
Abstract
Objective
The study aimed to retrospectively evaluate the positive yield rate of a custom 212-gene next-generation sequencing (NGS) panel, the JAX ActionSeq™ assay, used in molecular profiling of solid tumors for precision medicine.
Methods
We evaluated 261 cases tested over a 24-month period including cancers across 24 primary tissue types and report on the mutation yield in these cases.
Results
Thirty-three of the 261 cases (13%) had no detectable clinically significant variants. In the remaining 228 cases (87%), we identified 550 clinically significant variants in 88 of the 212 genes, with four of fewer clinically significant variants being detected in 62 of 88 genes (70%). TP53 had the highest number of variants (125), followed by APC (47), KRAS (47), ARID1A (20), PIK3CA (20) and EGFR (18). There were 38 tier I and 512 tier II variants, with two genes having only a tier I variant, seven genes having both a tier I and tier II variant, and 79 genes having at least one tier II variant. Overall, the ActionSeq™ assay detected clinically significant variants in 42% of the genes included in the panel (88/212), 68% of which (60/88) were detected in more than one tumor type.
Conclusions
This study demonstrates that of the genes with documented involvement in cancer, only a limited number are currently clinically significant from a therapeutic, diagnostic and/or prognostic perspective.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Our study demonstrates that the yield of clinically significant variants in a > 200-gene panel is < 50%, suggesting that more studies are needed addressing the functionality and thereby the therapeutic potential of genomic alterations. |
Adaptation of tiered classification of genomic variants based on Association for Molecular Pathology consensus guidelines for variant interpretation in cancer (2017) has enabled evaluation and stratification of clinically significant variants in solid tumors. |
Identification of causal etiology for devising therapeutic strategies to target solid tumors should involve multi-dimensional testing including DNA-based NGS assays, RNA-based fusion analysis, tumor molecular burden and gene-expression analysis. |
1 Introduction
Cancer is a group of diseases involving uncontrolled cell growth and division that evolves and progresses due to the accumulation of somatic DNA changes, occasionally in the presence of inherited germline variants. The consequences of these changes lead to simultaneous tumor suppressor inactivation (e.g., TP53, PTEN, and NF1) and oncogenic activation (e.g., ERBB2, EGFR, and KRAS) resulting in a constitutively active cell cycle and uncontrolled proliferation [1,2,3]. Fortunately, the rise of molecular diagnostics has allowed for the detection and subsequent targeting of these oncogenic variants via specific targeted molecular therapies in certain tumor types. Genomic profiling has become a standard of care for melanoma, lung, breast, and colorectal cancers [4,5,6,7]. Furthermore, genomics-guided clinical trials are underway to evaluate the efficacy of approved and investigational molecular targeted therapies for cancers harboring specific genomic profiles [8, 9]. In this regard, precision oncology is focused on decoding comprehensive genomic tumor profiles to identify targetable genomic variants specific to the patient’s tumor that offer better clinical outcomes than standard care [10, 11].
Traditionally, low-throughput methods limited diagnostics laboratories to testing a single variant per assay. However, massively parallel next-generation sequencing (NGS) greatly expanded the content and throughput of genomic testing and dramatically changed the molecular diagnostics landscape [12]. Advances in NGS technologies have resulted in genomic sequencing becoming faster and cheaper and achieving higher throughput, all of which have helped increase genomic testing in the clinic. Therefore, the clinical community is rapidly developing NGS testing ranging from targeted gene panels to comprehensive genome-wide platforms [13,14,15,16]. Although whole exome sequencing (WES) and whole genome sequencing (WGS) have delivered comprehensive gene alteration profiles, their utility is more relevant to constitutional disorders, particularly in the identification of genotype–phenotype associations, as they provide more information than can be practically analyzed and interpreted for relevance in cancer diagnostics. Thanks to the large-scale genomic studies, including The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC), key driver-gene mutations have been identified in various tumor types [17, 18]. Accordingly, cancer-associated genes can be selected for NGS-based gene panel testing, which has become the first choice in routine cancer care [19]. However, the clinical actionability of sequenced genes remains a key challenge for clinicians and researchers, due to the constantly changing landscape of clinical trials and molecular therapies [20].
