologies. A combination of association mapping with physical mapping helps in identifying the gene sequences underlying a particular trait. This further helps in better understanding of the variation at the sequence level which in turn helps to identify the genomic location responsible for different traits in closely related crops through comparative genome mapping.
References
Abdurakhmonov IY, Abdukarimov A (2008) Application of association mapping to understand the genetic diversity of plant germplasm resources. Int J Plant Genomics. doi:10.1155/2008/574927
Agrama HA, Tuinstra MR (2003) Phylogenetic diversity and relationships among sorghum accessions using SSRs and RAPDs. Afr J Biotechnol 10:334–340
Akimoto M, Shimamoto Y, Morishima H (1999) The extinction of genetic resources of Asian wild rice, Oryza rufipogon Griff.: a case study in Thailand. Genet Resour Crop Evol 46:419–425
Aldrich PR, Doebley J, Schertz KF, Stec A (1992) Patterns of allozyme variation in cultivated and wild Sorghum bicolor. Theor Appl Genet 85:451–460
Ali ML, Rajewski JF, Baenziger PS, Gill KS, Eskridge KM, Dweikat L (2008) Assessment of genetic diversity and relationship among a collection of US sweet sorghum germplasm by SSR markers. Mol Breed 21:497–509
Balyan HS, Gupta PK, Rustgi S, Bandopadhyay R, Goyal A, Singh R, Kumar A, Kumar N, Sharma S (2005) Development and use of SSRs of bread wheat for genetic and physical mapping and transferability to the species of Triticum–Aegilops complex. Czech J Genet Plant Breed 41:141–144
Barnaud A, Deu M, Garine E, McKey D, Joly H (2007) Local genetic diversity of sorghum in a village in northern Cameroon: structure and dynamics of landraces. Theor Appl Genet 114:237–248
Barro-Kondombo Sagnard F, Chantereau J, Deu M, Brocke KV, Durand P, Goze E, Zongo JD (2010) Genetic structure among sorghum landraces as revealed by morphological variation and microsatellite markers in three agroclimatic regions of Burkina Faso. Theor Appl Genet 120:1511–1523
Bhattramakki D, Dong JM, Chhabra AK, Hart GE (2000) An integrated SSR and RFLP linkage map of Sorghum bicolor (L) Moench. Genome 43:988–1002
Billot C, Rivallan R, Sall MN, Fonceka D, Deu M, Glaszmann JC, Noyer JL, Rami JK, Risterucci AM, Wincker P, Ramu P, Hash CT (2012) A reference microsatellite kit to assess for genetic diversity of Sorghum bicolor (Poaceae). Am J Bot 6:e245–e260
Billot C, Ramu P, Bouchet S, Chantereau J, Deu M, Gardes L, Noyer JL, Rami JF, Rivallan R, Li Y, Lu P, Wang T, Folkertsma RT, Arnaud E, Upadhyaya HD, Glaszmann JC, Hash CT (2013) Massive sorghum collection genotyped with SSR markers to enhance use of global genetic resources. PLoS ONE 8(4):e59714. doi:10.1371/journal.pone.0059714
Bouchet S, Pot D, Deu M, Rami J-F, Billot C, Perrier X, Rivallan R, Gardes L, Xia L, Wenzl P, Kilian A, Glaszmann J-C (2012) Genetic structure, linkage disequilibrium and signature of selection in sorghum: lessons from physically anchored DArT markers. PLoS One 7(3):e33470. doi:10.1371/journal.pone.0033470
Brown SM, Hopkins MS, Mitchell SE, Senior ML, Wang TY, Duncan RR, Gonzalez-Candelas F, Kresovich S (1996) Multiple methods for the identification of polymorphic simple sequence repeats (SSRs) in sorghum [Sorghum bicolor (L.) Moench]. Theor Appl Genet 93:190–198
Caniato FF, Guimarães CT, Schaffert RE, Alves VM, Kochian LV, Borem A, Klein PE, Magalhaes JV (2007) Genetic diversity for aluminum tolerance in sorghum. Theor Appl Genet 114:863–876
