
Abstract
In recent years, bispecific antibodies (BsAbs) have emerged as a promising therapeutic strategy against tumors. BsAbs can recruit and activate immune cells, block multiple signaling pathways, and deliver therapeutic payloads directly to tumor sites. This review provides a comprehensive overview of the recent advances in the development and clinical application of BsAbs for the treatment of solid tumors. We discuss the different formats, the unique mechanisms of action, and the clinical outcomes of the most advanced BsAbs in solid tumor therapy. Several studies have also analyzed the clinical progress of bispecific antibodies. However, this review distinguishes itself by exploring the challenges associated with bispecific antibodies and proposing potential solutions. As the field progresses, BsAbs hold promise to redefine cancer treatment paradigms and offer new hope to patients with solid tumors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Bispecific antibodies (BsAbs) can enhance the specificity and efficacy of cancer treatments by combining different mechanisms of action in a single molecule. |
Currently, over 100 types of BsAbs for solid tumors are in clinical trials. |
In treating solid tumors, BsAbs face significant challenges, such as the inhibitory nature of the tumor microenvironment. |
1 Background
Cancer ranks as a primary cause of mortality globally, affecting countries across the entire economic spectrum [1]. Surgery, radiotherapy, and chemotherapy represent the traditional pillars in cancer treatment [2]. Recently, immunotherapy has revolutionized both the therapeutic landscape and the outlook for various cancers. It encompasses both passive strategies—such as monoclonal antibodies, adoptive cell transfer, and cytokines, and active strategies—including cancer vaccines, oncolytic viruses, and checkpoint inhibitors. The effectiveness of immunotherapies varies widely, influenced by factors related to the patient, the tumor, or the specific treatment modalities employed [3, 4]. In this review, we turn our attention to BsAbs, the next-generation strategy of antibody-target cancer immunotherapy, particularly in the context of solid tumors.
Natural human antibodies are bivalent monospecific antibodies, i.e., they have two identical binding sites [5]. Since the 1980s, researchers have generated bispecific antibodies and gradually introduced them into clinical trials for tumor therapy [6, 7]. BsAbs are capable of simultaneously binding to two distinct antigens, which are often used in the mechanism of engaging immune cells such as T cells and natural killer (NK) cells to kill cancer cells. In addition to this, it can also be used to shield immune checkpoints, or to deliver payloads to tumors [8].
While BsAbs have demonstrated remarkable clinical success in treating hematological cancers, their impact on solid tumors—which constitute 90% of all cancer cases—has yet to be fully realized in clinical settings [9]. A significant obstacle to the efficacy of BsAbs in advanced solid tumors is the inhibitory nature of the tumor microenvironment (TME). This environment severely restricts T-cell function, leading to compromised immune responses [10].
Currently, 14 BsAbs have received global approval, with 12 endorsed by the US Food and Drug Administration (FDA). Specifically, five BsAbs—cadonilimab for cervical cancer, amivantamab for non-small-cell lung cancer (NSCLC), tarlatamab for extensive-stage small-cell lung cancer (ES-SCLC), tebentafusp for unresectable or metastatic uveal melanoma, and catumaxomab (withdrawn) for malignant ascites—are tailored for solid tumors. Seven BsAbs, including blinatumomab, mosunetuzumab, glofitamab, epcoritamab, teclistamab, talquetamab, and elranatamab, target hematologic malignancies, addressing various leukemias and lymphomas. The other two approved BsAbs, faricimab and ozoralizumab, have not been developed for cancer treatment [11].
2 Bispecific Antibodies
2.1 Bispecific Antibody Format
The basic structure of a natural antibody consists of four polypeptide chains divided into two identical heavy chains and two light chains. These four chains form a Y-shaped molecular structure through disulfide bonds and non-covalent interactions. Antibodies can be categorized into fragment crystallizable (Fc) and fragment antigen-binding (Fab) regions based on regional functionality. The Fc region can be recognized by the Fc receptor to mediate the killing response of immune cells or the complement effect [12]. The Fab region contains one antibody-binding variable region and one constant region. The single-chain variable fragments (scFvs) are obtained when connecting the heavy chain variable region to the light chain variable region with a linker [13].
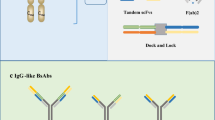
Bispecific antibodies, which are engineered antibodies with two different binding sites, are a new strategy for treating tumors [5]. The formats of bispecific antibodies can be broadly categorized into fragment-based types and Fc-based types. Brinkmann et al. provide a comprehensive and detailed catalog of over 100 different forms of bispecific antibodies [14]. Figure 1 illustrates several representative formats of bsAbs.

Several representative formats of bispecific antibodies (BsAbs). The formats of BsAbs are categorized according to the presence or absence of the Fc region. IgG Immunoglobulin G, scFv single-chain variable fragment, VH the variable region of a heavy chain, VL the variable region of a light chain, BiTE bispecific t-cell engagers, DART dual affinity re-targeting proteins, TandAb tandem diabodies, Fab fragment antigen-binding, Fc fragment crystallizable, DVD-Ig the dual variable domain immunoglobulin, KiH knobs-into-holes, Common LC common light chain
2.1.1 Fragment-Based Bispecific Antibodies
Fragment-based bispecific antibodies represent a minimalistic approach to designing bispecific molecules that combine multiple antigen-binding moieties, such as antibody fragments, within a single molecule. These fragments are designed without an Fc region, circumventing the chain-association issues commonly encountered with traditional antibody formats [15]. This design allows for relatively simple production through the co-expression of one or two polypeptide chains in lower eukaryotic and prokaryotic expression systems. The lack of an Fc region simplifies the structure and reduces production costs while maintaining high yields [16]. The modular nature of this approach allows for the customization of valency and specificity, supporting diverse therapeutic applications. However, the lack of an Fc region means these antibodies do not benefit from the neonatal Fc receptor’s (FcRn) protective mechanisms, leading to a shorter circulation time in the body [17].
The fragment-based bispecific antibodies mainly include bispecific T-cell engagers (BiTE), dual-affinity re-targeting (DART) proteins, tandem diabodies (TandAbs), and so on [18]. BiTEs (Fig. 1a) consist of two scFvs linked together: one targets a tumor-associated antigen, and the other binds to CD3 on T cells. This design enables BiTEs to bridge the gap between T cells and cancer cells, facilitating the direct killing of tumor cells by the immune system. DARTs (Fig. 1b) are characterized as a disulfide-linked diabody, meaning that it consists of two polypeptide chains arranged in a VH-linker-VL configuration that form a heterodimer. These chains are covalently connected to each other by a disulfide bond at their opposing C-termini. This complex structure enhances molecular stability [19]. TandAbs (Fig. 1c) comprise two diabodies for simultaneous targeting of two antigens, promoting clustering and potent signaling. There are also other formats such as tandem VHHs, F(ab)2 (Fig. 1d), scFv-Fabs (Fig.1e), etc.
2.1.2 Fc-Based Bispecific Antibodies
Fc-based bispecific antibodies are engineered to include the Fc region of immunoglobulins, which confers several beneficial properties, such as an extended serum half-life and improved stability [20]. For therapeutic purposes, incorporating an Fc region into antibodies is often advantageous, except in cases where a smaller size and shorter half-life are required. To enhance their biological and physicochemical characteristics, numerous engineering techniques have been employed on the Fc region [21]. Fc-based bispecific antibodies can be divided into two main categories: symmetric and asymmetric.
Symmetric BsAbs are designed to closely resemble native antibodies but vary in size and architecture. In the design of symmetric Fc-based bispecific antibodies, additional antigen-binding fragments are integrated into immunoglobulin G (IgG)’s heavy or light chains, either at the N- or C-termini (Fig. 1f, g). These fragments can be scFv [22] or directly fused Fv without linkers, as seen in dual-variable domain-IgGs (DVD-Ig) (Fig. 1h) [23]. Alternatively, domain antibodies or other scaffold molecules may replace the scFv for antigen binding [24,25,26]. Adding these binding elements to traditional IgGs can significantly impact the molecule’s physicochemical characteristics, influenced by the properties of the added Fvs and their attachment sites [27].
The creation of asymmetric BsAbs often involves sophisticated engineering techniques to ensure the preferential formation of the desired Fc heterodimers. This can include modifications at various domains of the antibody molecule, such as the CH3 domain, to promote specific heavy chain pairing [28] or the use of common light chains to address issues with light chain mispairing [29].
The knobs-into-holes (KiH) strategy (Fig. 1k) is a CH3-engineering strategy widely used for formation of Fc heterodimer. One heavy chain (the “knob” side) is engineered to have a small amino acid protrusion, while the corresponding area on the other heavy chain (the “hole” side) is engineered to have a complementary cavity. When the two halves of the bispecific antibody are mixed, the knob of one heavy chain fits into the hole of the other heavy chain [30]. This interaction helps to ensure that the correct heavy chains pair with each other, promoting the assembly of the intended bispecific antibody rather than mispaired antibody forms. From Atwell et al.’s study, altering Thr366 to the bulkier Tyr (T366Y) in one CH3 domain and Tyr407 to the smaller Thr (Y407T) in the other CH3 domain resulted in 92% heterodimer formation. A similar efficiency in heterodimer production was also observed with the T366W/Y407A mutation pair [31]. Fusing scFv or scFab to the N-terminus of an Fc heterodimer is a good way to avoid mismatches [32, 33].
The common light chain (common LC) approach (Fig. 1l) involves using a single light chain that pairs with two different heavy chains to form the functional regions of the antibody. The approach can reduce the complexity and potential for mispairing of chains and enhance the stability and solubility of the bispecific antibody. In antibodies of common LC design, the heavy chain plays a more crucial role in antigen binding compared to the light chain. The light chain can be replaced without losing the antibody’s ability to bind to its target [34].
The CrossMab format (Fig. 1m) presents a sophisticated solution to the issue of light chain mispairing often encountered in the production of asymmetric IgG bispecific antibodies. This approach involves swapping the CH1 and CL domains within one of the Fab arms of the asymmetric IgG, enabling distinct Fab regions to be produced without the traditional Fd-LC pairing [35].
An early method for producing such asymmetric BsAbs involved the creation of quadromas through the fusion of two hybridomas, leading to the production of what is known as hybrid-IgG antibodies (Fig. 1j). This process leads to the production of cells that express two different heavy and light chains, which can randomly assemble into 16 possible combinations, only one of which will have the desired bispecificity [36].
2.2 Mechanisms of Bispecific Antibodies in Tumor Therapy
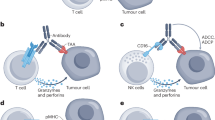
Bispecific antibodies have great potential in tumor therapy. There are several working mechanisms of BsAbs that can be categorized into immune cell engagement, tumor-associated antigen (TAA) targeting, immune checkpoint blockade, and payload delivery (Fig. 2).

Mechanisms of bispecific antibodies (BsAbs) in tumor therapy. a BsAbs engage immune cells (such as T cells or NK cells) with tumor cells by binding to CD3 or CD16 on immune cells and targeting tumor-associated antigens (TAAs) on tumor cells, mediating the release of cytokines by immune cells and triggers antibody-dependent cellular cytotoxicity (ADCC). b An example of TAAs targeting BsAbs: a BsAb targets HER2 and HER3, blocking the downstream signaling pathway. c BsAbs inhibit immune checkpoints like PD-L1 on cancer cells or PD-L1 and CTLA-4 on T cells, blocking inhibitory signals that prevent T cells from attacking tumor cells, thereby modulating the immune responses. d BsAbs can serve as precision delivery vehicles for therapeutic payloads, directly conveying drugs to tumor cells and thus aiding in their treatment
2.2.1 Immune Cell Engagement
Immune cell engagement is the most used mechanism for bispecific antibodies. The application of bispecific antibodies that engage immune cells has been under investigation for over three decades [37]. The heightened interest in cancer immunotherapy is propelling the advancement of bispecific antibodies that engage immune cells [38]. The immune cells engaging BsAbs work by binding to a TAA on cancer cells with one arm and an effector cell, often a T cell or NK cell, with the other, bridging the immune cell to the tumor for targeted attack.
Engagement of CD3+ T cells represents the most common approach in the utilization of bispecific antibodies for immune cell engagement. Additionally, targeting CD16 to engage NK cells and CD64 for the activation of phagocytic cells presents viable strategies [39].
2.2.2 Tumor-Associated Antigen Targeting
These BsAbs focus on inhibiting cancer cell growth by targeting specific tumor-associated antigens (TAAs). By binding to different epitopes or receptors on the tumor cell surface, they can block critical signaling pathways necessary for tumor growth and survival, such as the HER2/HER3 pathway.
MCLA-128, a BsAb targeting HER2 and HER3, acts by inhibiting the interaction between HER3 and its ligand, NRG1. This ligand, NRG1, facilitates the heterodimerization of HER3 with HER2, triggering downstream signaling pathways such as PI3K-AKT and ERK, which are crucial for tumor growth. By blocking NRG1 binding, MCLA-128 interrupts the heterodimerization and these signaling pathways [40].
2.2.3 Immune Checkpoint Blockade
Immune checkpoints are crucial inhibitory pathways within the immune system, essential for preserving self-tolerance and adjusting the strength and duration of immune responses in peripheral tissues [41].
BsAbs in this category aim to modulate the immune system’s response to cancer by blocking inhibitory signals that prevent T cells from attacking tumor cells. They target checkpoint proteins such as PD-1 or CTLA-4, enhancing the immune system’s ability to fight cancer.
MEDI5752 is a monovalent bispecific antibody targeting PD-1 and CTLA-4. In preclinical studies, MEDI5752 preferentially saturated CTLA-4 on PD-1+ tumor-infiltrating lymphocytes (TILs) at significantly lower concentrations than required for PD-1- cells, indicating a targeted mechanism that may reduce toxicity. The results suggest that MEDI5752 could offer an improved therapeutic index over the combination of bivalent αPD-1 and αCTLA-4 mAbs, potentially benefiting cancer treatment with a more favorable safety profile [42].
2.2.4 Payload Delivery
The delivery of therapeutic agents through antibodies has been clinically implemented [43]. Leveraging BsAbs for payload delivery represents an innovative strategy. The payloads can be drugs or radioactive payloads.
ZW-49 is an innovative bispecific antibody-drug conjugate, built on the BsAb zanidatamab, and is coupled with an auristatin toxin via a protease-sensitive linker. ZW-49 has shown promising results in its first-in-human trial [44].
[225Ac]-FPI-2068 is a bispecific IgG-based targeted alpha therapy (TAT) designed to deliver actinium-225 to various solid tumors that co-express EGFR-cMET [45].
TF2 is a tri-Fab BsAb that targets the carcinoembryonic antigen (CEA) with two of its binding sites and the histamine-succinyl-glycine (HSG) with the third. HSG is a synthetic hapten that can be labelled with various radionuclides [46]. Therefore, the BsAbs combined with HSG labelled with radionuclides are suitable for diagnostic imaging.