To address the need for genomic testing in precision oncology, The Jackson Laboratory (JAX) Clinical Genomics Laboratory developed the JAX ActionSeq™ assay, an NGS-based 212 cancer-associated gene panel for the identification of protein-coding mutations, copy number alterations (CNAs), and structural rearrangements for solid tumor evaluation. Here we evaluate all cases sent to our clinical laboratory for testing over a 24-month period and report the clinical characteristics and results for 261 specimens across 24 primary tissue types. Using these data, we characterized clinically actionable variants as well as genomic features that were shared among different tumor types.
2 Materials and Methods
2.1 Case Cohort
The current study is a retrospective review of 383 formalin-fixed paraffin-embedded (FFPE) samples sent to JAX’s Clinical Laboratory Improvement Amendments (CLIA) lab for testing using the ActionSeq™ assay, a custom 212-gene NGS panel, and reports on results from 261 cases.
2.2 ActionSeq™: Process and Metrics Established for Clinical Testing Based on Analytical Validation
ActionSeq™ incorporates a targeted-enrichment sequencing assay for a 212-gene DNA-based hybrid capture panel (Supplementary Table 1; see the Electronic Supplementary Material) for which all coding exons ± 10 bps are sequenced. Five-micron section slides (ten unstained slides and one stained with hematoxylin and eosin) are reviewed for neoplastic content (established cut-off is ≥ 30%) and areas of tumor marked off by a pathologist to enable macro-dissection for nucleic acid extraction. Samples which did not meet the neoplastic content cut-off criteria were either processed under deviations (≥ 20% to 30%) or failed for not meeting specimen requirements (≤ 20%).
Genomic DNA was extracted from macrodissection-enriched FFPE tissue sections (5-µm sections) using the all prep kit (Qiagen). Fifty nanograms of DNA that passed quality metrics (A260/A280 ratio between 1.6 and 1.8) was used as input for enrichment of target exons by hybrid-capture (Roche Nimblegen). Illumina NextSeq 500 sequencers generated 150-bp paired-end sequence reads with a mean coverage ≥ 500X. Variant analysis was performed using the ActionSeq™ Genome Analytics (AGA) pipeline, developed at JAX, which was CLIA validated to establish robustness and reproducibility, addressing the analytical sensitivity and specificity discussed below. The AGA pipeline links an in-house quality control (QC) Toolkit, Burrows-Wheeler Aligner (BWA) (http://bio-bwa.sourceforge.net/bwa.shtml#13), Picard (http://broadinstitute.github.io/picard/), Genome Analysis Toolkit (GATK) [21], LoFreq [22], and COpy Number Targeted Resequencing Analysis (CONTRA) [23] for QC, alignment, and variant discovery. Variants were called against human genome build GRCh38. The analytical sensitivity for single nucleotide variants (SNVs) was determined as 5% and that for indels as 8% mutant alleles. The analytical sensitivity for copy number variants (CNVs) was determined to be eight copies for amplifications and zero copies (homozygous deletions) for losses. A minimum depth of coverage of 130× was required for reporting SNVs and indels (insertions and deletions up to 50-bp in length) at the established analytical sensitivity.
2.3 Variant Interpretation and Clinical Reporting
Evidence of association between genomic variants and potential response to therapy (including clinical trials), prognostic and/or diagnostic outcomes is obtained from peer-reviewed literature, clinical practice guidelines, Food and Drug Administration (FDA) labels, publicly available databases and the JAX Clinical Knowledgebase (CKB). Information from these sources is curated into Variant Explorer (VariEx; Precision Health Software, Inc.), the JAX Clinical Laboratory’s patient knowledgebase, and the clinical significance of genomic variants is interpreted in the context of each patient’s molecular/disease profile. The ActionSeq™ report reflects the variants determined to be clinically relevant at the time of reporting. Variants are classified into four tiers based on the 2017 joint consensus guidelines of the Association for Molecular Pathology, American Society of Clinical Oncology and College of American Pathologists (AMP/ASCO/CAP) on interpretation of sequence variants in cancer [24]. The four tiers include strong clinical significance (tier I), potential clinical significance (tier II), unknown clinical significance (tier III) and benign or likely benign variants (tier IV).