Casa AM, Mitchell SE, Hamblin MT, Sun H, Bowers JE, Paterson AH, Aquadro CF, Kresovich S (2005) Diversity and selection in sorghum: simultaneous analyses using simple sequence repeats. Theor Appl Genet 111:23–30
Casa AM, Pressoir G, Brown PJ, Mitchell SE, Rooney WL, Tuinstra MR, Franks CD, Kresovich S (2008) Community resources and strategies for association mapping in sorghum. Crop Sci 48:30–40
Cho YG, Ishii T, Temnykh S, Chen X, Lipovich L, McCouch SR, Park WD, Ayres N, Cartinhour S (2000) Diversity of microsatellites derived from genomic libraries and GenBank sequences in rice (Oryza sativa L.). Theor Appl Genet 100:713–722
Cui YX, Xu GW, Magill CW, Schertz KF, Hart GE (1995) RFLP-based assay of Sorghum bicolor (L) Moench genetic diversity. Theor Appl Genet 90:787–796
da Silva J, Solis-Gracia N (2006) Development of simple sequence repeat markers from genes related to stress resistance in sugarcane. Subtrop Plant Sci 58:5–11
de Oliveira AC, Richter T, Bennetzen JL (1996) Regional and racial specificities in sorghum germplasm assessed with DNA markers. Genome 39:579–587
Deu M, Gonzalez-de-Leon D, Glaszmann J-C, Degremont I, Chantereau J, Lanaud C, Hamon P (1994) RFLP diversity in cultivated sorghum in relation to racial differentiation. Theor Appl Genet 88:838–844
Deu M, Hamon P, Chantereau J, Dufour P, D’Hont A, Lanaud C (1995) Mitochondrial DNA diversity in wild and cultivated sorghum. Genome 38:635–645
Deu M, Rattunde F, Chantereau J (2006) A global view of genetic diversity in cultivated sorghums using a core collection. Genome 49:168–180
Deu M, Sagnard F, Chantereau J, Calatayud C, Hérault D, Mariac C, Pham J-L, Vigouroux Y, Kapran I, Traore PS, Mamadou A, Gerard B, Ndjeunga J, Bezancon G (2008) Niger-wide assessment of in situ sorghum genetic diversity with microsatellite markers. Theor Appl Genet 116:903–913
Deu M, Sagnard F, Chantereau J, Calatayud C, Vigouroux Y, Pham JL, Mariac C, Kapran I, Mamadou A, Gerard B, Ndjeunga J, Bezancon G (2010) Spatio-temporal dynamics of genetic diversity in Sorghum bicolor in Niger. Theor Appl Genet 120:1301–1313
Dje` Y, Forcioli D, Ater M, Lefe`bvre C, Vekemans X (1999) Assessing population genetic structure of sorghum landraces from north-western Morocco using allozyme and microsatellite markers. Theor Appl Genet 99:157–163
Dje` Y, Heuertz M, Lefe`bvre C, Vekemans X (2000) Assessment of genetic diversity within and among germplasm accessions in 408 cultivated sorghum using microsatellite markers. Theor Appl Genet 100:918–925
Doggett H (1988) Sorghum, 2nd edn. Longman Scientific and Technical, London
Ellis JR, Burke JM (2007) EST-SSRs as a resource for population genetic analyses. Heredity 99:125–132
Eujayl I, Sorrells ME, Wolters P, Baum M, Powell W (2002) Isolation of EST-derived microsatellite markers for genotyping the A and B genomes of wheat. Theor Appl Genet 104:399–407
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotyping data: linked loci and correlated allele frequencies. Genetics 164:1565–1587
Folkertsma RT, Rattunde FH, Chandra S, Soma Raju W, Hash CT (2005) The pattern of genetic diversity of guinea-race Sorghum bicolor (L.) Moench landraces as revealed with SSR markers. Theor Appl Genet 111:399–409
Ghebru B, Schmidt RJ, Bennetzen JL (2002) Genetic diversity of Eritrean sorghum landraces assessed with simple sequence repeat (SSR) markers. Theor Appl Genet 105:229–236
Gupta PK, Rustgi S, Sharma S, Singh R, Kumar N, Balyan HS (2003) Transferable EST-SSR markers for the study of polymorphism and genetic diversity in bread wheat. Mol Genet Genomics 270:315–323
Harlan JR (1992) Crops and man. ASA, Madison