2.3 Clinical Trials of Bispecific Antibodies for Solid Tumors
Bispecific antibodies have emerged as a promising therapeutic approach in the treatment of solid tumors due to their ability to simultaneously engage two different targets. This dual-targeting strategy can enhance the specificity and efficacy of cancer treatment by combining different mechanisms of action in a single molecule.
To date, over 100 kinds of BsAbs for solid tumors are in clinical trials. In this review, we will systematically examine the current state of these clinical trials (Table 1), organizing our discussion based on the sequence of targets.
2.3.1 HER2
Human epidermal growth factor receptor 2 (HER2) is a transmembrane tyrosine kinase receptor belonging to the epidermal growth factor receptor (EGFR/ErbB) family. HER2 plays a key role in the regulation of cell growth, differentiation, and survival [47]. In many cancer types, especially breast cancer, HER2 is activated by gene amplification or overexpression, which is closely associated with cancer progression and prognosis [48]. HER2 is a very popular target in bispecific antibody research for tumor therapy.
2.3.1.1 TAA Targeting
Zanidatamab (ZW25), KN026, KM257, MBS301 and TQB2930 are bispecific antibodies designed to target two non-overlapping epitopes of HER2. This kind of BsAb shows better binding and an enhanced ADCC effect to kill tumor cells than the HER2 monoclonal antibodies trastuzumab and pertuzumab.
NCT02892123 was a Phase 1 dose-escalation and expansion clinical trial evaluating zanidatamab in patients with locally advanced or metastatic HER2-expressing or HER2-amplified solid tumors. Parts 1 (to identify the maximum tolerated dose, optimal biological dose, or recommended dose) and 2 (to evaluate safety and tolerability) have been completed. Part 1 followed a 3 + 3 dose-escalation design, with intravenous doses ranging from 5 mg/kg to 30 mg/kg administered every 1, 2, or 3 weeks. In Part 1 (n = 46), no dose-limiting toxicities were detected and the maximum tolerated dose was not reached. In Part 2, among the 83 patients evaluated, 31 (37%) achieved a confirmed objective response. Part 3 of the study, focusing on zanidatamab in combination with chemotherapy, is ongoing [49]. In a Phase 2 clinical trial (NCT04466891), zanidatamab has shown a meaningful clinical benefit and a manageable safety profile in patients with treatment-refractory, HER2-positive biliary tract cancer. Eighty-seven patients across 32 sites in nine countries were enrolled from 15 September 2020 to 16 March 2022. Among 80 patients in cohort 1 (HER2-postive), 33 (41.3%) achieved a confirmed objective response. Grade 3 treatment-related adverse events (TRAEs) occurred in 16 (18%) patients, with diarrhea and decreased ejection fraction being the most common. No grade 4 TRAEs or treatment-related deaths were reported [50].
KN026 is currently undergoing a number of clinical trials in China and the USA in various phases, with indications that include breast cancer and gastric/gastro-esophageal junction cancer, among others. In a completed Phase 2 clinical trial (NCT03925974), a total of 45 patients were enrolled from 17 June 2019 to 23 August 2021, including 27 patients classified within the high-level HER2 expression cohort, 14 patients within the low-level HER2 expression cohort, and four patients who exhibited no HER2 expression. Patients received KN026 at varying dosages. The results showed an ORR of 56% in the high-level HER2 cohort, with a median DOR of 9.7 months. The low-level HER2 cohort had a 14% ORR. The most common grade 3 treatment-emergent adverse events were gastrointestinal disorders. KN026 demonstrated promising antitumor activity and a favorable safety profile, particularly in patients with high-level HER2 expression [51]. In another completed Phase 2 clinical trial (NCT04521179), KN026 was combined with another anti-PD-L1 × CTLA-4 BsAb KN046 in the treatment of HER2-positive solid tumors. This combination aims to enhance antitumor activity by simultaneously inhibiting key pathways in tumor growth and immune evasion. Twenty-six patients with HER2-positive tumors were enrolled. Patients received KN026 at a dose of 30 mg/kg every 3 weeks, with an initial loading dose on days 1 and 8 of cycle 1, and KN046 at a dose of 5 mg/kg every 3 weeks. The combination treatment showed promising efficacy, particularly in CRC patients, with a confirmed ORR of 53.8% across all patients and an ORR of 53.3% in CRC patients specifically. The mDOR and mPFS were also notable, especially in CRC patients, indicating a significant therapeutic impact. The treatment was generally well tolerated, with most TRAE being grade 1 or 2. The most common TRAEs included infusion-related reactions and increases in liver enzymes. Severe TRAEs were rare, and there were no treatment-related deaths [52].
MM-111 is a BsAb targeting the HER2/HER3 heterodimer. A preclinical trial showed that MM-111 with trastuzumab synergistically inhibits tumor growth by blocking heregulin (HRG)-mediated activation of HER3 and subsequent signaling through the AKT and ERK pathways [53]. In a completed dose-escalation Phase 1 clinical trial (NCT00911898), 20 patients were enrolled and treated with MM-111. Maximum tolerated dose (MTD) was not attained as no patients experienced a dose-limiting toxicity. Another completed Phase 1 clinical trial (NCT01304784) investigated the safety of combining MM-111 with standard-of-care HER2-targeting regimens in patients with advanced HER2-positive cancer. Testing various drug combinations across five arms, the study involved 86 patients and identified dose-limiting toxicities, without reaching a maximum tolerated dose in most arms [54]. A Phase 2 clinical trial (NCT01774851) assessed MM-111 combined with paclitaxel and trastuzumab for advanced HER2+ gastric/esophageal cancer. However, the trial was halted early due to lack of efficacy [55].
Zenocutuzumab (MCLA-128) is a novel engineered anti-HER2 × HER3 BsAb with modified CH3 and common light chain design [56]. In a Phase 1/2 study (NCT02912949), 116 patients with various kinds of solid tumors (breast, colorectal, endometrium, gastric, lung, ovarian, and others) were enrolled. The study developed a pharmacokinetic model which described MCLA-128 kinetics with a two-compartment model featuring linear and non-linear clearance. Fat-free mass significantly influenced linear clearance and distribution, accounting for variability in pharmacokinetics. Tumor burden impacted non-linear clearance. Simulations supported flat dosing of 750 mg bi-weekly as it provided consistent exposure across patients, affirming its suitability for treating solid tumors with minimal impact from body size parameters on drug disposition [56].
YH32367 (ABL105) is a BsAb targeting HER2 × 4-1BB. 4-1BB, also known as CD137 or TNFSF9, is a member of the tumor necrosis factor receptor (TNFR) superfamily that is expressed on activated T cells, NK cells, and other immune cells. It acts as a potent co-stimulatory molecule, enhancing the survival, proliferation, and effector functions of T cells upon activation. 4-1BB binds to its ligand, 4-1BBL, present on antigen-presenting cells, facilitating powerful immune activation and potential antitumor effects. Agonistic antibodies targeting 4-1BB have shown potential in enhancing tumor-specific immunity by biasing signals towards CD8+ T cells, thereby promoting their survival and acquisition of potent cytolytic properties [57]. The BsAb is designed to overcome HER2 resistance. In the preclinical study, in vitro YH32367 stimulated IFN-γ secretion and tumor cell lysis. In vivo, it demonstrated superior tumor eradication and long-term immunity in mouse models compared to trastuzumab and a 4-1BB agonistic antibody. A 4-week toxicity study in monkeys revealed no adverse effects, indicating a favorable safety profile [58]. A Phase 1/2 clinical trial (NCT05523947) in HER2-positive solid tumors is in the recruiting stage.
2.3.1.2 Immune Cell Engagement
M802 and GBR1302 are novel bispecific antibodies targeting HER2 and CD3, designed to engage T-cells against HER2-expressing tumor cells.
M802 was produced by a KiH strategy. It has an anti-HER2 monovalent unit and an anti-CD3 single chain unit bound to the Fc region. In a preclinical trial, M802 demonstrated enhanced cytotoxicity compared to Herceptin against HER2-positive tumor cells, including those with low HER2 expression or Herceptin resistance [59]. M802 is undergoing a Phase 1 clinical trial NCT04501770.
GBR1302 has an anti-HER2 Fab arm and an anti-CD3 scFv arm bound to the Fc region. A Phase 1 clinical trial (NCT02829372) was performed to assess the safety, tolerability, and preliminary efficacy of GBR1302 for treating patients with HER2-postive cancers. Clinical and PD data suggest T-cell activation at higher GBR1302 dosages [60].
2.3.1.3 Immune Checkpoint Blockade
IMM2902 is a novel recombinant anti-HER2 × CD47-SIRPα bispecific mAb-Trap fusion protein [61]. CD47, often termed the “don’t eat me” signal, is a transmembrane protein that plays a critical role in the immune system’s ability to distinguish between self and foreign cells [62]. It is ubiquitously expressed on the surface of healthy cells and delivers a signal that inhibits phagocytosis through its interaction with signal-regulatory protein alpha (SIRPα) on the surface of macrophages and dendritic cells. CD47 is overexpressed in many types of cancer cells to avoid immune surveillance and destruction [63], including HER2-postive cancer cells. The co-expression of HER2 and CD47 has been notably observed in recurrent BC patients with poor prognoses compared to their primary tumors [61]. With its high affinity to HER2 and CD47 on tumor cell membrane, IMM2902 is capable of targeting HER2-positive cancer cells and reactivating the tumor-engulfing ability of macrophages by interrupting the CD47-SIRPα interaction simultaneously [64]. At present the clinical trials of IMM2902, NCT05076591 and NCT05805956 are in the recruiting stage.
2.3.1.4 Payload Delivery
ZW-49 is a bispecific antibody drug conjugate based on zanidatamab. In a Phase 1 clinical trial (NCT03821233), 76 patients with a median age of 59 years, primarily suffering from gastric (28%) and breast (22%) cancers, were included. Most of them (70%) had previously received HER2-targeted therapy. Adverse events were mostly mild to moderate, with keratitis, alopecia, and diarrhea being the most common. A confirmed objective response rate (ORR) of 28% and a disease control rate of 72% among the 29 patients evaluated for response at the 2.5 mg/kg tri-weekly dose were reported, indicating the potential of ZW-49 as an effective treatment for various cancer types [44].
2.3.2 EGFR
EGFR, also known as HER1 or ErbB1, was the first member of the ErbB family [65]. It is a transmembrane tyrosine kinase receptor, crucial for cell growth, differentiation, and survival. Upon binding with its ligands, such as epidermal growth factor (EGF) and transforming growth factor-alpha (TGF-α), EGFR dimerizes and activates its tyrosine kinase domain, leading to autophosphorylation and triggering various downstream signaling pathways, including the MAPK, AKT, and JNK pathways [66]. EGFR is overexpressed in many cancers, such as NSCLC, colorectal cancer, and head and neck squamous cell carcinoma.
2.3.2.1 TAA Targeting
Amivantamab, MCLA-129, EMB-01 (bafisontamab) and HS-20117 are BsAbs targeting EGFR and c-MET. c-MET, also known as hepatocyte growth factor receptor (HGFR), is a tyrosine kinase receptor involved in cell growth, motility, and morphogenesis, particularly in cancer progression and metastasis [67]. A preclinical study demonstrated that amivantamab showed significant antitumor activity against EGFR Exon 20 insertion mutations in NSCLC across various models [68]. Amivantamab effectively blocks ligand-induced phosphorylation of EGFR and c-MET, surpassing the inhibition achieved by antibodies targeting a single receptor. In NSCLC models driven by EGFR or c-MET, amivantamab induces tumor regression by inhibiting signaling, downmodulating receptors, and leveraging Fc-driven effector functions. Combination with a third-generation EGFR tyrosine kinase inhibitor (TKI) leads to complete, durable regression of lung xenograft tumors [69]. In a Phase 1 clinical trial (NCT02609776) in NSCLC patients, it achieved a 40% overall response rate, including three complete responses, and a median progression-free survival (PFS) of 8.3 months. The most common adverse events were rash, infusion-related reactions, and paronychia [70]. Recently, NCT02609776 evaluated the combination of amivantamab and lazertinib (laz) in treatment-naïve NSCLC patients. After a median follow-up of 33.6 months, 50% remained progression-free on treatment. Median durations of response (DORs), PFS, and overall survival (OS) were not reached, showcasing the combination’s durability [71]. The findings highlight amivantamab’s efficacy for EGFR-mutant NSCLC patients, leading to the FDA granting it breakthrough therapy designation on 10 March 2020 [72]. Amivantamab received accelerated approval from the FDA on 21 May 2021 for the treatment of adult patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations whose disease has progressed on or after platinum-based chemotherapy.
MCLA-158 (petosemtamab) is a BsAb targeting EGFR and LGR5. LGR5, also known as GPR49, is a member of the class A Rhodopsin-like family of G-protein coupled receptors (GPCRs) [73]. It plays a critical role in regulating Wnt signaling, which is vital for cell proliferation, differentiation, and embryonic development. In a Phase 1 dose-escalation clinical trial (NCT03526835), 33 CRC patients were enrolled and treated with MCLA-158 at over 11 dose levels (5–1,500 mg, flat dose). It was well tolerated with a recommended Phase 2 dose (RP2D) of 1,500 mg intravenously (IV) every 2 weeks. No dose-limiting toxicities occurred, with infusion-related reactions being the most common adverse event [74].
SI-B001 is a tetravalent BsAb targeting EGFR and HER3. In a preclinical study, SI-B001 demonstrated antitumor efficacy in various xenograft models. Through pharmacokinetic (PK) and PK/pharmacodynamic (PD) modeling and simulation, effective doses were estimated for different cancers (16 mg/kg per week for colon cancer, 5–7 mg/kg per week for head and neck cancer, and 5–6 mg/kg per week for esophageal cancer), utilizing data from animal studies [75]. In a clinical trial NCT05044897 patients who progressed after anti-PD-1/L1 and platinum-based chemotherapy received SI-B001 at 16 mg/kg IV weekly. Another clinical trial NCT05054439 involved patients with similar progression criteria, treated with SI-B001 at 12 mg/kg IV weekly plus chemotherapy, differentiated by prior paclitaxel exposure. The results revealed an ORR of 22.2% in NCT05044897, while NCT05054439 showed an ORR of 45.5%, with higher responses in the paclitaxel-naïve group. Median PFS varied across groups, with manageable and tolerable toxicity profiles. These findings suggest SI-B001 plus chemotherapy, particularly with paclitaxel, might offer improvements in ORR and DCR for this patient population [76].