3 Results
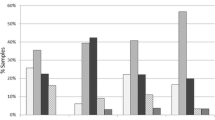
We developed and validated for clinical use a custom 212-gene NGS panel, the JAX ActionSeq™ assay, for solid tumor evaluation. Over a 24-month span, we received 383 samples for testing using our custom panel and successfully processed 261 samples (Fig. 1a). Of the 383 samples, 122 were not processed, as a result of pre-analytical failures. A majority of the failures, 63% (77 of 122), were due to low neoplastic content (0–20%), which was lower than the acceptance criteria for our clinical assay (≥ 30%), while 25% (30/122) failed due to inadequate DNA quality (passing A260/A280 ratio between 1.6–1.8) and 12% (15/122) due to insufficient DNA quantity/data quality; less than 50 ng which was the input of our assay, or less than 500X mean target coverage (Fig. 1a). At least one actionable variant was detected in 87% (228 of 261) of the processed cases. Sixteen percent of those cases (37 of 228) resulted in tier I actionable variants and 84% (191 of 228) had tier II actionable variants (Fig. 1b).

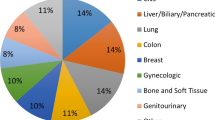
Case cohort information: a samples received for clinical testing (top light grey bar) with reasons for pre-analytical failure of samples (pie chart); b mutation yield of the JAX ActionSeq™ panel; and c primary tissue types evaluated in the cohort
Our case cohort was comprised of 24 unique tissue types (Table 1, Fig. 1c). Of the 261 specimens processed, the most common pathological diagnosis was cancer of unknown primary (CUP) (n = 50), which included metastatic CUP (n = 23), serous carcinomas (n = 8), and adenocarcinoma of unknown primary (n = 6). Other well represented specimen types included those of colorectal (n = 36), brain (n = 28), ovarian (n = 23), and lung (n = 22) primaries, while those of bone (n = 1), cervical (n = 1), tonsillar (n = 1), and soft tissue (n = 1) primaries were present to a much lesser extent.
Analysis of mutational yield revealed that 33 of the 261 cases (13%) had no detectable clinically significant variants (tier I or tier II). In the remaining 228 cases, 550 clinically significant variants (an average of approximately three variants per gene) were identified in 88 of the 212 genes (41.5%) encompassing the ActionSeq™ panel. Of the variants called, there were 229 missense variants, 102 nonsense variants, 82 amplifications and 15 deletions across 32, 28, 31, and nine genes, respectively (Fig. 2a). Stratification of genes based on the total number of clinically significant variants detected that 26 genes had five or more clinically significant variants called, of which TP53 (125), APC (47), KRAS (47), ARID1A (20), PIK3CA (20), and EGFR (18) appeared to be driver genes, as they harbored the highest number of genomic aberrations (Fig. 2b). TP53, the gene shown in our cohort to have the highest number of clinically significant variants reported (Fig. 2b), most commonly had missense variants (82), followed by 23 frameshift and 15 nonsense variants, respectively. Seven of the 26 genes with five or more clinically significant variants were comprised of only one class of variant, while the remaining 19 genes were characterized by a more heterogeneous mixture of variant classes, lending to the complexity of underlying biomolecular pathways involved in tumorigenesis (Fig. 2b). Additionally, 62 of the 88 genes (70%) contained four or fewer clinically significant variants, with AKT1, CDK4, and CTNNB1 amongst those with four variants/gene. There were 28 genes for which only one variant was identified, including AKT2, MET, SMO, SYK, and TNK2 (Supplementary Table 2; see the Electronic Supplementary Material).

Mutation yield in the cohort: a breakdown of different types of variants identified; b genes with ≥ 5 clinically significant variants detected; c number of tier I and tier II variants identified
Reporting of variants called from NGS pipelines involves their evaluation based on several criteria to classify variants according to clinical actionability, with the tiering system ranging from tier I (strong clinical significance) to tier IV (benign or likely benign). Of the 550 clinically significant variants identified in our cohort, 38 (~ 7%) were tier I and 512 (~ 93%) were classified as tier II (potential clinical significance) (Supplementary Table 3). Across the 88 total genes identified with actionable variants, just two genes (2%), MSH2 and MSH6, had only tier I variants, 79 genes (90%) had only tier II variants, and seven genes (8%) had a combination of both tier I and tier II variants (Fig. 2c). Of all the genes with both tier I and tier II variants detected, KRAS topped the list, with 47 variants (Supplementary Table 3), indicating a high level of clinical significance for cancer types characterized by this variant. A total of 1513 variants of uncertain significance (VUS) (tier III) were identified in 198 of the 212 genes, with 83 genes having tier I, II and III variants. Five of the 212 genes (HRAS, PTEN, PTPN11, ID3 and POLB) had only tier I and tier II variants and no VUS.