Jayashree B, Ramu P, Prasad P, Kassahun B, Hash CT, Chandra S, Hoisington DA, Varshney RK (2006) A database of simple sequence repeats from cereal and legume expressed sequence tags mined in silico: survey and evaluation. In Silico Biol 6:0054 (http://www.bioinfo.de/isb/2006/06/0054/)
Ji GS, Song YF, Liu GQ, Du RH, Hao FW (2011) Genetic analysis of sorghum resources from China using SSRs. J SAT Agric Res 9 (http://ejournal.icrisat.org/Volume9/Sorghum_Millets/Genetic_analysis_Ji.pdf)
Kantety RV, Rota ML, Matthews DE, Sorrells ME (2002) Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol Biol 48:501–510
Kimura M, Crow JF (1964) The numbers of alleles that can be maintained in a finite population. Genetics 49:725–738
Kong L, Dong J, Hart GE (2000) Characteristics, linkage-map positions, and allelic differentiation of Sorghum bicolour (L.) Moench DNA simple-sequence repeats (SSRs). Theor Appl Genet 101:438–448
Leigh F, Lea V, Law J, Wolters P, Powell W, Donini P (2003) Assessment of EST- and genomic microsatellite markers for variety discrimination and genetic diversity studies in wheat. Euphytica 133:359–366
Li M, Yuyama N, Luo L, Hirata M, Cai H (2009) In silico mapping of 1758 new SSR markers developed from public genomic sequences for sorghum. Mol Breed 24:41–47
Liu K, Muse SV (2005) Powermarker: integrated analysis environment for genetic markers data. Bioinformatics 21(9):2128–2129
Mace ES, Buhariwalla HK, Crouch JH (2003) A high throughput DNA extraction protocol for molecular breeding programs. Plant Mol Biol Rep 21:459a–459h
Mace ES, Xia L, Jordan DR, Halloran K, Parh DK, Huttner E, Wenzl P, Kilian A (2008) Dart markers: diversity analysis and mapping in Sorghum bicolor. BMC Genomics 9:26. doi:10.1186/1471-2164-9-26
Mariac C, Robert T, Allinne C, Remigereau MS, Luxereau A, Tidjani M, Seyni O, Bezancon G, Pham JL, Sarr A (2006) Genetic diversity and gene flow among pearl millet crop/weed complex: a case study. Theor Appl Genet 113:1003–1014
Menkir A, Goldsbrough P, Ejeta G (1997) RAPD based assessment of genetic diversity in cultivated races of sorghum. Crop Sci 37:564–569
Menz MA, Klein RR, Unruh NC, Rooney WL, Klein PE, Mullet JE (2004) Genetic diversity of public inbreds of sorghum determined by mapped AFLP and SSR markers. Crop Sci 44:1236–1244
Mian MAR, Saha MC, Hopkins AA, Wang Z-Y (2005) Use of tall fescue EST-SSR markers in phylogenetic analysis of cool-season forage grasses. Genome 48:637–647
Murray SC, Rooney WL, Hamblin MT, Mitchell SE, Kresovich S (2009) Sweet sorghum genetic diversity and association mapping for brix and height. Plant Genome 2:48–62
Ng’uni D, Geleta M, Bryngelsson T (2011) Genetic diversity in sorghum (Sorghum bicolor (L.) Moench) accessions of Zambia as revealed by simple sequence repeats (SSR). Hereditas 148:52–62
Ngugi K, Onyango CM (2012) Analysis of the molecular diversity of Kenyan sorghum germplasm using microsatellites. J Crop Sci and Biotech 15:189–194
Ohta T, Kimura M (1973) A model of mutation appropriate to estimate the number of electrophoretically detectable alleles in a finite population. Genet Res 22:201–204
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M, Tang H, Wang X, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA, Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, Wang Y, Zhang L, Carpita NC, Freeling M, Gingle AR, Hash CT, Keller B, Klein P, Kresovich S, McCann MC, Ming R, Peterson DG, Mehboob-ur-Rahman Ware D, Westhoff P, Mayer KFX, Messing J, Rokhsar DS (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457:551–556