2.3.2.2 Immune Cell Engagement
AFM-24 is a novel anti EGFR × CD16a BsAb. It features a tetravalent IgG(H)-scFv format, with the anti-EGFR scFv attached to the C-termini of the silenced Fc region of the anti-CD16a IgG [77]. In a preclinical study, AFM24 was effective in inducing antibody-dependent cellular cytotoxicity and phagocytosis in vitro, showing potency across a range of EGFR expression levels and mutational statuses. In cynomolgus monkeys, AFM24 was well tolerated, with no significant adverse effects observed [77]. In a Phase 1 dose-escalation clinical trial (NCT05099549), six patients (five microsatellite stable colorectal and one head and neck cancer) were enrolled and received treatment with AFM24 in combination with SNK01 cell therapy. Preliminary results from this study indicate no dose-limiting toxicities, with most adverse events being grade 1/2 infusion reactions. Early findings suggest stable disease in some patients, supporting further investigation of this combination therapy’s tolerability and potential efficacy [78].
REGN7075, an IgG4-based co-stimulatory EGFR × CD28 BsAb, is designed to bridge EGFR-positive tumor cells with CD28-positive T cells and to provide amplified T-cell receptor-CD3 complex-mediated T-cell activation within the tumor by leveraging the CD28 co-stimulation pathway [79]. In genetically humanized immunocompetent mouse models, REGN7075, when used in combination with an anti-PD-1 antibody, demonstrated enhanced antitumor activity compared to the use of either agent alone [80]. A Phase 1/2 clinical trial (NCT04626635) evaluating the safety, tolerability, pharmacokinetics, and preliminary antitumor activity of REGN7075 alone and in combination with cemiplimab in solid tumor patients is ongoing. As of September 2022, 30 patients have been treated, with the study continuing to enroll participants [81].
2.3.2.3 Payload Delivery
M1231 is a pioneering bispecific antibody-drug conjugate (ADC) targeting EGFR and MUC-1, fused with a novel hemiasterlin-related microtubule inhibitor payload. The bispecific antibody structure comprises an anti-MUC-1 scFv and an anti-EGFR Fab attached to an Fc region [82]. MUC-1, also known as mucin-1, is a glycoprotein found on the epithelial cell surface. It plays a crucial role in forming a protective mucous barrier and is involved in cellular signaling. MUC-1 is overexpressed in various kinds of cancer cells [83]. A Multi-scale Systems Pharmacology model was developed to address how the ADC’s behavior and interaction with physiological systems lead to the release of its payload inside tumor cells, effectively inhibiting tumor growth. The M1231 internalization and lysosomal trafficking in the MUC1 and EGFR-expressing cancer cell lines MDA-MB-468 and OVCAR-3 was quantified through an in vitro model. Preclinical studies in mice and pharmacokinetics in monkeys helped predict an effective human dose range of 2.4–4.3 mg/kg every 3 weeks, guiding the first-in-human trial’s design (NCT04695847) [84]. The trial has been completed, but currently there are no posted results. M1231 was discontinued in June 2023.
[225Ac]-FPI-2068 is a bispecific IgG-based TAT designed to deliver actinium-225 to various solid tumors that co-express EGFR-cMET [45]. A Phase 1 clinical trial (NCT06147037) is at the recruiting stage.
2.3.3 CEA
CEA, also known as carcinoembryonic antigen-associated cell adhesion molecule 5 (CEACAM5), is a glycoprotein involved in cell adhesion [85]. It is typically present at very low levels in the blood of healthy adults. However, CEA levels can increase significantly in certain types of cancers, making it a valuable tumor marker, especially for colorectal cancer. Elevated CEA levels may also be found in other malignancies such as breast, lung, gastric, pancreatic, and ovarian cancers [86].
2.3.3.1 Immune Cell Engagement
BA1202, RO6958688 (cibisatamab), and MEDI-565 (AMG211) are anti-CEA × CD3 BsAbs of different designs.
BA1202 adopts a novel butterfly-shaped asymmetric Fc-based antibody structure, with one end binding bivalently with high affinity to CEA, and the other end binding monovalently with relatively low affinity to CD3. BA1202 effectively eradicates CEA-positive tumors in preclinical models, showing efficacy at low doses and good safety in toxicity studies [87]. A clinical trial (NCT05909241) has been initiated.
RO6958688 is a BsAb utilizing “knobs-in-holes” technology for its asymmetric Fc structure, where one arm consists of a CEA-binding Fab, and the other arm combines a CEA-binding Fab and a CD3-binding Fab in a unique “head-to-tail” configuration. This allows it to bivalently bind to CEA on tumor cells and monovalently to CD3 on T cells. In vitro and in vivo efficacy experiments, complemented by histology, confocal, and intravital imaging studies, have shown that RO6958688 effectively mediates T-cell-dependent tumor cell lysis by facilitating stable crosslinking of multiple T cells to individual tumor cells [88]. In two dose-escalation Phase 1 clinical trials (NCT02324257 and NCT02650713), RO6958688 was given as monotherapy or in combination with atezolizumab 1,200 mg in adult patients with advanced CEA+ solid tumors. A 5% partial response in NCT02324257 and a 20% response in NCT02324257 were observed. Common adverse events included pyrexia, infusion-related reactions, and diarrhea. Grade ≥ 3 events were primarily infusion-related reactions and diarrhea. There were five DLTs in NCT02324257 and one DLT in NCT02650713, which were likely linked to tumor inflammation [89].
MEDI-565 (AMG211) is a single-chain BsAb of BiTE structure [90]. In a preclinical study, it was tested both in vitro and in vivo, showing specific binding to CEA-positive cells and efficient T-cell-mediated tumor cell lysis. The potency of MEDI-565 was independent of mutations common in colorectal adenocarcinomas, such as KRAS, BRAF, PI3KCA, and TP53 [91]. A completed Phase 1 clinical trial (NCT01284231) evaluated MEDI-565 in gastrointestinal adenocarcinomas. Administered intravenously in 28-day cycles, the study aimed to determine the MTD, assessing pharmacokinetics, antidrug antibodies, and antitumor activity in 39 patients. Dose-limiting toxicities included hypoxia and cytokine-release syndrome, with the MTD established at 5 mg with dexamethasone. Although no objective responses were observed, 28% of patients had stable disease [92]. MEDI-565 was discontinued in 2019.
2.3.3.2 Payload Delivery
TF2 is a tri-Fab BsAb targeting CEA and HSG. In a Phase 2 clinical trial (NCT02587247), 11 CRC patients with no reported adverse effects from BsAb and peptide injections were enrolled. The patients received treatment with 120 nmol of 150 MBq of TF2 followed by 68Ga-IMP-288. Immuno-PET was positive in nine out of 11 patients [93]. The feasibility of the BsAb was also investigated in breast cancer patients with 68 Ga-IMP-288 (NCT01730612) [94], medullary thyroid carcinoma patients with 68 Ga-IMP-288 (NCT01730638), and SCLC patients with 177Lu-IMP-288(NCT01221675). These studies suggested that immuno-PET using TF2 and IMP288 was a safe and feasible approach, offering promising diagnostic performance for several cancers.
2.3.4 EpCAM
EpCAM is a transmembrane glycoprotein involved in cell signaling, adhesion, and proliferation. Predominantly expressed on the surface of epithelial tissues, EpCAM plays a crucial role in cell growth and differentiation [95]. EpCAM is involved in several signaling pathways that relate to cancer cell growth, including the regulation of intramembrane proteolysis (RIP)-mediated signaling and activation of Wnt signaling [96]. Its overexpression is often observed in various cancers, including colorectal, breast, gastric, prostate, ovarian, and lung cancer, making it a target for cancer diagnostics and therapy [97].
Catumaxomab is a trifunctional bispecific antibody produced using quadroma technology, combining mouse IgG2a and rat IgG2b antibodies. It has two antigen-binding sites: one that targets CD3 on T cells and another that binds to the EpCAM antigen on tumor cells. Additionally, its Fc region has the capability of selectively engaging and activating accessory cells expressing Fcγ receptors I, IIa, or III [98]. In preclinical studies, catumaxomab has shown anti-tumor activity in vitro [99,100,101,102,103] and in vivo [104]. A Phase 1/2 clinical trial was conducted to assess the safety and effectiveness of treating ovarian cancer patients with malignant ascites [105]. In a Phase 2 clinical trial, the pharmacokinetics of catumaxomab were explored in patients suffering from malignant ascites [106]. In a pivotal Phase 2/3 clinical trial (NCT00836654), 258 patients with malignant ascites due to epithelial cancer, including ovarian and non-ovarian cancers, were involved. Patients were randomized into two groups: one receiving catumaxomab treatment alongside paracentesis (170 patients) and a control group receiving paracentesis alone (88 patients). Catumaxomab was administered through four intraperitoneal infusions at doses of 10, 20, 50, and 150 μg within 11 days. The results demonstrated that median puncture-free survival was markedly longer in the catumaxomab group, indicating effective management of malignant ascites. The treatment was also associated with a reduction in ascites symptoms, offering a quality-of-life improvement for patients. Additionally, the adverse events were manageable [107]. A Phase 1 study was conducted to assess the safety and tolerability of intravenous catumaxomab treatment in NSCLC patients. Including stage IB-IV NSCLC patients with at least one prior therapy, the study identified dose-limiting toxicities related to liver enzyme elevation, determining the maximum tolerated dose. The findings suggest that a starting dose of 5 µg of catumaxomab, preceded by 40 mg dexamethasone and antihistamines, is viable for further treatments in NSCLC patients [108]. A Phase 1 study evaluated catumaxomab’s efficacy for peritoneal carcinomatosis from gastrointestinal cancer via intraperitoneal infusions in 24 patients. The maximum tolerated dose was identified, with fever, vomiting, abdominal pain, skin toxicity, and nausea being the most common adverse events. A post hoc analysis revealed significantly longer median survival for study participants compared to those receiving conventional chemotherapy [109]. In a Phase 2 clinical trial (NCT01504256), combining catumaxomab with chemotherapy for treating peritoneal carcinomatosis in gastric cancer patients proved to be a viable and well-tolerated strategy [110]. Catumaxomab received European Medicines Agency (EMA) approval in 2009 as the first bispecific antibody or trifunctional antibody to be approved [111]. Its journey in the EU market concluded in 2017 following discontinuation of sales in 2014, attributed to its lackluster market performance [112]. Despite its withdrawal, catumaxomab remains the subject of ongoing clinical trials aiming to further unlock its therapeutic potential.
MT110 (solitomab) is a novel anti EpCAM × CD3 BsAb of BiTE structure. MT110 has been shown to trigger a broad activation of both CD4- and CD8-positive T cells, with CD8 cells primarily driving tumor cell lysis. The antibody demonstrated strong efficacy in preclinical models, completely preventing tumor growth with early treatment and eradicating established tumors with later dosing [113]. In a Phase 1 dose-escalation study (NCT00635596), 65 patients with solid tumors (colorectal, ovarian, gastric, lung, and prostate) received MT110 via continuous IV infusion across various doses. The study encountered dose-limiting toxicities, including severe diarrhea and increased liver enzymes, leading to a maximum tolerated dose of 24 mg/day. One unconfirmed partial response was observed [114]. MT110 was discontinued in 2015.
Two further EpCAM × CD3 BsAbs are BA3182 and M701, for which clinical studies have been initiated. A Phase 1 trial (NCT05808634) is going to evaluate BA3182 in adenocarcinoma and a Phase 2 trial (NCT06266091) is going to evaluate M701 for treating malignant ascites caused by gastrointestinal or ovarian cancer.
2.3.5 GD2
GD2 is a disialoganglioside, a type of glycolipid found on the surface of cells, particularly in the nervous system and on tumors of neuroectodermal origin, such as neuroblastoma, and melanoma [115]. GD2’s presence on the neuroblastoma cell membrane is universal across all primary tumors, without relation to the tumor’s stage. It is highly abundant, with an estimated count ranging up to 106 molecules per cell [116]. Due to its limited expression in normal tissues and high expression on certain tumor cells, GD2 has become a target for cancer immunotherapy.
Nivatrotamab is a novel GD2-targeted bispecific antibody that combines a humanized anti-CD3 component (huOKT3 scFv) with a humanized anti-GD2 part (hu3F8), linked at the carboxyl end of the IgG1 light chain. In a Phase 1/2 clinical trial (NCT02173093), the potential of GD2-targeted bispecific antibody armed T cells (GD2BATs) was explored in treating patients with GD2-positive tumors, including neuroblastoma, osteosarcoma, and desmoplastic small round cell tumor. Twelve patients were enrolled. They received bi-weekly infusions of GD2BATs alongside IL-2 and GM-CSF. The trial demonstrated that such treatment is feasible and safe, with some patients showing signs of tumor response or stable disease [117]. Nivatrotamab was also involved in two clinical trials, NCT04750239 and NCT03860207, which progressed to the stage of patient recruitment and preliminary data collection. However, both trials were ultimately terminated due to shifts in business priorities.
2.3.6 GPC3
Glypican-3 (GPC3) is a cell surface oncofetal protein highly expressed in various pediatric solid embryonal tumors, including hepatoblastomas, Wilms’ tumors, rhabdoid tumors, certain germ cell tumor subtypes, and some rhabdomyosarcomas. GPC3 plays a crucial role in cell growth and differentiation by interacting with growth factors and activating the canonical Wnt/β-catenin pathway, which is often overexpressed in these malignancies [118].
ERY974 is an anti-GPC3 × CD3 BsAb. The combined effect of ERY974 with chemotherapy has been confirmed on non-inflamed tumors in a xenograft model. While ERY974 alone showed limited antitumor effects due to restricted T-cell infiltration to the tumor-stromal boundary, combination therapy enhanced efficacy by promoting deeper T-cell infiltration into the tumor and increasing ERY974 distribution within the tumor. Additionally, ERY974 boosted the cytotoxicity of capecitabine by inducing thymidine phosphorylase expression in tumors, aiding in the drug’s activation [119]. A Phase 1 study (NCT02748837) explored ERY974 in patients with advanced solid tumors expressing GPC3, aiming to determine the maximum tolerated dose of ERY974. The study enrolled 29 patients across dose levels up to 0.81 μg/kg, identifying TRAEs including cytokine-release syndrome (CRS) and pyrexia. The study observed biologic activity with one partial response in esophageal cancer and stable disease in four patients [120]. No further developments have been made since 2019.
2.3.7 5T4
The 5T4 oncofetal antigen, also known as trophoblast glycoprotein (TBPG), is a target for cancer immunotherapy due to its restricted expression in normal tissues but broad expression across many cancers. It is a 72 kD, heavily N-glycosylated protein with several leucine-rich repeats, often associated with protein-protein interactions. The expression of 5T4 is linked with processes like epithelial mesenchymal transition, enhancing chemotaxis and affecting Wnt signaling pathways, which facilitate cancer cell spread [121].