Overall, the ActionSeq™ assay detected clinically actionable variants in 42% of the genes included in the panel (88 of 212), 68% of which (60 of 88) were detected in more than one tumor type (Supplementary Table 4). No variants (actionable or VUS) were detected in ten of the 212 genes, AKT3, CCND2, DDR1, FH, MYD88, NOTCH4, PRKAA1, SEM1, SMARCB1 and STAT3 (4.7%). Twenty-four different cancer types were analyzed in this cohort (Fig. 1c), demonstrating the level of sample diversity seen in the modern clinical genomics laboratory. Cancers of the unknown primary, the most prevalent tumor type processed, had the greatest mutational distribution, followed by lung and gynecological specimens. Variants in the TP53 gene were detected in all but two cancer sub-groups (bone; skin and soft tissue), encompassing 23% (125 of 550) of all the clinically significant variants identified in the study. Variants in the TP53 gene often lead to inactivation of the p53 protein involved in tumor suppression [25], resulting in such variants being potentially classified as tier II variants.
4 Discussion
The development of precision oncology has forced diagnostic laboratories to abandon single-variant assays in favor of mid-to-large scale genomic testing [26, 27]. The wide variety of FDA-approved small molecule inhibitors, as well as a constantly growing clinical trial landscape, requires a diverse yet clinically relevant testing menu [28, 29]. While WES may initially be a tempting response to this problem, issues regarding high costs and massive amounts of data to interpret make targeted NGS panels a more practical option [30]. Here we described the mutational yield of a custom NGS panel developed at JAX and utilized over a 24-month period for the detection of clinically actionable variants in solid tumors.
A common hurdle for many diagnostic laboratories is minimizing the number of specimen failures [31]. Regarding the specimens unsuccessfully processed in the ActionSeq™ test system, all failures were due to pre-analytical metrics (Fig. 1a). Interestingly, the most common reason for specimen failure was due to low total neoplastic content with samples that failed due to low neoplastic content of less than 20% tumor. Insufficient yields (< 50 ng) and poor quality of DNA (suboptimal A260/A280 ratio) were other common reasons for sample failure, highlighting that inadequate specimens remain an obstacle in genomic testing. Review of the two commonly used hybridization-based commercial panels indicate that the JAX ActionSeq™ was comparable on all parameters reviewed: FoundationOne ≥ 25% neoplastic content, 50–1000 ng DNA input and > 500X coverage; Tempus ≥ 20% neoplastic content and 100 ng DNA input.
Review of the ActionSeq™ panel has revealed the absence of critical mismatch repair (MMR) genes MLH1, PMS2, and MSH3, which were added to subsequently upgrade the panel to the ActionSeq™ 2.0 test system, which also includes the evaluation of microsatellite instability (MSI) and tumor mutational burden (TMB). The high proportion of non-synonymous variants (missense, nonsense, frameshift, etc.) compared to CNAs is consistent with previously published literature [20]. While variant types differed among genes, oncogenes tended to have activating variants (SNVs, in-frame insertion/deletions) and most tumor suppressors harbored loss of function variants (nonsense and frameshift variants) (Fig. 2b, Supplementary Table 2). Overall, the mutational frequencies detected by the ActionSeq™ assay were consistent with previously published literature [20, 32]. Of note, TP53 variants were detected in approximately 50% of cases, which is consistent with other studies that have seen deleterious TP53 variants in approximately 40–50% of solid tumors [20, 32]. Further, the most frequently observed pathogenic variants on the ActionSeq™ panel were common drivers of colorectal (KRAS, APC), brain (EGFR), gynecological (ARID1A, PIK3CA), and lung (EGFR, KRAS) cancers, which represent the most commonly sequenced primaries [33,34,35,36,37]. The distribution of these tier I and II variants throughout the cohort (Fig. 2c, Supplementary Table 3) highlights the need to interpret the clinical significance of variants in the context of the primary tumor. For example, KRAS G12 V represents a tier I classification in colorectal cancer due to conferred resistance to cetuximab [38]. However, the same variant in ovarian cancer does not provide eligibility for FDA-approved therapies nor confer resistance to common treatment options, making it a tier II scenario. In contrast, FDA approval of pembrolizumab for MMR defective cancers, regardless of primary, results in all deleterious variants in the MMR genes being classified as tier I [39, 40], as seen in this study where MSH2 and MSH6 represent the only genes with just tier I variants (Supplementary Tables 3 and 4). The high number of VUS, threefold greater than the clinically significant variants (1513 vs 550), suggests that the clinical utility of larger NGS panels is yet to be maximized for precision medicine in oncology.