Perrier X, Jacquemoud-Collet JP (2006) DARwin software. http://darwin.cirad.fr/darwin
Perrier X, Flori A, Bonnot F (2003) Methods for data analysis. In: Hamon P, Seguin M, Perrier X, Glaszmann JC (eds) Genetic diversity of cultivated tropical plants. Science Publishers, Inc. and CIRAD, Montpellier, pp 31–63
Pritchard J, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotyping data. Genetics 155:945–959
Ramu P, Kassahun B, Senthilvel S, Kumar CA, Jayashree B, Folkertsma RT, Reddy LA, Kuruvinashetti MS, Haussmann BIG, Hash CT (2009) Exploiting rice–sorghum synteny for targeted development of EST-SSRs to enrich the sorghum genetic linkage map. Theor Appl Genet 119:1193–1204
Sagnard F, Deu M, Dembe′le′ D, Leblois R, Toure′ L, Diakite′ M, Calatayud C, Vaksma M (2011) Genetic diversity, structure, gene flow and evolutionary relationships within the Sorghum bicolor wild–weedy–crop complex in a western African region. Theor Appl Genet 123:1231–1246
Saha MC, Mian MAR, Eujayl I, Zwonitzer JC, Wang L, May GD (2004) Tall fescue EST-SSR markers with transferability across several grass species. Theor Appl Genet 109:783–791
Schloss SJ, Mitchell SE, White GM, Kukatla R, Bowers JE, Paterson AH, Kresovich S (2002) Characterization of RFLP clone sequences for gene discovery and SSR development in Sorghum bicolor (L.) Moench. Theor Appl Genet 105:912–920
Senthilvel S, Jayashree B, Mahalakshmi V, Kumar PS, Nakka S, Nepolean T, Hash CT (2008) Development and mapping of simple sequence repeat markers for pearl millet from data mining of expressed sequence tags. BMC Plant Biol 8:119
Smith JSC, Kresovich S, Hopkins MS, Mitchell SE, Dean RE, Woodman WL, Lee M, Porter K (2000) Genetic diversity among elite sorghum inbred lines assessed with simple sequence repeats. Crop Sci 40:226–232
Snow AA, Pilson D, Rieseberg LH, Paulsen M, Pleskac N, Reagon MR, Wolf DE, Selbo SM (2003) A Bt transgene reduces herbivory and enhances fecundity in wild sunflowers. Ecol Appl 13:279–286
Srinivas G, Satish K, Murali Mohan S, Nagaraja Reddy R, Madhusudhana R, Balakrishna D, Venkatesh Bhat B, Howarth CJ, Seetharama N (2008) Development of genic-microsatellite markers for sorghum staygreen QTL using a comparative genomic approach with rice. Theor Appl Genet 117:283–296
Srinivas G, Satish K, Madhusudhana R, Seetharama N (2009) Exploration and mapping of microsatellite markers from subtracted drought stress ESTs in Sorghum bicolor (L.) Moench. Theor Appl Genet 118:703–717
Studer B, Asp T, Frei U, Hentrup S, Meally H et al (2008) Expressed sequence tag-derived microsatellite markers in perennial ryegrass (Lolium perenne L.). Mol Breed 21:533–548
Tao Y, Manners JM, Ludlow MM, Henzell RG (1993) DNA polymorphisms in grain sorghum (Sorghum bicolor (L.) Moench). Theor Appl Genet 86:679–688
Taramino G, Tarchini R, Ferrario S, Lee M, Pe′ ME (1997) Characterization and mapping of simple sequence repeats (SSRs) in Sorghum bicolor. Theor Appl Genet 95:66–72
Tesso T, Kapran I, Grenier C, Snow A, Sweeney P, Pedersen J, Marx D, Bothma G, Ejeta G (2008) The potential for crop-to-wild gene flow in sorghum in Ethiopia and Niger: a geographic survey. Crop Sci 48:1425–1431
Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422
Upadhyaya HD, Wang Y-H, Sharma S, Singh S (2012) Association mapping of height and maturity across five environments using the sorghum mini core collection. Genome 55:471–479
Uptmoor R, Wenzel W, Friedt W, Donaldson G, Ayisi K, Ordon F (2003) Comparative analysis on the genetic relatedness of Sorghum bicolor accessions from Southern Africa by RAPDs, AFLPs and SSRs. Theor Appl Genet 106:1316–1325