2.3.7.1 TAA Targeting
ALG.APV-527, designed using the ADAPTIR bispecific platform, targets both 5T4 and 4-1BB. It combines two distinct scFvs, one targeting 5T4 and the other targeting 4-1BB, both linked to the hinge and Fc-domain of an IgG1 molecule. In preclinical models, ALG.APV-527 demonstrated potent antitumor activity without significant systemic toxicity. This effect was observed across various cancer types expressing the 5T4 antigen. The treatment effectively activated T cells within the tumor microenvironment, leading to tumor regression [122]. The Phase 1/2 clinical trial (NCT05934539) is at the recruiting stage.
2.3.7.2 Immune Cell Engagement
GEN1044 is a bispecific antibody targeting CD3 on T cells and 5T4 on tumor cells. In vitro, the antibody demonstrated dose-dependent cytotoxicity, dependent on the interaction between T cells and 5T4-expressing tumor cells, with robust T-cell activation and cytokine release. In vivo, it showed antitumor activity in humanized mouse models of breast, prostate, and lung cancers, alongside biomarkers indicative of T-cell activation [123]. A Phase 1/2 clinical trial (NCT04424641) evaluating GEN1044 in patients with malignant solid tumors was terminated after reaching the MTD.
2.3.8 HLA-G
HLA-G is a non-classical HLA class I molecule known for its immunomodulatory properties, initially identified at the maternal-fetal interface, indicating its role in maintaining fetal-maternal immune tolerance. It is also found in specific immune-privileged tissues and plays a crucial role in various processes including organ transplantation, viral infections, cancer progression, and autoimmunity. HLA-G can inhibit immune system cells through its tolerogenic function, aiding in tumor escape. Polymorphisms in the HLA-G gene, particularly in the 3’UTR region, affect its expression and have been linked with different tumor types and autoimmune diseases [124]. HLA-G, typically exhibiting restricted expression in normal tissues, is found across a diverse range of human cancers [125].
JNJ-78306358 is a BsAb developed using Zymeworks’ Azymetric™ and EFECT™ therapeutic platforms [126]. It targets HLA-G and CD3, redirecting T cells to kill HLA-G-expressing tumor cells. JNJ-78306358 combines high-affinity binding to HLA-G with weaker affinity to CD3ε, facilitating specific tumor cell destruction while minimizing interaction with Fcγ receptors. Its efficacy and T-cell activation capabilities have been demonstrated in vitro and in humanized mouse models [127]. A Phase 1 study (NCT04991740) explored JNJ-78306358 treatment in patients with solid tumors expressing HLA-G, including renal cell carcinoma, ovarian cancer, and colorectal cancer. The study, involving 39 participants, evaluated safety, pharmacokinetics, pharmacodynamics, and preliminary efficacy. Common adverse events included CRS, liver enzyme elevation, and injection site reactions. Dose escalation was hindered by CRS-related toxicities, leading to a decision to halt further escalation without establishing a recommended Phase 2 dose [125].
2.3.9 DLL4 × VEGF
DLL4 (delta-like ligand 4) and VEGF (vascular endothelial growth factor) are crucial in angiogenesis, the process of new blood vessel formation [128]. DLL4 is part of the Notch signaling pathway, playing a pivotal role in vascular development and tumor angiogenesis by regulating the sprouting and branching of blood vessels. VEGF, on the other hand, is a key regulator of vascular permeability and angiogenesis, promoting the growth of blood vessels, particularly in the context of cancer, where it supports tumor growth by enhancing blood supply. As shown in previous studies, blocking the DLL4 signaling pathway in blood vessels leads to increased VEGF levels and endothelial cell overproliferation. Consequently, targeting both DLL4 and VEGF simultaneously could overcome resistance to DLL4 inhibitors and enhance antitumor effects [129]. Research in ovarian cancer models has demonstrated that the combined inhibition of VEGF and DLL4 offers significantly greater antitumor efficacy compared to using either agent alone. This approach suggests a promising strategy for improving cancer treatment outcomes by simultaneously disrupting these key angiogenic pathways [130]. Currently, two bispecific antibodies targeting both DLL4 and VEGF, navicixizumab (OMP-305B83) and ABL001 (CTX-009, NOV1501), are undergoing clinical trials.
Navicixizumab is an IgG-like BsAb. In a Phase 1a clinical trial (NCT02298387), 66 patients received various doses in a 3 + 3 dose-escalation format, leading to the selection of 7.5 mg/kg for the expansion cohort, with a focus on ovarian cancer for further trials. Most common TRAEs were hypertension and headache, with preliminary signs of antitumor activity observed, particularly in ovarian cancer. The maximum tolerated dose was not reached, but doses up to 7.5 mg/kg were manageable with acceptable safety profiles [131]. Navicixizumab was further explored in a Phase 1b study (NCT03030287), assessing its combination with paclitaxel for treating patients with platinum-resistant ovarian cancer. Enrolling 44 patients, it tested navicixizumab doses alongside paclitaxel, finding no dose-limiting toxicities and identifying common adverse events. The study reported an overall ORR of 43.2%, with promising results especially in patients previously untreated with bevacizumab [132].
A Phase 1a study (NCT03292783) explored the safety and efficacy of ABL001 in patients with advanced cancers. Conducted with a classic 3 + 3 dose-escalation design, ABL001 showed no dose-limiting toxicities up to 7.5 mg/kg, with common adverse events including hypertension and anemia. Preliminary results indicate promising antitumor activity, particularly in a gastric cancer patient, without identifying a MTD [133]. A Phase 2 study (NCT04492033) further evaluated the efficacy and safety of ABL001 combined with paclitaxel or irinotecan in advanced solid tumor patients, focusing on biliary tract cancer. With 24 patients enrolled, the study reported a promising ORR, especially in second- and third-line (2L/3L) settings, despite a high rate of TRAEs [134].
2.3.10 DLL3
DLL3 (delta-like ligand 3) is an inhibitory ligand in the Notch signaling pathway [135]. It is highly expressed on the surface of SCLC cells, while its expression in normal tissues is minimal [136]. This makes DLL3 a promising therapeutic target for SCLC.
Tarlatamab, also known as AMG 757, is a first-in-class anti DLL3 × CD3 bispecific antibody featuring a half-life extended bispecific T-cell engager (HLE BiTE) molecule [137]. In preclinical SCLC models, tarlatamab promotes tumor regression [138]. In a Phase 1 study (NCT03319940), 107 patients were recruited and received tarlatamab in dose-exploration (0.003–100 mg; n = 73) and expansion (100 mg; n = 34) cohorts. The ORR was 23.4%, with two complete responses and 23 partial responses. The median duration of response was 12.3 months. The disease control rate was 51.4%. The median PFS was 3.7 months, and the median OS was 13.2 months. The MTD was not reached. Treatment-related adverse events of any grade occurred in 90.7% of patients, with 30.8% experiencing grade 3 or higher events. Grade 5 pneumonitis was reported in 1% of patients. The most common TRAE was CRS [137]. Tarlatamab showed manageable safety and encouraging response durability. In May 2024, tarlatamab was approved by the FDA for the treatment of SCLC patients [139].
2.3.11 PSMA
PSMA, also known as glutamate carboxypeptidase II, is a type II transmembrane protein associated with folate hydrolase activity, primarily produced by cells in the prostatic epithelium [140]. Its expression significantly increases in androgen-independent prostate cancers, making it a distinct target for cancer therapy due to its limited expression in non-prostatic tissues [141].
Several BsAbs targeting PSMA and CD3, including acapatamab, pasotuxizumab, CC-1, ES414, HPN424, and JNJ-63898081, are currently being explored in clinical trials.
Acapatamab, also known as AMG 160, incorporates a BiTE (bispecific T-cell engager) structure attached to an Fc region, enhancing its half-life [142]. In preclinical studies, it showed potent and specific activity against PSMA-expressing prostate cancer cells and exhibited significant tumor regression in mCRPC xenograft models [143]. A Phase 1 study assessed acapatamab in patients with mCRPC who were refractory to standard therapies. Administered IV every 2 weeks across various dosages, acapatamab primarily caused CRS, with the most severe cases observed in the first treatment cycle. Despite this, CRS became less frequent and severe in subsequent cycles. The study found promising preliminary efficacy, including prostate-specific antigen responses and radiographic partial responses, alongside notable T-cell activation and increased cytokine production post-treatment initiation. However, treatment-emergent antidrug antibodies were detected in a significant number of patients, affecting serum exposures in some cases [144]. Acapatamab was discontinued in 2023.
Pasotuxizumab, or BAY2010112, is constructed in the BiTE format. A Phase 1 study (NCT01723475) explored pasotuxizumab for mCRPC refractory to standard therapies. Involving 16 patients across five dosing cohorts, the trial aimed to determine safety, MTD, and observe antitumor activity. Despite all participants experiencing adverse events, the therapy showed dose-dependent clinical activity, including PSA declines and significant tumor regressions in some cases [145]. No developments have been made since 2021.
ES414, or APVO414, is constructed in the ADAPTIR™ format, which consists of an Fc part and two scFv fragments for each specificity [146]. While the preclinical research showed promise for APVO414 in treating mCRPC, the clinical trial was halted due to significant immunogenicity concerns. This was evidenced by the development of anti-drug antibodies (ADAs) in a notable proportion of patients, even after adjusting the administration method to continuous infusion [147]. Consequently, the development of APVO414 was discontinued.
2.3.12 PD-1, PD-L1, and CTLA-4
Immune checkpoint blockade refers to a cancer treatment strategy using therapeutic antibodies to block regulatory checkpoints that typically inhibit immune responses [148]. This approach aims to activate the immune system’s ability to fight cancer by disrupting the mechanisms cancer cells use to evade immune detection. Particularly, inhibitors targeting PD-1/PD-L1 and CTLA-4 have shown promising results, leading to their approval for treating specific cancers [149]. Merging monoclonal antibodies that inhibit immune checkpoints into bispecific antibodies via antibody engineering represents a cutting-edge and potent strategy.
LY3434172 and IBI318 are both IgG-like BsAbs targeting PD-1 and PD-L1. LY3434172 has shown significant antitumor effectiveness in human xenograft models at doses much lower than those required by either of its parent antibodies or their combination [150]. The Phase 1 clinical trial for LY3434172 in advanced solid tumors (NCT03936959) has concluded, although the results have yet to be published and there is no reported further development. The clinical trial for IBI318 in NSCLC (NCT04777084) is at the recruiting stage.
LY3415244 is an anti-PD-L1 × TIM-3 BsAb. TIM-3, or T-cell immunoglobulin and mucin domain–containing molecule 3, functions as a negative regulator of T cells that secrete interferon-gamma. A Phase 1 multicenter open-label study (NCT03752177) enrolled 12 patients with advanced solid tumors across four cohorts, with doses escalating from 3 mg to 70 mg every 2 weeks. The study encountered significant immunogenicity issues, leading to an early termination. All patients developed treatment-emergent antidrug antibodies (TE-ADAs), with some experiencing clinically significant anaphylactic infusion-related reactions [151], no further development has been undertaken.
ABL501 is a BsAb targeting PD-L1 and LAG-3, designed with an anti-PD-L1 scFv attached to the C-terminus of an anti-LAG-3 IgG heavy chain. LAG-3, or lymphocyte-activation gene 3, is a protein functioning as an immune checkpoint receptor. ABL501 effectively inhibits both LAG-3 and PD-L1 pathways, enhancing effector CD4+ and CD8+ T cell activation. In a humanized mouse model, ABL501, compared to its parental antibodies, significantly reduced tumor growth and enhanced tumor infiltration by CD8+ T cells. It also increased the production of key effector molecules like IL-2, IFN-g, and granzyme B in CD8+ T cells within the tumor [152]. A Phase 1 clinical trial (NCT05101109) for ABL501 is at the recruiting stage.
IBI322, designed from the “knobs-into-holes” strategy, is a BsAb that simultaneously targets PD-L1 and CD47, adopting a “1 + 2” format. IBI322 demonstrated effective tumor cell binding and potentiation of immune responses in vitro, with significant tumor growth inhibition observed in vivo. The treatment was well tolerated in non-human primates, indicating a favorable safety profile [153]. In Phase 1 studies (NCT04328831, NCT04912466), 58 patients with solid tumors were enrolled, and were administered IBI322. A single dose-limiting toxicity was observed; common TRAEs were anemia and thrombocytopenia. Among patients treated with active doses, 20% achieved partial response and 35% stable disease [154]. A Phase 2 clinical trial (NCT05296603) is currently underway to investigate the combined effectiveness of IBI322 and lenvatinib in treating patients with extensive stage SCLC.
MCLA-145, FS222, ES101, ATG-101, ABL-503, and PRS-344 are anti PDL1 × 4-1BB BsAbs. MCLA-145 shows high affinity for both 4-1BB and PD-L1. It activates T cells, even under suppressive conditions, enhances T-cell memory, and overcomes immunosuppression. The treatment was well tolerated in non-human primates [155]. A Phase 1 clinical trial (NCT03922204) explores MCLA-145 for treating advanced or metastatic solid tumors. Thirty-four patients have been enrolled, experiencing mostly fatigue and neutropenia as TRAEs. Preliminary evidence of antitumor activity and dose-dependent exposure were observed, with pharmacodynamic and clinical activity noted at doses of 25 mg and above [156].
FS222 is a tetravalent IgG-like BsAb. In vitro and in vivo experiments demonstrated that FS222 effectively activates T cells and exhibits potent antitumor activity in colorectal cancer models without significant toxicity. The antibody was shown to have mAb-like pharmacokinetics and was effective in syngeneic mouse tumor models [157]. A Phase 1 clinical trial (NCT05159388) for FS222 is enrolling patients with specific solid tumor types who have progressed on standard-of-care therapy [158]. There have been no updates since 2022.
PRS-344 utilizes a novel antibody-Anticalin fusion design. The anti-4-1BB Anticalin protein is fused to the C-terminus of the anti-PD-L1 IgG heavy chain. In the preclinical study, PRS-344 demonstrated high affinity for its targets, blocking the PD-1/PD-L1 interaction effectively and inducing T-cell activation dependent on PD-L1 presence. PRS-344 showed dose-dependent efficacy in non-clinical mouse models, including those resistant to anti-PD-L1 therapy [159]. A Phase 1 clinical trial (NCT05159388) for PRS-344 is at the recruiting stage.
XmAb23104 is an innovative BsAb featuring an anti-ICOS Fab linked to an anti-PD-1 scFv, along with an Fc region [160]. ICOS, a co-stimulatory molecule, becomes rapidly upregulated on activated CD4 and CD8 T cells, indicating its potential role in regulating adaptive T-cell responses [161]. A Phase 1 study (NCT03752398) evaluated XmAb23104 in patients with advanced solid tumors. The dose-escalation phase did not identify an MTD but selected a dose of 10 mg/kg for expansion based on safety, pharmacokinetics, and clinical activity. The drug was generally well tolerated, with manageable TRAEs. Preliminary efficacy included partial responses and stable disease [162]. A Phase 2 clinical trial (NCT05695898) planned to investigate the combination of XmAb23104 with XmAb22841, a CTLA-4 × LAG-3 BsAb, in patients with melanoma who have not responded to standard immune checkpoint inhibitors [163]. XmAb23104 was discontinued in 2023.