In conclusion, the ActionSeq™ test system detected variants of clinical significance for 87% of the sequenced cases. While updating the panel may decrease the percentage of future cases with no clinically significant variants detected, it also highlights that precision oncology requires a multi-faceted approach in which changes in gene expression, large-scale genomic rearrangements, and other potential clinical markers are also interrogated.
5 Study Limitations
Panel design represents a critical aspect of NGS test development in that an appropriate balance needs to be reached between covering potential clinical variants and keeping the panel to a manageable size [28]. We found that although ActionSeq™ contains 212 “actionable” genes, clinically actionable variants (tiers I and II) were observed in only 88 of these genes throughout the entire cohort. We also observed that in the 212-gene panel, of the ten genes which had neither tier I nor tier II or tier III variants, only four genes (DDR1, MYD88, PRKAA1 and SEM1) did not have a strong clinical significance in solid tumors. While this may be due to a variety of factors, including a relatively small cohort (n = 261) across a comprehensive distribution of cancer primaries, these data suggest that periodic review of a test’s performance characteristics are critical to maintain appropriate clinical effectiveness.
References
Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–24.
Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37.
King CR, Kraus MH, Aaronson SA. Amplification of a novel v-erbB-related gene in a human mammary carcinoma. Science. 1985;229(4717):974–6.
Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, Jenkins RB, Kwiatkowski DJ, Saldivar JS, Squire J, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol. 2013;8(7):823–59.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16.
Ellis MJ, Perou CM. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013;3(1):27–34.
Weng W, Feng J, Qin H, Ma Y. Molecular therapy of colorectal cancer: progress and future directions. Int J Cancer. 2015;136(3):493–502.
Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–36.
Qin BD, Jiao XD, Liu K, Wu Y, He X, Liu J, Qin WX, Wang Z, Zang YS. Basket trials for intractable cancer. Front Oncol. 2019;9:229.
Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793–5.
Lowy DR, Collins FS. Aiming high-changing the trajectory for cancer. N Engl J Med. 2016;374(20):1901–4.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–31.
Singh RR, Patel KP, Routbort MJ, Reddy NG, Barkoh BA, Handal B, Kanagal-Shamanna R, Greaves WO, Medeiros LJ, Aldape KD, et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn. 2013;15(5):607–22.
Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, Kalyana-Sundaram S, Sam L, Balbin OA, Quist MJ, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011;3(111):111ra121.
Nakagawa H, Fujita M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018;109(3):513–22.
Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, Garcia EP, Jia Y, Davineni P, Abo RP, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight. 2016;1(19):e87062.
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM, Cancer Genome Atlas Research N. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45(10):1113–20.
Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, Bhan MK, Calvo F, Eerola I, International Cancer Genome C, et al. International network of cancer genome projects. Nature. 2010;464(7291):993–8.
Nagahashi M, Shimada Y, Ichikawa H, Kameyama H, Takabe K, Okuda S, Wakai T. Next generation sequencing-based gene panel tests for the management of solid tumors. Cancer Sci. 2019;110(1):6–15.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–13.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8.
Wilm A, Aw PP, Bertrand D, Yeo GH, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012;40(22):11189–201.