Varshney RK, Thiel T, Stein N, Langridge P, Graner A (2002) In silico analysis of frequency and distribution of microsatellites in ESTs of some cereal species. Cell Mol Biol Lett 7:537–546
Varshney RK, Grosse I, Hähnel U, Siefken R, Prasad M, Stein N, Langridge P, Altschmied L, Graner A (2006) Genetic mapping and BAC assignment of EST-derived SSR markers shows non-uniform distribution of genes in barley genome. Theor Appl Genet 113:239–250
Varshney RK, Thiel T, Sretenovic-Rajicic T, Baum M, Valkoun J, Guo P, Grando S, Ceccarelli S, Graner A (2008) Identification and validation of a core set of informative genic SSR and SNP markers for assaying functional diversity in barley. Mol Breed 22:1–13
Wang ML, Barkley NA, Yu J-K, Dean RE, Newman ML, Sorrells ME, Pederson GA (2005) Transfer of simple sequence repeat (SSR) markers from major cereal crops to minor grass species for germplasm characterization and evaluation. Plant Genet Resour Charact Util 3:45–57
Wang ML, Mosjidis JA, Morris JB, Dean RE, Jenkins TM, Paderson GA (2006) Genetic diversity of Crotalaria germplasm assessed through phylogenetic analysis of EST-SSR markers. Genome 49:707–715
Wang ML, Zhu C, Barkley NA, Chen Z, Erpelding JE, Murray SC, Tuinstra MR, Tesso T, Pederson GA, Yu J (2009) Genetic diversity and population structure analysis of accessions in the US historic sweet sorghum collection. Theor Appl Genet 120:13–23
Yu J, Dake TM, Singh S, Benscher D, Li W, Gill B, Sorrells ME (2004) Development and mapping of EST-derived simple sequence repeat (SSR) markers for hexaploid wheat. Genome Res 47:805–818
Zhang LY, Ravel C, Bernard M, Balfourier F, Leroy P, Feuillet C, Sourdille P (2006) Transferable bread wheat EST-SSRs can be useful for phylogenetic studies among the Triticeae species. Theor Appl Genet 113:407–418
Zheng L-Y, Guo X-S, He B, Sun L-J, Peng Y, Dong S–S, Liu T-F, Jiang S, Ramachandran R, Liu C-M, Jing H-C (2011) Genome-wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor). Genome Biol 12:R114
Zhu C, Gore M, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. Plant Genome 1:5–20
Acknowledgments
We thank the two anonymous reviewers for their helpful suggestions and comments. This work is a part of the Ph.D. thesis of PR. The research fellowship provided to PR by the Council of Scientific and Industrial Research (CSIR), New Delhi, India is greatly acknowledged. Work reported here was supported by the grant from the Generation Challenge Programme (GCP) of the CGIAR.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by H.-C. Jing.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental table S1: List of 384 accessions included in reference set of sorghum and their passport information.
Supplemental table S2: Data matrix of allele calls used in the present study for 384 accessions across 40 EST-SSR markers. Missing data points were scored as numerical “zero (0)″s.
Supplemental figure S3: Dendrogram of reference set without their wild relatives. Cluster analysis with (Fig. 2) and without (supplemental figure S3) wild accessions suggest that they have greater similarity in grouping patterns according to their races within geographic origins.
Rights and permissions
About this article
Cite this article
Ramu, P., Billot, C., Rami, JF. et al. Assessment of genetic diversity in the sorghum reference set using EST-SSR markers. Theor Appl Genet 126, 2051–2064 (2013). https://doi.org/10.1007/s00122-013-2117-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-013-2117-6