Ivonescimab, or AK112, is a first-in-class BsAb targeting PD-1 and VEGF. A Phase 2 study evaluated AK112 in combination with chemotherapy in patients with advanced NSCLC across three cohorts based on prior treatments and genetic mutations. The study showed promising antitumor efficacy and a manageable safety profile across all cohorts, with significant improvements in ORR and PFS, suggesting AK112 plus chemotherapy as a potential superior treatment option for advanced NSCLC [164].
Anti-PD-1 × CTLA-4 BsAbs include MEDI5752 as discussed above, SI-B003, AK104 (cadonilimab), MGD-019 (lorigerlimab), and XmAb20717 (vudalimab).
SI-B003 is a tetravalent BsAb. A Phase 1 clinical trial (NCT04606472) evaluated SI-B003 in patients with solid tumors who had failed standard therapy. Administered every other week, it showed a median PFS of 3.7 months across 60 patients. Among 56 evaluable patients, the ORR was 16.1%, with a disease control rate of 50.0%. Notably, the treatment demonstrated activity in patients previously treated with PD-1/L1 inhibitors, with a manageable safety profile [165].
As mentioned in Sect. 1 (Background), AK104 (cadonilimab) has been approved for cervical cancer. In the preliminary study, AK104 shows promising antitumor effects in specific cancers and has a more favorable safety profile compared to separate treatments with anti-PD-1 and anti-CTLA-4 antibodies [166]. A Phase 2 clinical trial (NCT04868708) investigated AK104 combined with standard chemotherapy as first-line treatment for recurrent/metastatic cervical cancer, involving three cohorts with different doses and combinations. The trial highlighted promising efficacy and manageable toxicity of AK104 [167]. Investigation with AK104 extends to combination treatments with various agents across different clinical trials, including lenvatinib (NCT04728321), chiauranib (NCT05505825), bevacizumab (NCT05930665), AK119 (NCT05559541), AK117 (NCT05235542), AK112 (NCT06196775), and chemotherapy or chemoradiotherapy (NCT05430906, NCT05377658, NCT05522894, NCT06310473, NCT05587374).
MGD019 is a tetravalent bispecific Fc-bearing DART molecule [168]. MGD019 was evaluated in a Phase 1 study (NCT03761017) for its safety, pharmacokinetics, and antitumor effects in four tumor-specific cohorts. Administered at 6 mg/kg intravenously every 3 weeks, preliminary results from the mCRPC cohort highlight its manageable safety profile and encouraging antitumor activity, with an ORR of 25.7% [169]. There is also an ongoing Phase 1 study (NCT05293496) exploring the combination of MGD019 with anti-B7-H3 MGC018 in patients with advanced solid tumors [170].
XmAb20717 utilizes Xencor’s XmAb® Bispecific Fc Domain as the framework, featuring two antigen-binding sites attached to a Fc region [171]. In a Phase 1 study (NCT03517488), 34 patients across six dose cohorts received XmAb20717, showing generally tolerable safety profiles and signs of pharmacodynamic activity indicative of dual PD-1/CTLA-4 blockade. Among treated patients, there was one reported complete response in melanoma previously unresponsive to pembrolizumab [172]. A Phase 2 study aims to explore the potential benefits of combining vudalimab with chemotherapy or targeted agents in enhancing antitumor immunity across different mCRPC subgroups, with enrollment already underway [173].
CDX-527 is a BsAb combining anti-PD-L1 and CD27 targeting domains in an IgG-scFv format. CD27 is a key immunostimulatory molecule that enhances T-cell activation, effector function, and survival. In preclinical studies, CDX-527 was found to enhance antigen-specific T-cell responses significantly more than the combination of its parental antibodies in human CD27 transgenic mice [174]. CDX-527 was evaluated in a Phase 1 trial for safety, pharmacokinetics, pharmacodynamics, and clinical activity in patients with advanced solid tumors. Doses up to 1 mg/kg have been well tolerated, with no severe adverse events or discontinuations due to adverse effects [175]. CDX-527 was discontinued in 2022.
KN046 (erfonrilimab) is a first-in-class humanized PD-L1/CTLA-4 BsAb. It combines an anti-PD-L1 single domain antibody and an anti-CTLA-4 single domain antibody, integrated with a Fc region [176]. In a Phase 1 clinical trial (NCT03529526), including various solid tumors, patients were enrolled across multiple dosing regimens. The majority experienced TRAEs, mostly mild or moderate, with some grade ≥ 3 events reported. Among 25 evaluable patients, objective responses were observed [177]. A Phase 2 clinical trial (NCT04324307) explores KN046 combined with nab-paclitaxel/gemcitabine for advanced pancreatic ductal adenocarcinoma (PDAC). Early results from 17 patients show a 55.6% ORR and 88.9% disease control rate, with manageable side effects [178]. As mentioned earlier, KN046 was combined with KN026 for treatment of HER2-postive solid tumors in a Phase 2 clinical trial (NCT04521179). The trial achieved a notable outcome, with a confirmed ORR of 53.8% [52]. KN046 is undergoing Phase 3 trials for NSCLC (NCT04474119) and pancreatic cancers (NCT05149326).
2.3.13 B7-H3 (CD276)
B7-H3, also known as CD276, is a member of the B7 family, identified as a promising target for antibody-based immunotherapy due to its overexpression in various cancers, limited heterogeneity, and high frequency across different cancer types. It plays a dual role in the immune system and tumorigenesis, including inhibiting tumor antigen-specific immune responses and promoting tumor growth and invasion through non-immunological functions. Despite its potential, the development of B7-H3 blocking antibodies has been limited, mainly because the B7-H3 receptor remains unknown [179].
For T cells to be fully activated within the tumor environment, they must receive stimulation through their T-cell receptor (TCR, Signal 1) and also engage with co-stimulatory receptors such as CD28 (Signal 2). XmAb808 is a tumor-selective XmAb® 2 + 1 BsAb targeting B7-H3 on tumor cells and the CD28 co-receptor on T cells [180]. By delivering “Signal 2” within the tumor microenvironment, XmAb808 is expected to boost antitumor responses when combined with other treatments such as CD3-targeting bispecific T-cell engagers and immune checkpoint blockers [181]. A Phase 1 first-in-human clinical trial (NCT05585034) aims to study the safety, tolerability, and pharmacokinetics of XmAb808, as well as its initial efficacy and pharmacodynamic effects in combination with pembrolizumab in patients with specific advanced solid tumors.
CC-3 is a B7-H3xCD3 BsAb leveraging an IgG format with a UCHT-1-derived low-affinity anti-CD3 sequence to minimize side effects. It has shown promising in vitro efficacy, activating T cells, fostering proliferation and memory, and efficiently lysing tumor cells [182]. In vivo, CC-3 exhibited strong therapeutic effectiveness, both in a lung metastasis model and in eliminating established tumors in immunocompromised mice, without toxicity in the absence of target cells [183]. A Phase 1 first-in-human clinical trial (NCT05999396) is designed to assess the safety and effectiveness of CC-3 in treating patients with colorectal cancer.
MGD009 is a B7-H3xCD3 BsAb in the DART format. A Phase 1 clinical trial (NCT03406949) explores the combination of MGD009 and an anti-PD-1 MGA012 in patients with B7-H3-expressing unresectable, locally advanced, or metastatic solid tumors [184]. MGD009 was discontinued in April 2022.
2.3.14 GP100
Glycoprotein 100 (Gp-100) is a protein that is highly expressed in both melanocytes and melanoma [185].
Tebentafusp, also known as IMCgp100, is a first-in-class anti gp-100 × CD3 bispecific antibody featuring the immune-mobilizing monoclonal T-cell receptors against cancer (ImmTAC) [186]. In preclinical studies, tebentafusp demonstrated a high affinity for the pHLA target, allowing it to detect cells with low levels of HLA-A-gp100280–288 on their surface [187]. In a Phase 1 study (NCT01211262), 31 patients received tebentafusp ranging from 5 ng/kg to 900 ng/kg. The MTD was determined to be 600 ng/kg weekly. Confirmed partial responses were seen in four patients (two with uveal melanoma and two with cutaneous melanoma, including patients refractory to checkpoint inhibitors), and 12 patients had stable disease. Common adverse events of any grade included rash (100%), pruritus (64%), pyrexia (50%), and periorbital edema (46%). Grade 3 or 4 related adverse events were rash (23%), hypotension (6%), and lymphopenia (8%). Tebentafusp demonstrated a favorable safety profile and durable responses in both cutaneous and uveal melanoma [188]. In January 2022, tebenfafusp received approval by the FDA for the treatment of unresectable or metastatic uveal melanoma patients [189].
2.3.15 Other Targets
In addition to the targets reviewed above, numerous other promising targets for solid tumor BsAbs are currently being explored in clinical trials. These include B7-H4 (ABL-103, GEN1047), ENPP3 (JNJ-87890387), CLDN6 (XmAb541), CLDN18.2 (AK132, AZD5863), STEAP1 (AMG509), ICAM-1, CD38 (VP301), ROR-1 (EMB-07), mesothelin (NI-8001), OX40 (EMB-09), TGF-β (Y101D), LILRB2 (SPX-303), TIGIT (HLX301, AZD2936), and ITL4 (CDX-585). Each of these targets represents a unique approach to cancer therapy, with BsAbs designed to engage various mechanisms of action.
There are also some valuable targets being explored at the preclinical development stage. For example, GPA33, a type II transmembrane protein, is particularly notable for its significant overexpression in androgen-independent prostate cancers. MGD007, an anti-GPA33 × CD3 BsAb in DART format, demonstrated promising antitumor activity in preclinical studies [190].
2.4 Challenges and Prospects for Solid Tumor Bispecific Antibodies
Bispecific antibodies have revolutionized cancer therapy, especially through their ability to redirect the immune system to combat cancer cells. However, their clinical application faces multiple challenges that necessitate comprehensive research and technological innovation to overcome.
2.4.1 Safety and Tolerability Issues
While BsAbs can activate T cells to fight tumors, this may also cause overactivation of the immune system, leading to severe immune-related adverse events such as CRS and autoimmune reactions. These adverse events are serious and can significantly impact patient safety. Therefore, it is crucial to develop strategies that optimize treatment regimens to minimize these risks while maintaining therapeutic efficacy.
2.4.1.1 Cytokine-Release Syndrome
Cytokine-release syndrome (CRS) is a potentially life-threatening condition characterized by the rapid release of large amounts of cytokines into the bloodstream [191]. Symptoms can range from mild, flu-like symptoms to severe reactions, including high fever, hypotension, and multi-organ failure. Managing CRS often requires immediate medical intervention and may involve the use of immunosuppressive agents like corticosteroids or tocilizumab, an IL-6 receptor antagonist.
2.4.1.2 Autoimmune Reactions
Autoimmune reactions occur when the immune system mistakenly attacks the body’s own tissues. In the context of BsAb therapy, this can lead to a range of complications, depending on the tissues or organs affected. These reactions can be challenging to predict and manage, necessitating close monitoring and potentially requiring long-term immunosuppressive treatment.
2.4.1.3 Catumaxomab Case
Catumaxomab, the pioneering bispecific or trifunctional antibody to receive approval, encountered a critical safety issue due to its unmodified Fc region. This Fc region engaged off-target with FcγR-expressing Kupffer cells in the liver, initiating immune responses even in the absence of TAA, which led to severe hepatotoxicity. Tragically, this adverse effect resulted in a patient’s death [15, 192]. In response to this incident, contemporary CD3-targeting BsAbs either completely lack the Fc region or incorporate mutations within the Fc domains to eliminate interactions with Fcγ receptors and the complement component C1q, aiming to reduce off-target effects and mitigate the risk of severe immune-related adverse events [10, 15].
2.4.2 Selectivity and Specificity
Although BsAbs aim to improve therapy selectivity by targeting specific tumor markers, tumor heterogeneity and low-level expression of markers in normal tissues may lead to nonspecific binding and toxicity issues. Tumor heterogeneity refers to the existence of diverse cancer cell populations within the same tumor. This diversity can occur at multiple levels, including genetic, phenotypic, and functional variations among cells [193]. This means that not all cancer cells within a tumor express the target antigens uniformly. Consequently, some tumor cells may escape recognition and destruction by BsAbs, leading to incomplete therapeutic efficacy. Additionally, if the targeted markers are also present, albeit at lower levels, in normal tissues BsAbs may bind nonspecifically, resulting in off-target effects and toxicity. Designing BsAbs with greater selectivity and specificity is a current research focus.
One promising approach to overcoming these challenges is the development of simultaneous multiple interaction t-cell Engagers (SMITEs). SMITEs represent an innovative subclass of bispecific antibodies designed to augment the performance of traditional BiTEs. Structurally, SMITEs consist of two distinct BiTE molecules, each targeting specific epitopes. One BiTE is engineered to target a tumor-associated antigen (TAA) alongside CD3, while the other is focused on engaging CD28, which may target the same or a different antigen [194]. This multi-targeting approach can increase the specificity of BsAbs and mitigate the risk of immune evasion by tumors.
Additionally, combining BsAbs with other therapies, such as checkpoint inhibitors, chemotherapy, or targeted therapies, can address the challenge posed by tumor heterogeneity and enhance treatment efficacy. This combination strategy ensures more comprehensive tumor cell clearance.
2.4.3 Immunogenicity
Immunogenicity is a significant concern in the development of bispecific antibodies (BsAbs) due to their heterogeneous nature. Patients may develop immune responses against these therapies, leading to reduced therapeutic efficacy and potential adverse reactions. The immune system can recognize the foreign elements of BsAbs, such as non-human sequences or novel structural configurations, triggering the production of anti-drug antibodies (ADAs) [195]. These ADAs can neutralize the therapeutic antibodies or accelerate their clearance from the body, thereby diminishing their clinical benefits.
Reducing immunogenicity is crucial for the clinical success of BsAbs and other alternative high-affinity protein-binding scaffolds that incorporate non-human sequences. Several strategies have been developed to minimize the immunogenic potential of these therapies.
2.4.3.1 Humanization of Antibodies
By modifying the antibody sequences to more closely resemble human proteins, the risk of immunogenicity can be reduced [196]. This involves grafting the antigen-binding regions of a non-human antibody onto a human antibody framework, resulting in chimeric or humanized antibodies that are less likely to be recognized as foreign by the patient’s immune system.
2.4.3.2 Use of “Inert” IgG Isotypes
Selecting IgG isotypes that are less likely to provoke an immune response can help mitigate immunogenicity. These “inert” isotypes are engineered to avoid interaction with immune cells that could trigger adverse reactions, thereby enhancing the safety profile of the BsAbs [197].
2.4.3.3 Screening for Low Immunogenic Potential
Meticulously screening monoclonal antibody (mAb) candidates to ensure they have lower immunogenic potential is another effective approach [197]. This involves using advanced computational tools and in vitro assays to predict and evaluate the immunogenicity of antibody candidates before clinical development.