Li J, Lupat R, Amarasinghe KC, Thompson ER, Doyle MA, Ryland GL, Tothill RW, Halgamuge SK, Campbell IG, Gorringe KL. CONTRA: copy number analysis for targeted resequencing. Bioinformatics. 2012;28(10):1307–13.
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, Tsimberidou AM, Vnencak-Jones CL, Wolff DJ, Younes A, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19(1):4–23.
Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer. 2011;2(4):466–74.
Goel G. Molecular characterization and biomarker identification in colorectal cancer: Toward realization of the precision medicine dream. Cancer Manag Res. 2018;10:5895–908.
Jonna S, Giaccone G, Subramaniam DS. Understanding molecular diagnostic technology in oncology through the lens of lung cancer. Discov Med. 2018;26(141):21–9.
Schildgen V, Schildgen O. The lonely driver or the orchestra of mutations? How next generation sequencing datasets contradict the concept of single driver checkpoint mutations in solid tumours - NSCLC as a scholarly example. Semin Cancer Biol. 2019;58:22–8.
El-Deiry WS, Goldberg RM, Lenz HJ, Shields AF, Gibney GT, Tan AR, Brown J, Eisenberg B, Heath EI, Phuphanich S, et al. The current state of molecular testing in the treatment of patients with solid tumors, 2019. CA Cancer J Clin. 2019;69(4):305–43.
Horak P, Frohling S, Glimm H. Integrating next-generation sequencing into clinical oncology: strategies, promises and pitfalls. ESMO Open. 2016;1(5):e000094.
Al-Kateb H, Nguyen TT, Steger-May K, Pfeifer JD. Identification of major factors associated with failed clinical molecular oncology testing performed by next generation sequencing (NGS). Mol Oncol. 2015;9(9):1737–43.
Perri F, Pisconti S, Della Vittoria Scarpati G. P53 mutations and cancer: a tight linkage. Ann Transl Med. 2016;4(24):522.
Schell MJ, Yang M, Teer JK, Lo FY, Madan A, Coppola D, Monteiro AN, Nebozhyn MV, Yue B, Loboda A, et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat Commun. 2016;7:11743.
Crobach S, Ruano D, van Eijk R, Schrumpf M, Group P, Fleuren G, van Wezel T, Morreau H. Somatic mutation profiles in primary colorectal cancers and matching ovarian metastases: Identification of driver and passenger mutations. J Pathol Clin Res. 2016;2(3):166–74.
Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 2010;12(9):675–84.
Takeda T, Banno K, Okawa R, Yanokura M, Iijima M, Irie-Kunitomi H, Nakamura K, Iida M, Adachi M, Umene K, et al. ARID1A gene mutation in ovarian and endometrial cancers (Review). Oncol Rep. 2016;35(2):607–13.
Femi OF. Genetic alterations and PIK3CA gene mutations and amplifications analysis in cervical cancer by racial groups in the United States. Int J Health Sci (Qassim). 2018;12(1):28–32.
Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66(8):3992–5.
Kwok G, Yau TC, Chiu JW, Tse E, Kwong YL. Pembrolizumab (Keytruda). Hum Vaccin Immunother. 2016;12(11):2777–89.
Zhao P, Li L, Jiang X, Li Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J Hematol Oncol. 2019;12(1):54.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of Interest
The authors, Pavalan Selvam, Meng-Chang Hsiao, Gregory Omerza, Daniel Bergeron, Shannon Rowe, Jasmina Uvalic, Melissa Soucy, Michael Peracchio, Shelbi Burns, Bridgette Meyers, Matthew Prego, Qian Nie, Guruprasad Ananda, Harshpreet Chandok, Kevin Kelly, Andrew Hesse, and Honey V. Reddi, declare no relevant conflicts of interest.
Ethical Approval and Informed Consent
Review by institutional review committee was deemed not necessary because only data collected during regular patient testing were used.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Selvam, P., Hsiao, MC., Omerza, G. et al. Mutation Yield of a Custom 212-Gene Next-Generation Sequencing Panel for Solid Tumors: Clinical Experience of the First 260 Cases Tested Using the JAX ActionSeq™ Assay. Mol Diagn Ther 24, 103–111 (2020). https://doi.org/10.1007/s40291-019-00435-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-019-00435-9