2.4.3.4 Deimmunization Techniques
Employing deimmunization techniques, such as epitope removal or modification, can further reduce the immunogenicity of BsAbs. By identifying and modifying specific amino acid sequences that are recognized by the immune system, the overall immunogenic profile of the antibody can be improved [198].
2.4.4 Tissue Penetration
For solid tumors, the efficacy of BsAbs significantly hinges on their ability to navigate and penetrate the complex tumor microenvironment (TME). An inflamed TME, characterized by interferon-γ signaling, is associated with favorable clinical responses to immunotherapies. However, the TME of solid tumors presents considerable barriers that can impede the effectiveness of therapeutic agents, including BsAbs. These barriers are often categorized into two primary types: “immune-excluded” and “immune desert” environments [199].
The “immune-excluded” TME is characterized by the presence of immune cells that are unable to effectively infiltrate the tumor mass. Instead, these immune cells, such as T cells and other lymphocytes, are often found surrounding the tumor or trapped within the stroma (the connective tissue component of the tumor) [200]. Factors such as transforming growth factor-beta (TGF-β) and other cytokines can create an immunosuppressive environment that discourages immune cell infiltration. TGF-β, in particular, is known to inhibit T-cell function and promote the differentiation of immunosuppressive cell types like regulatory T cells (Tregs). Increased angiogenesis, or the formation of new blood vessels, is another hallmark of the immune-excluded TME. While angiogenesis supplies the tumor with necessary nutrients, it also creates a dense and disorganized vascular network that can act as a physical barrier, preventing immune cells and therapeutic agents from effectively reaching the tumor cells.
On the other hand, the “immune desert” TME is characterized by a complete or near-complete absence of immune cells within the tumor [200]. These tumors may exhibit traits such as enhanced fatty acid metabolism, which can provide energy for rapid tumor growth and survival in a nutrient-deprived environment. Additionally, neuroendocrine characteristics may be present, further contributing to an immunosuppressive and resistant phenotype [201]. These adaptations make it challenging for immune cells to recognize and attack the tumor, thereby limiting the efficacy of immunotherapies, including BsAbs.
To enhance the therapeutic impact of BsAbs in such challenging environments, current research explores the potential of combination therapies. These include the integration of TME-modulating agents or other immunotherapeutic approaches aimed at altering the TME to be more conducive to effective BsAb intervention [46]. For instance, targeting TGF-β signaling pathways can potentially reduce the immunosuppressive barrier [202], allowing for better penetration and function of BsAbs.
2.4.5 Resistance
As therapy progresses, tumor cells can develop resistance to BsAbs through various mechanisms, thereby reducing the effectiveness of the treatment. Understanding and overcoming these resistance mechanisms is critical for the sustained efficacy of BsAb therapies. Resistance mechanisms to immune-based therapies arise from multiple factors, including tumor-intrinsic factors, the TME, and prior treatment-related immune suppression [203].
Tumor-intrinsic factors contributing to resistance include genetic mutations, loss of tumor antigen expression, changes in the major histocompatibility complex (MHC) molecules, and the expression of immune checkpoints [204, 205]. Tumor cells may downregulate the expression of TAAs that BsAbs target, reducing their visibility to the immune system. Alterations in MHC molecules, which present antigens on the surface of tumor cells, can impair T-cell recognition. These changes can result from genetic mutations or epigenetic modifications that affect MHC expression or function. Additionally, tumor cells can evade the immune system by expressing immune checkpoint proteins, such as PD-L1, which inhibit T-cell activity.
The TME-induced resistance may involve both cellular components (Tregs, myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), particularly the M2 phenotype) and secreted factors (IL-10, TGF-β, COX2, and PGE2) [206, 207]. These elements collectively create an immunosuppressive environment that hampers the effectiveness of BsAb therapies.
To counteract these resistance mechanisms, advanced combinatorial treatments are being explored [208].
2.4.6 Production and Cost
The production of BsAbs presents challenges due to their complex structure, which often requires sophisticated manufacturing processes. BsAbs are typically composed of two different antibody fragments, which need to be correctly assembled and maintained in a stable configuration. This complexity not only makes the production process more intricate but also increases the risk of manufacturing errors and product inconsistencies.
Developing more efficient and cost-effective production methods is crucial for making BsAb therapies more widely available. Strategies to achieve this can be explored, including optimizing cell lines and expression systems, streamlining purification processes, and exploring alternative production platforms such as microbial fermentation.
The future of solid tumors BsAbs lies in the development of more specific, effective, and adaptable therapeutic strategies that can overcome the challenges posed by tumor complexity and heterogeneity. Numerous innovative strategies are being explored to enhance the application and effectiveness of BsAbs in treating solid tumors, including bispecific T-cell engaging receptors (TCERs), BiTE-armed oncolytic viruses, BsAb-armed CAR-T cells, etc. Through continued research and innovation, BsAbs have the potential to significantly improve outcomes for patients with solid tumors.
2.5 Conclusion
The landscape of cancer treatment has been notably enhanced by the development and clinical integration of BsAbs, particularly in the realm of solid tumors. These innovative therapeutic agents, characterized by their ability to simultaneously target two distinct antigens, have demonstrated significant potential to overcome the limitations of traditional therapies and improve patient outcomes. The clinical success of BsAbs in hematologic malignancies has set a promising precedent for their application in solid tumors, despite the intrinsic challenges posed by the tumor microenvironment and the heterogeneity of solid malignancies.
The current generation of BsAbs targeting solid tumors, including those directed against EGFR, HER2, and PD-L1, among others, exemplifies the strides made in precision medicine. These treatments not only provide a more targeted attack on cancer cells but also engage the immune system in a more effective and regulated manner. However, the journey from bench to bedside is fraught with challenges, including overcoming the immunosuppressive tumor microenvironment, ensuring selective targeting to minimize off-target effects, and managing the unique toxicity profiles associated with dual targeting.
Emerging strategies, such as combining BsAbs with other therapeutic modalities, optimizing BsAb formats for better tissue penetration and reduced immunogenicity, and leveraging novel targets within the tumor microenvironment, hold promise for enhancing the efficacy and safety of BsAbs in solid tumors. Furthermore, the exploration of personalized medicine approaches, including the use of biomarkers to predict response and tailor therapy, is critical for maximizing the therapeutic potential of BsAbs.
As we advance, the continuous evolution of BsAbs will necessitate a multidisciplinary effort encompassing innovative drug design, comprehensive preclinical testing, and adaptive clinical trial designs. The ultimate goal is to fully realize the potential of BsAbs in providing durable, effective, and safe treatments for patients with solid tumors. The progress made thus far underscores the transformative potential of BsAbs, heralding a new era in cancer therapy where precision, efficacy, and patient safety are paramount.
References
Torre LA, et al. Global cancer incidence and mortality rates and trends—an update. Cancer Epidemiol Biomarkers Prevent. 2016;25(1):16–27.
Roy PS, Saikia BJ. Cancer and cure: a critical analysis. Indian J Cancer. 2016;53(3):441–2.
Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science (New York, NY). 2013;342(6165):1432–3.
Dougan M, Dranoff G, Dougan SK. Cancer immunotherapy: beyond checkpoint blockade. Annu Rev Cancer Biol. 2019;3:55–75.
Suurs FV, et al. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol Ther. 2019;201:103–19.
Karpovsky B, Titus J, Stephany DA, Segal DM. Production of target-specific effector cells using hetero-cross-linked aggregates containing anti-target cell and anti-Fc gamma receptor antibodies. J Exp Med. 1985;160(6):1686–701.
Perez PH, Robert W, Shaw S, Bluestone JA, Segal DM. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature. 1985;316(6026):354–6.
Patrick Chames DB. Bispecific antibodies for cancer therapy The light at the end of the tunnel? Landes Biosci. 2009;1(6):539–47.
Rader C. Bispecific antibodies in cancer immunotherapy. Curr Opin Biotechnol. 2020;65:9–16.
Middelburg J, et al. Overcoming challenges for CD3-bispecific antibody therapy in solid tumors. Cancers (Basel). 2021;13(2):287.
Bispecific antibodies—current status and prospects. https://www.biochempeg.com/article/252.html. Accessed 9 Mar 2022.
Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12(4):278–87.
Ma H, O’Kennedy R. The structure of natural and recombinant antibodies. Methods Mol Biol. 2015;1348:7–11.
Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9(2):182–212.
Labrijn AF, et al. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18(8):585–608.
Birch JR, Racher AJ. Antibody production. Adv Drug Deliv Rev. 2006;58(5–6):671–85.
Lowe D, et al. Aggregation, stability, and formulation of human antibody therapeutics. Adv Protein Chem Struct Biol. 2011;84:41–61.
Shim H. Bispecific antibodies and antibody-drug conjugates for cancer therapy: technological considerations. Biomolecules. 2020;10(3):360.
Johnson S, et al. Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J Mol Biol. 2010;399(3):436–49.
Kang TH, Jung ST. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp Mol Med. 2019;51(11):1–9.
Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. 2018;9(1):63–73.
Coloma MJ, Morrison S. Design and production of novel tetravalent bispecific antibodies. Nat Biotechnol. 1997;15(2):159–63.
Wu C, et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat Biotechnol. 2007;25(11):1290–7.
Hambach J, et al. Targeting multiple myeloma with nanobody-based heavy chain antibodies, bispecific killer cell engagers, chimeric antigen receptors, and nanobody-displaying AAV vectors. Front Immunol. 2022;13:1005800.
Yu F, et al. An affibody-adalimumab hybrid blocks combined IL-6 and TNF-triggered serum amyloid A secretion in vivo. MAbs. 2014;6(6):1598–607.
Shen J, et al. Single variable domain antibody as a versatile building block for the construction of IgG-like bispecific antibodies. J Immunol Methods. 2007;318(1–2):65–74.
Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10(5):301–16.
Davis JH, et al. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng Des Sel. 2010;23(4):195–202.
Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, Carter P. An efficient route to human bispecific IgG. Nat Biotechnol. 1998;16(7):677–81.
Ridgway JB, Presta L, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9(7):617–21.
Atwell S, et al. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol. 1997;270(1):26–35.
Gunasekaran K, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285(25):19637–46.
Choi H-J, et al. A heterodimeric Fc-based bispecific antibody simultaneously targeting VEGFR-2 and Met exhibits potent antitumor activity. Mol Cancer Ther. 2013;12(12):2748–59.
Sampei Z, et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS ONE. 2013;8(2): e57479.
Schaefer W, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A. 2011;108(27):11187–92.
Staerz UD, Bevan M. Hybrid hybridoma producing a bispecific monoclonal antibody that can focus effector T-cell activity. Proc Natl Acad Sci U S A. 1986;83(5):1453–7.
Staerz UD, Kanagawa O, Bevan MJ. Hybrid antibodies can target sites for attack by T cells. Nature. 1985;314(6012):628–31.
Wu Z, Cheung NV. T cell engaging bispecific antibody (T-BsAb): from technology to therapeutics. Pharmacol Ther. 2018;182:161–75.
Fucà G, et al. Immune cell engagers in solid tumors: promises and challenges of the next generation immunotherapy. ESMO Open. 2021;6(1): 100046.
Schram AM, et al. A phase II basket study of MCLA-128, a bispecific antibody targeting the HER3 pathway, in NRG1 fusion-positive advanced solid tumors. J Clin Oncol. 2020;38(15_suppl):TPS3654.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64.
Dovedi SJ, et al. Design and efficacy of a monovalent bispecific PD-1/CTLA4 antibody that enhances CTLA4 blockade on PD-1+ activated T cells. Cancer Discov. 2021;11(5):1100–17.
Moek KL, et al. The antibody-drug conjugate target landscape across a broad range of tumour types. Ann Oncol. 2017;28(12):3083–91.
Jhaveri K, et al. 460MO Preliminary results from a phase I study using the bispecific, human epidermal growth factor 2 (HER2)-targeting antibody-drug conjugate (ADC) zanidatamab zovodotin (ZW49) in solid cancers. Ann Oncol. 2022;33:S749–50.
Fusion Pharmaceuticals Announces Presentation of Preclinical Data Supporting FPI-2068, a Novel Targeted Alpha Therapy for EGFR-cMET Expressing Cancers. Available from https://www.prnewswire.com/news-releases/fusion-pharmaceuticals-announces-presentation-of-preclinical-data-supporting-fpi-2068-a-novel-targeted-alpha-therapy-for-egfr-cmet-expressing-cancers-301954807.html. Accessed 12 Oct 2023.
Antonarelli G, et al. Research and clinical landscape of bispecific antibodies for the treatment of solid malignancies. Pharmaceuticals (Basel). 2021;14(9):889.
Slamon DJ, Clark G, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–82.
Lopez-Albaitero A, et al. Overcoming resistance to HER2-targeted therapy with a novel HER2/CD3 bispecific antibody. Oncoimmunology. 2017;6(3): e1267891.
Meric-Bernstam F, et al. Zanidatamab, a novel bispecific antibody, for the treatment of locally advanced or metastatic HER2-expressing or HER2-amplified cancers: a Phase 1, dose-escalation and expansion study. Lancet Oncol. 2022;23(12):1558–70.
Harding JJ, et al. Zanidatamab for HER2-amplified, unresectable, locally advanced or metastatic biliary tract cancer (HERIZON-BTC-01): a multicentre, single-arm, Phase 2b study. Lancet Oncol. 2023;24(7):772–82.
Xu J, et al. KN026 (anti-HER2 bispecific antibody) in patients with previously treated, advanced HER2-expressing gastric or gastroesophageal junction cancer. Eur J Cancer. 2023;178:1–12.
Gong J, et al. Efficacy and safety of KN026 in combination with KN046 in patients with locally advanced unresectable or metastatic HER2-positive other solid tumors. J Clin Oncol. 2023;41(16_suppl):3621–3621.
Zhang B, et al. Abstract 4633: MM-111, a bispecific HER2 and HER3 antibody, synergistically combines with trastuzumab and paclitaxel in preclinical models of gastric cancer. Cancer Res. 2013;73(8_Supplement):4633.
Richards DA, et al. A Phase 1 study of MM-111, a bispecific HER2/HER3 antibody fusion protein, combined with multiple treatment regimens in patients with advanced HER2-positive solid tumors. J Clin Oncol. 2014;32(15_suppl):651.
Denlinger CS, et al. Randomized Phase 2 study of paclitaxel (PTX), trastuzumab (T) with or without MM-111 in HER2 expressing gastroesophageal cancers (GEC). J Clin Oncol. 2016;34(15_suppl):4043.
de Vries Schultink AHM, et al. Population pharmacokinetics of MCLA-128 a HER2/HER3 bispecific monoclonal antibody in patients with solid tumors. Clin Pharmacokinet. 2020;59(7):875–84.
Singh R, et al. 4-1BB immunotherapy: advances and hurdles. Exp Mol Med. 2024;56(1):32–9.
Lee E, et al. Abstract 1139: a novel HER2/4-1BB bispecific antibody, YH32367 (ABL105) exhibits the optimal efficacy and superior safety profile through tumor-directed 4–1BB agonism. Cancer Res. 2022;82(12_Supplement):1139.
Yu S, et al. A novel asymmetrical anti-HER2/CD3 bispecific antibody exhibits potent cytotoxicity for HER2-positive tumor cells. J Exp Clin Cancer Res. 2019;38(1):355.
Wermke M, Alt J, Kauh J, Back J, Salhi Y, Reddy V, Barve M, Ochsenreither S. Preliminary results from a phase I study of GBR 1302, a bispecific antibody T-cell engager, in HER2 positive cancers. Ann Oncol. 2018;29:408–9.
Zhang B, Li S, Chen D, Liu D, Guo H, Yang C, Zhang L, Zhang W, Tu X, Peng L, Zhao G, Zhang R, Gan FX, Tian W, Zhang F, Song Y. Abstract 2938: preclinical development of a novel bispecific mAb-Trap fusion protein, IMM2902, targeting both HER2 and CD47 as cancer immunotherapy. Cancer Res. 2023;83:2938.
Jaiswal S, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–85.
Weiskopf K, Weissman IL. Macrophages are critical effectors of antibody therapies for cancer. MAbs. 2015;7(2):303–10.
Meng Y, et al. Preliminary results of a phase I, first-in-human, dose escalation study of IMM2902 in patients with HER2-expressing advanced solid tumors. J Clin Oncol. 2023;41(16): e15185.
Xu MJ, Johnson DE, Grandis JR. EGFR-targeted therapies in the post-genomic era. Cancer Metastasis Rev. 2017;36(3):463–73.
Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160–74.
Drilon A, et al. Targeting MET in lung cancer: will expectations finally be MET? J Thorac Oncol. 2017;12(1):15–26.
Yun J, et al. Antitumor activity of amivantamab (JNJ-61186372), an EGFR-MET bispecific antibody, in diverse models of EGFR Exon 20 insertion-driven NSCLC. Cancer Discov. 2020;10(8):1194–209.
Moores SL, et al. A novel bispecific antibody targeting EGFR and cMet is effective against EGFR inhibitor-resistant lung tumors. Cancer Res. 2016;76(13):3942–53.
Park K, et al. Amivantamab in EGFR Exon 20 insertion-mutated non-small-cell lung cancer progressing on platinum chemotherapy: initial results from the CHRYSALIS phase I study. J Clin Oncol. 2021;39(30):3391–402.
Lee S-H, et al. Amivantamab and lazertinib in treatment-naïve EGFR-mutated advanced non–small-cell lung cancer (NSCLC): long-term follow-up and ctDNA results from CHRYSALIS. J Clin Oncol. 2023;41(16_suppl):9134.
Remon J, et al. EGFR exon 20 insertions in advanced non-small cell lung cancer: a new history begins. Cancer Treat Rev. 2020;90: 102105.
Morgan RG, Mortensson E, Williams AC. Targeting LGR5 in colorectal cancer: therapeutic gold or too plastic? Br J Cancer. 2018;118(11):1410–8.
Argiles G, et al. Phase I dose-escalation study of MCLA-158, a first-in-class bispecific antibody targeting EGFR and LGR5, in metastatic colorectal cancer (CRC). J Clin Oncol. 2021;39(3_suppl):62.
Xue J, et al. Prediction of human pharmacokinetics and clinical effective dose of SI-B001, an EGFR/HER3 Bi-specific monoclonal antibody. J Pharm Sci. 2020;109(10):3172–80.
Zhao Y, et al. SI-B001 plus chemotherapy in patients with locally advanced or metastatic EGFR/ALK wild-type non-small cell lung cancer: A phase II, multicenter, open-label study. J Clin Oncol. 2023;41(16_suppl):9025.
Wingert S, et al. Preclinical evaluation of AFM24, a novel CD16A-specific innate immune cell engager targeting EGFR-positive tumors. MAbs. 2021;13(1):1950264.
El-Khoueiry AB, et al. AFM24 in combination with autologous NK cells (SNK01) in patients with advanced/metastatic epidermal growth factor receptor (EGFR) expressing solid tumors: Initial results from the Phase 1 dose-escalation study. JCO Glob Oncol. 2023;9(Supplement_1):26.
Lakhani N, et al. 535 A phase I/II study of REGN7075 (EGFRxCD28 costimulatory bispecific antibody) in combination with cemiplimab (anti–PD-1) in patients with advanced solid tumors. J Immunother Cancer. 2021;9(Suppl 2):A565–A565.
Waite JC, et al. Tumor-targeted CD28 bispecific antibodies enhance the antitumor efficacy of PD-1 immunotherapy. Sci Transl Med. 2020;12(549):eaba2325.
Segal NH, et al. A phase 1/2 study of REGN7075 (EGFRxCD28 costimulatory bispecific antibody) in combination with cemiplimab (anti-PD-1) in patients with advanced solid tumors: trial-in-progress update. J Clin Oncol. 2023;41(4_suppl):TPS277.
Bispecific Antibody Drug Conjugates (ADCs): Emerging Trends. Available from https://www.biochempeg.com/article/290.html. Accessed 11 Aug 2022.
Gao T, Cen Q, Lei H. A review on development of MUC1-based cancer vaccine. Biomed Pharmacother. 2020;132:110888.
Zutshi A, et al. Abstract 5423: translational PK/PD/efficacy modeling and efficacious human dose prediction for a first-in-class MUC1-EGFR (M1231) bispecific antibody drug conjugate. Cancer Res. 2022;82(12_Supplement):5423.
Beauchemin N, Arabzadeh A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 2013;32(3–4):643–71.
Hammarström S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol. 1999;9(2):67–81.
Wang N, et al. An optimal antitumor response by a novel CEA/CD3 bispecific antibody for colorectal cancers. Antibody Therap. 2021;4(2):90–100.
Bacac M, Klein C, Umana P. CEA TCB: A novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunology. 2016;5(8): e1203498.
Tabernero J, et al. Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: Preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J Clin Oncol. 2017;35(15_suppl):3002.
Lutterbuese R, et al. Potent control of tumor growth by CEA/CD3-bispecific single-chain antibody constructs that are not competitively inhibited by soluble CEA. J Immunother (Hagerstown, Md, 1997). 2009;32(4):341–52.
Oberst MD, et al. CEA/CD3 bispecific antibody MEDI-565/AMG 211 activation of T cells and subsequent killing of human tumors is independent of mutations commonly found in colorectal adenocarcinomas. MAbs. 2014;6(6):1571–84.
Pishvaian MJ, et al. Phase 1 dose escalation study of MEDI-565, a bispecific T-cell engager that targets human carcinoembryonic antigen (CEA), in patients with advanced gastrointestinal (GI) adenocarcinomas. J Clin Oncol. 2016;34(4_suppl):320.
Touchefeu Y, et al. Promising clinical performance of pretargeted immuno-PET with anti-CEA bispecific antibody and gallium-68-labelled IMP-288 peptide for imaging colorectal cancer metastases: a pilot study. Eur J Nucl Med Mol Imaging. 2021;48(3):874–82.
Rousseau C, et al. Initial clinical results of a novel immuno-PET theranostic probe in human epidermal growth factor receptor 2-negative breast cancer. J Nucl Med. 2020;61(8):1205–11.
Gires O, et al. Expression and function of epithelial cell adhesion molecule EpCAM: where are we after 40 years? Cancer Metastasis Rev. 2020;39(3):969–87.
Keller L, Werner S, Pantel K. Biology and clinical relevance of EpCAM. Cell Stress. 2019;3(6):165–80.
Went P, et al. Frequent high-level expression of the immunotherapeutic target Ep-CAM in colon, stomach, prostate and lung cancers. Br J Cancer. 2006;94(1):128–35.
Linke R, Klein A, Seimetz D. Catumaxomab: clinical development and future directions. MAbs. 2010;2(2):129–36.
Ruf P, et al. Characterisation of the new EpCAM-specific antibody HO-3: implications for trifunctional antibody immunotherapy of cancer. Br J Cancer. 2007;97(3):315–21.
Riesenberg R, et al. Lysis of prostate carcinoma cells by trifunctional bispecific antibodies (alpha EpCAM x alpha CD3). J Histochem Cytochem. 2001;49(7):911–7.
Zeidler R, et al. The Fc-region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br J Cancer. 2000;83(2):261–6.
Zeidler R, et al. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol (Baltimore, Md, 1950). 1999;163(3):1246–52.
Schmitt M, et al. Opsonization with a trifunctional bispecific (alphaCD3 x alphaEpCAM) antibody results in efficient lysis in vitro and in vivo of EpCAM positive tumor cells by cytotoxic T lymphocytes. Int J Oncol. 2004;25(4):841–8.
Ruf P, Lindhofer H. Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood. 2001;98(8):2526–34.
Burges A, et al. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM x anti-CD3 antibody: a phase I/II study. Clin Cancer Res. 2007;13(13):3899–905.
Ruf P, et al. Pharmacokinetics and in vivo stability of intraperitoneally administered therapeutic antibody catumaxomab. J Clin Oncol. 2008;26(15_suppl):14006.
Heiss MM, et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int J Cancer. 2010;127(9):2209–21.
Sebastian M, et al. Treatment of non-small cell lung cancer patients with the trifunctional monoclonal antibody catumaxomab (anti-EpCAM x anti-CD3): a phase I study. Cancer Immunol Immunother CII. 2007;56(10):1637–44.
Ströhlein M, et al. Peritoneal carcinomatosis immunotherapy with the trifunctional anti-EpCAM x anti-CD3 antibody catumaxomab in patients with colon, gastric, or pancreatic cancer: long-term results after a 2-year follow-up. J Clin Oncol. 2009;27(15_suppl):3033.
Knödler M, et al. Randomised phase II trial to investigate catumaxomab (anti-EpCAM × anti-CD3) for treatment of peritoneal carcinomatosis in patients with gastric cancer. Br J Cancer. 2018;119(3):296–302.
Seimetz D, Lindhofer H, Bokemeyer C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev. 2010;36(6):458–67.
AGENCY EM. Removab: withdrawal of the marketing authorisation in the European Union. Available from: https://www.ema.europa.eu/en/documents/public-statement/public-statement-removab-withdrawal-marketing-authorisation-european-union_en.pdf. Accessed 10 July 2017.
Brischwein K, et al. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol. 2006;43(8):1129–43.
Kebenko M, et al. A multicenter Phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE®) antibody construct, in patients with refractory solid tumors. Oncoimmunology. 2018;7(8): e1450710.
Sait S, Modak S. Anti-GD2 immunotherapy for neuroblastoma. Expert Rev Anticancer Ther. 2017;17(10):889–904.
Wu ZL, et al. Expression of GD2 ganglioside by untreated primary human neuroblastomas. Cancer Res. 1986;46(1):440–3.
Yankelevich M, et al. Phase I study of OKT3 x hu3F8 bispecific antibody (GD2Bi) armed T cells (GD2BATs) in GD2-positive tumors. J Clin Oncol. 2019;37(15_suppl):2533.
Ortiz MV, et al. Immunotherapeutic targeting of GPC3 in pediatric solid embryonal tumors. Front Oncol. 2019;9:108.
Sano Y, et al. Combination of T cell-redirecting bispecific antibody ERY974 and chemotherapy reciprocally enhances efficacy against non-inflamed tumours. Nat Commun. 2022;13(1):5265.
Safran H, et al. Abstract CT111: results of a phase 1 dose escalation study of ERY974, an anti-glypican 3 (GPC3)/CD3 bispecific antibody, in patients with advanced solid tumors. Cancer Res. 2021;81(13):CT111.
Stern PL, Harrop R. 5T4 oncofoetal antigen: an attractive target for immune intervention in cancer. Cancer Immunol Immunother. 2017;66(4):415–26.
Nelson MH, et al. The bispecific tumor antigen-conditional 4–1BB x 5T4 agonist, ALG.APV-527, mediates strong T-cell activation and potent antitumor activity in preclinical studies. Mol Cancer Ther. 2023;22(1):89–101.
Kemper KG, Gielen E, Houtkamp M, Boross P, Burm S, Poot S, Engelberts P, Goeij B, Satijn D, Sasser K, Breij E. 704 Preclinical mechanism of action and pharmacodynamic biomarker studies of DuoBody®-CD3x5T4 in vitro and in vivo in solid cancer models. J Immunother Cancer. 2020;8:746.
Martin-Villa JM, et al. HLA-G: too much or too little? Role in cancer and autoimmune disease. Front Immunol. 2022;13: 796054.
Geva R, et al. 1045P Safety and preliminary clinical activity of JNJ-78306358 (JNJ-358), an HLA-G and CD3 bispecific antibody, for the treatment of advanced stage solid tumor. Ann Oncol. 2023;34:S633–4.
Zymeworks announces second Janssen bispecific antibody to begin clinical development utilizing Azymetric™ and EFECT™ therapeutic platforms. Available from https://www.biospace.com/article/releases/zymeworks-announces-second-janssen-bispecific-antibody-to-begin-clinical-development-utilizing-azymetric-and-efect-therapeutic-platforms/. Accessed 01 Dec 2021.
Obermajer N, et al. Abstract ND07: JNJ-78306358: a first-in-class bispecific T cell redirecting HLA-G antibody. Cancer Res. 2022;82(12):ND07.
Chung AS, Lee J, Ferrara N. Targeting the tumour vasculature: insights from physiological angiogenesis. Nat Rev Cancer. 2010;10(7):505–14.
Sainson RCA, Harris AL. Anti-Dll4 therapy: can we block tumour growth by increasing angiogenesis? Trends Mol Med. 2007;13(9):389–95.
Huang J, et al. Dll4 inhibition plus aflibercept markedly reduces ovarian tumor growth. Mol Cancer Ther. 2016;15(6):1344–52.
Jimeno A, et al. A first-in-human phase 1a study of the bispecific anti-DLL4/anti-VEGF antibody navicixizumab (OMP-305B83) in patients with previously treated solid tumors. Invest New Drugs. 2019;37(3):461–72.
Fu S, et al. Phase Ib study of navicixizumab plus paclitaxel in patients with platinum-resistant ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2022;40(23):2568–77.
Lee J, et al. Phase 1a study results investigating the safety and preliminary efficacy of ABL001 (NOV1501), a bispecific antibody targeting VEGF and DLL4 in metastatic gastrointestinal (GI) cancer. J Clin Oncol. 2019;37(15_suppl):3023.
Oh D-Y, et al. CTX-009 (ABL001), a bispecific antibody targeting DLL4 and VEGF A, in combination with paclitaxel in patients with advanced biliary tract cancer (BTC): a phase 2 study. J Clin Oncol. 2023;41(4_suppl):540.
Sabari JK, et al. Unravelling the biology of SCLC: implications for therapy. Nat Rev Clin Oncol. 2017;14(9):549–61.
Owen DH, et al. DLL3: an emerging target in small cell lung cancer. J Hematol Oncol. 2019;12(1):61.
Paz-Ares L, et al. Tarlatamab, a first-in-class DLL3-targeted bispecific T-cell engager, in recurrent small-cell lung cancer: an open-label, phase I study. J Clin Oncol. 2023;41(16):2893–903.
Giffin MJ, et al. AMG 757, a half-life extended, DLL3-targeted bispecific T-Cell engager, shows high potency and sensitivity in preclinical models of small-cell lung cancer. Clin Cancer Res. 2021;27(5):1526–37.
FDA U. FDA grants accelerated approval to tarlatamab-dlle for extensive stage small cell lung cancer. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-tarlatamabdlle-extensive-stage-small-cell-lung-cancer?utm_medium=email&utm_source=govdelivery. Accessed 16 May 2024.
Haberkorn U, et al. New strategies in prostate cancer: prostate-specific membrane antigen (PSMA) ligands for diagnosis and therapy. Clin Cancer Res. 2016;22(1):9–15.
Wang F, et al. Advances in PSMA-targeted therapy for prostate cancer. Prostate Cancer Prostat Dis. 2022;25(1):11–26.
Heitmann JS, et al. Bispecific antibodies in prostate cancer therapy: current status and perspectives. Cancers (Basel). 2021;13(3):549.
Deegen P, et al. The PSMA-targeting half-life extended BiTE therapy AMG 160 has potent antitumor activity in preclinical models of metastatic castration-resistant prostate cancer. Clin Cancer Res. 2021;27(10):2928–37.
Dorff T, et al. A phase I study of acapatamab, a half-life extended, PSMA-targeting bispecific T-cell engager for metastatic castration-resistant prostate cancer. Clin Cancer Res. 2024;30(8):1488–500.
Hummel H-D, et al. Phase 1 study of pasotuxizumab (BAY 2010112), a PSMA-targeting bispecific T cell engager (BiTE) immunotherapy for metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2019;37(15_suppl):5034.
Technology Overview. Available from: https://aptevotherapeutics.com/our-technology/. Accessed 7 Apr 2024.
Aptevo Therapeutics and MorphoSys End Joint Development and Commercialization Agreement for MOR209/ES414. Available from: https://www.globenewswire.com/news-release/2017/08/31/1106420/0/en/Aptevo-Therapeutics-and-MorphoSys-End-Joint-Development-and-Commercialization58Agreement-for-MOR209-ES414.html. Accessed 31 Aug 2017.
Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol. 2020;20(1):25–39.
Darvin P, et al. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50(12):1–11.
Kotanides H, et al. Bispecific targeting of PD-1 and PD-L1 enhances T-cell activation and antitumor immunity. Cancer Immunol Res. 2020;8(10):1300–10.
Hellmann MD, et al. Safety and immunogenicity of LY3415244, a bispecific antibody against TIM-3 and PD-L1, in patients with advanced solid tumors. Clin Cancer Res. 2021;27(10):2773–81.
Sung E, et al. LAG-3xPD-L1 bispecific antibody potentiates antitumor responses of T cells through dendritic cell activation. Mol Ther. 2022;30(8):2800–16.
Wang Y, et al. Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced antitumor activity and limited toxicity. Cancer Immunol Immunother. 2021;70(2):365–76.
Wang J, et al. Abstract CT513: Phase I study of IBI322 (anti-CD47/PD-L1 bispecific antibody) monotherapy therapy in patients with advanced solid tumors in China. Cancer Res. 2022;82(12):CT513.
Geuijen C, et al. A human CD137×PD-L1 bispecific antibody promotes antitumor immunity via context-dependent T cell costimulation and checkpoint blockade. Nat Commun. 2021;12(1):4445.
Prenen H, et al. 136P Phase I dose escalation study of MCLA-145, a bispecific antibody targeting CD137 and PD-L1 in solid tumors. Ann Oncol. 2021;32:S1436.
Lakins MA, et al. FS222, a CD137/PD-L1 tetravalent bispecific antibody, exhibits low toxicity and antitumor activity in colorectal cancer models. Clin Cancer Res. 2020;26(15):4154–67.
Garralda E, et al. 165TiP a first-in-human phase I study of FS222, a CD137/PD-L1 tetravalent bispecific antibody, in patients with advanced malignancies. Ann Oncol. 2021;32:S1454.
Peper-Gabriel JK, et al. The PD-L1/4-1BB bispecific antibody-anticalin fusion protein PRS-344/S095012 elicits strong T-cell stimulation in a tumor-localized manner. Clin Cancer Res. 2022;28(15):3387–99.
Moore GEA. Anti-PD1 x anti-ICOS bispecific antibody XmAb23104 brings together PD1 blockade and ICOS costimulation to promote human T cell activation and proliferation. J Immunother Cancer. 2017;5:347.
Zhang X, et al. The role of ICOS in allergic disease: positive or negative? Int Immunopharmacol. 2022;103: 108394.
Akce M, et al. A Phase 1 multiple-ascending dose study to evaluate the safety and tolerability of XmAb23104 (PD-1 x ICOS) in subjects with selected advanced solid tumors (DUET-3). J Clin Oncol. 2022;40(16_suppl):2604.
Jacob S, Daud A. Phase Ib/II study of XmAb23104 (PD1 X ICOS) and XmAb22841 (CTLA-4 X LAG3) combination in metastatic melanoma refractory to prior immune checkpoint inhibitor therapy with and without CNS disease. J Clin Oncol. 2023;41(16):9595.
Zhao Y, et al. A phase II study of AK112 (PD-1/VEGF bispecific) in combination with chemotherapy in patients with advanced non-small cell lung cancer. J Clin Oncol. 2022;40(16_suppl):9019.
Wang Y, et al. SI-B003 (PD-1/CTLA-4) in patients with advanced solid tumors: a phase I study. J Clin Oncol. 2023;41(16): e14668.
Bai L, et al. Phase 2 study of AK104 (PD-1/CTLA-4 bispecific antibody) plus lenvatinib as first-line treatment of unresectable hepatocellular carcinoma. J Clin Oncol. 2021;39(15_suppl):4101.
Wang J, et al. A study of AK104 (an anti-PD1 and anti-CTLA4 bispecific antibody) combined with standard therapy for the first-line treatment of persistent, recurrent, or metastatic cervical cancer (R/M CC). J Clin Oncol. 2022;40(16_suppl):106.
Berezhnoy A, et al. Development and preliminary clinical activity of PD-1-guided CTLA-4 blocking bispecific DART molecule. Cell Rep Med. 2020;1(9): 100163.
Luke JJ, et al. Lorigerlimab, a bispecific PD-1×CTLA-4 DART molecule in patients (pts) with metastatic castration-resistant prostate cancer (mCRpc): a phase 1 expansion (exp) cohort. J Clin Oncol. 2023;41(6_suppl):155.
Powderly J, et al. 757 A phase 1/1b dose escalation and cohort expansion study of MGC018 in combination with lorigerlimab in patients with advanced solid tumors (AST). J Immunother Cancer. 2022;10(Suppl 2):A789.
Vudalimab (PD1 x CTLA4). Available from: https://xencor.com/pipeline/vudalimab/. Accessed 7 Apr 2024.
Hickingbottom B, et al. Preliminary safety and pharmacodynamic (PD) activity of XmAb20717, a PD-1 x CTLA-4 bispecific antibody, in a phase I dose escalation study of patients with selected advanced solid tumors. J Clin Oncol. 2020;38(15_suppl): e15001.
Stein MN, et al. A phase 2, multicenter, parallel-group, open-label study of vudalimab (XmAb20717), a PD-1 x CTLA-4 bispecific antibody, alone or in combination with chemotherapy or targeted therapy in patients with molecularly defined subtypes of metastatic castration-resistant prostate cancer. J Clin Oncol. 2022;40(16):5097.
Vitale LA, et al. Development of CDX-527: a bispecific antibody combining PD-1 blockade and CD27 costimulation for cancer immunotherapy. Cancer Immunol Immunother CII. 2020;69(10):2125–37.
Sanborn RE, et al. A phase 1 dose-escalation study of a PD-L1xCD27 bispecific antibody CDX-527 in patients with advanced malignancies. J Clin Oncol. 2021;39(15_suppl):2585.
Zhao Y, et al. Two-year follow-up from KN046 in combination with platinum doublet chemotherapy as first-line (1L) treatment for NSCLC: an open-label, multi-center phase 2 trial. EMSO. 2022. https://doi.org/10.1016/j.annonc.2022.07.1155.
Zhao H, et al. The preliminary efficacy and safety data of KN046 in patients failed on prior immune checkpoint inhibitors therapy. J Clin Oncol. 2020;38(15_suppl):3020.
Jin G, et al. Efficacy and safety of KN046 plus nab-paclitaxel/gemcitabine as first-line treatment for unresectable locally advanced or metastatic pancreatic ductal adenocarcinoma (PDAC). J Clin Oncol. 2021;39(15_suppl):4138.
Kontos F, et al. B7–H3: an attractive target for antibody-based immunotherapy. Clin Cancer Res. 2021;27(5):1227–35.
XmAb808 (B7-H3 x CD28). Available from: https://xencor.com/pipeline/xmab808/. Accessed 7 Apr 2024.
Bupathi M, et al. 764 A phase 1, first-in-human (FIH), open-label, dose-finding and expansion study of XmAb808, a B7H3 x CD28 bispecific antibody, in combination with pembrolizumab in patients with advanced solid tumors. J Immunother Cancer. 2023;11(Suppl 1):A859.
Lutz MS, et al. IgG-based B7–H3xCD3 bispecific antibody for treatment of pancreatic, hepatic and gastric cancer. Front Immunol. 2023;14:1163136.
Zekri L, et al. Abstract 2865: CC-3, an IgG-based B7–H3xCD3 bispecific antibody for targeting of gastrointestinal cancers. Cancer Res. 2022;82(12_Supplement):2865.
Shankar S, et al. A Phase 1, open label, dose escalation study of MGD009, a humanized B7–H3 x CD3 DART protein, in combination with MGA012, an anti-PD-1 antibody, in patients with relapsed or refractory B7-H3-expressing tumors. J Clin Oncol. 2018;36(15_suppl):TPS2601.
Jungbluth AA, et al. Expression of melanocyte-associated markers gp-100 and Melan-A/MART-1 in angiomyolipomas. An immunohistochemical and rt-PCR analysis. Virchows Arch. 1999;434(5):429–35.
Middleton MR, et al. Tebentafusp, a TCR/Anti-CD3 bispecific fusion protein targeting gp100, potently activated antitumor immune responses in patients with metastatic melanoma. Clin Cancer Res. 2020;26(22):5869–78.
Boudousquie C, et al. Polyfunctional response by ImmTAC (IMCgp100) redirected CD8+ and CD4+ T cells. Immunology. 2017;152(3):425–38.
Middleton MR, et al. Safety, pharmacokinetics and efficacy of IMCgp100, a first-in-class soluble TCR-antiCD3 bispecific t cell redirector with solid tumour activity: results from the FIH study in melanoma. J Clin Oncol. 2016;34(15_suppl):3016.
FDA, U., FDA approves tebentafusp-tebn for unresectable or metastatic uveal melanoma; 2022.
Moore PA, et al. Development of MGD007, a gpA33 x CD3-bispecific DART protein for T-cell immunotherapy of metastatic colorectal cancer. Mol Cancer Ther. 2018;17(8):1761–72.
Shimabukuro-Vornhagen A, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56.
Borlak J, Langer F, Spanel R, Schöndorfer G, Dittrich C. Immune-mediated liver injury of the cancer therapeutic antibody catumaxomab targeting EpCAM, CD3 and Fcγ receptors. Oncotarget. 2016;7(19):28059–74.
Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81–94.
Alsajjan R, Mason WP. Bispecific T-cell engagers and chimeric antigen receptor T-cell therapies in glioblastoma: an update. Curr Oncol (Toronto, ON). 2023;30(9):8501–49.
Enrico D, et al. Antidrug antibodies against immune checkpoint blockers: impairment of drug efficacy or indication of immune activation? Clin Cancer Res. 2020;26(4):787–92.
Rossotti MA, et al. Immunogenicity and humanization of single-domain antibodies. FEBS J. 2022;289(14):4304–27.
Brennan FR, et al. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. MAbs. 2010;2(3):233–55.
Zinsli LV, et al. Deimmunization of protein therapeutics—recent advances in experimental and computational epitope prediction and deletion. Comput Struct Biotechnol J. 2021;19:315–29.
Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res. 2016;22(8):1865–74.
Clifton GT, et al. Developing a definition of immune exclusion in cancer: results of a modified Delphi workshop. J Immunother Cancer. 2023;11(6): e006773.
Zheng G, et al. Immune desert in MMR-deficient tumors predicts poor responsiveness of immune checkpoint inhibition. Front Immunol. 2023. https://doi.org/10.3389/fimmu.2023.1142862.
Liu S, Ren J, ten Dijke P. Targeting TGFβ signal transduction for cancer therapy. Signal Transduct Target Ther. 2021;6(1):8.
Thakur A, Huang M, Lum LG. Bispecific antibody based therapeutics: strengths and challenges. Blood Rev. 2018;32(4):339–47.
Ansell SM, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–9.
Rooney MS, et al. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61.
Thakur A, et al. Microenvironment generated during EGFR targeted killing of pancreatic tumor cells by ATC inhibits myeloid-derived suppressor cells through COX2 and PGE2 dependent pathway. J Transl Med. 2013;11:35.
Thakur A, et al. A Th1 cytokine-enriched microenvironment enhances tumor killing by activated T cells armed with bispecific antibodies and inhibits the development of myeloid-derived suppressor cells. Cancer Immunol Immunother CII. 2012;61(4):497–509.
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (New York, NY). 2018;359(6382):1350–5.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
We thank the National Key R&D Program of China (2019YFA0904400), the Natural Science Foundation of Guangdong Province (2023A1515010549), the Science and Technology Development Fund of Macau (FDCT/0009/2023/RIC), the University of Macau (MYRG2022-00143-FHS, MYRG-GRG2023-00158-FHS-UMDF).
Conflicts of Interest
Yuheng Gu and Qi Zhao have no conflicts of interest to report.
Ethics Approval
Not applicable.
Data Availability
Not applicable.
Code Availability
Not applicable.
Consent
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Gu, Y., Zhao, Q. Clinical Progresses and Challenges of Bispecific Antibodies for the Treatment of Solid Tumors. Mol Diagn Ther (2024). https://doi.org/10.1007/s40291-024-00734-w
Accepted:
Published:
DOI: https://doi.org/10.1007/s40291-024-00734-w