
Abstract
Background
Next-generation sequencing of gene panels has supplanted single-gene testing for cancer molecular diagnostics in many laboratories. Considerations for the optimal number of genes to assess in a panel depend on the purpose of the testing.
Objective
To address the optimal size for the identification of clinically actionable variants in different-sized solid tumor sequencing panels.
Patients and methods
Sequencing results from 480 patients with a large, 315 gene, panel were compared against coverage of a medium, 161 gene, and small, 50 gene, panel.
Results
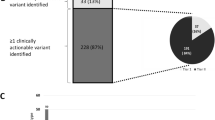
The large panel detected a total of 2072 sequence variants in 480 patient specimens; 61 (12.7%) contained variants for which there is therapy approved by the US Food and Drug Administration, 89 (18.5%) had variants associated with an off-label therapy, and 312 (65.0%) contained variants eligible for a genomically matched clinical trial. The small panel covered only 737 of the 2072 variants (35.5%) and somewhat fewer therapy-related variants (on-label 88.5%, off-label 60.7%). The medium-size panel included 1354 of the 2072 (65.3%) variants reported by the large panel. All 318 patients with a clinically actionable variant would have been identified by the medium panel.
Conclusions
The results demonstrate that a carefully designed medium size gene panel is as effective as a large panel for the detection of clinically actionable variants and can be run by most molecular pathology laboratories.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
In solid tumor profiling the optimal size of sequencing panel for the detection of actionable mutations is not defined. |
We compared the clinical results from a large panel (315 genes) to the coverage of a medium (161 genes) and a small (50 genes) panel. |
Our results indicate that the gene content of the medium-sized panel encompassed all actionable mutations of the large panel in our cohort with significantly less non-targetable results. |
1 Introduction
Genomic analysis of solid tumor tissue is the standard of care for several of the most common types of human malignancies, including non-small-cell lung carcinoma (NSCLC), colorectal carcinoma, and malignant melanoma [1,2,3]. Until recently this was generally performed by fluorescent in situ hybridization (FISH) and immunohistochemistry (IHC) for rearrangements [4,5,6] and copy number variations (CNVs) [7,8,9] and a wide array of single-gene assays (PCR, Sanger sequencing, pyrosequencing, etc.) for single nucleotide variants (SNVs) and small insertions and deletions (indels) [10,11,12]. While single-gene assays are relatively inexpensive and have a short turnaround time (TAT), they are limited in sensitivity and throughput (Sanger) or designed to detect only common mutations (PCR). The current guidelines published by the Association for Molecular Pathology (AMP), College of America Pathologists (CAP), and International Association for the Study of Lung Cancer (IASLC) recommend testing for mutation in four to eight genes for every patient with NSCLC [13]. This can lead to situations where multiple tests and/or platforms, each having a different tissue requirement, are required to analyze the tissue to determine appropriate therapy.
Next-generation sequencing (NGS) has begun to supplant single-gene assays as the test of choice for detecting SNVs and indels in some tumors, because this approach uses less tissue, is more cost efficient, and provides greater sequence coverage of the genes of interest across specimen types [14,15,16,17,18,19]. Additionally, CNVs and genomic rearrangements detected by NGS are highly concordant with both IHC and FISH [20,21,22]. The clinical NGS panels currently available interrogate different numbers of genes, ranging from a handful of tissue-specific targets [23] to whole-exome sequencing (WES) [24] and whole-genome sequencing (WGS) [25]. However, interrogating more genes increases cost and TAT, with clinical WGS (including matched non-tumor sample) currently costing up to US$10,000 per patient [26,27,28]. The largest non-exome panels contain over 1000 genes [29], many of which have no associated US Food and Drug Administration (FDA)-approved therapies, medically useful clinical trials, or prognostic implications. While WES/WGS has been shown to have no clinical benefit over a medium-sized (~ 150 genes) panel [30], and almost all large-scale trials attempting to show feasibility of molecular profiling use panels that have under 250 genes [31], the impression of many in the clinical practice community remains that “bigger is better.” Although some publications suggest that the information generated by large sequencing panels is of benefit [29, 32, 33], many clinicians are unprepared for the sheer volume of information generated by a large sequencing panel.
As a real-world example, the National Cancer Institute’s (NCI) ongoing NCI-MATCH trial was initially designed around using a medium-sized panel. The trial includes numerous treatment modalities and has grown to include dozens of arms with treatments designated for specific genomic variants [34]. Multiple testing paradigms were assessed and the Ion Oncomine™ Comprehensive Assay v1 (Thermo-Fisher Scientific, Waltham, MA, USA) was chosen and subsequently updated to version 3. These panels detect SNVs, indels, CNVs, and selected translocations in 143 genes (v1) and 161 genes (v3). This approach allowed for the conservation of tissue, as the assay only requires 20 ng each of DNA and RNA, while also allowing for a comprehensive interrogation of clinically relevant genetic variants. The aim of the present study was to test whether the additional information supplied by a large (300 + gene) NGS panel provides significantly greater clinical information than that provided by a more compact (~ 150 gene) panel.
2 Materials and Methods
Four hundred and eighty solid tumor NGS reports from a large commercial reference laboratory (Foundation Medicine, Cambridge, MA, USA) on samples from patients treated at Cedars-Sinai Medical Center from 2013 to 2017 were reviewed. Appropriate regulatory review for this study was completed by the Cedars Sinai Office of Research Compliance and Quality Improvement (IRB # Pro00051862).
All of the reference laboratory reports contained the results of an NGS panel that interrogated 315 genes, and reported SNVs, indels, CNVs, and translocations (Supplementary Table 1). The reference laboratory reports were analyzed to determine whether the reported genomic variants are contained in the Ion Oncomine™ Comprehensive Assay v3 (Thermo-Fisher Scientific) 161-gene panel (Supplementary Table 2) and the 50-gene Ion Oncomine™ Precision Assay (Thermo-Fisher Scientific) (Supplementary Table 3). Variants were assessed for pathogenicity by a precision oncology diagnostic team composed of molecular scientists, molecular pathologists, and oncologists. For variants with possible FDA approved, off-label, or clinical trial-based therapies, relevance to the indication provided by the FDA and/or National Comprehensive Cancer Network (NCCN) guidelines was evaluated with all currently available information (February 2020). A variant was considered as qualifying for a clinical trial if it was listed as an inclusion criterion for a currently recruiting clinical trial in the patient’s tumor type.
Finally, technical accuracy was assessed by running a subgroup of 55 representative cases (Supplementary Table 4) with the medium-sized (161 gene) panel. FFPE slides from the previously characterized blocks had DNA and RNA extracted with the AllPrep DNA/RNA FFEP kits running on Qiacubes (Qiagen Sciences Inc, Germantown, MD, USA). Automated library preparation and sequencing was performed on the Ion Oncomine™ Comprehensive Assay v3 running on the Ion Chef™ and S5XL™ NGS instruments (Thermo-Fisher Scientific). Sequence data were captured and analyzed with a combination of the Ion Torrent Suite Software v5.10.2, Ion Reporter Software v5.10.5, and an internally developed filtering pipeline. Discrepancies for SNVs and indels were confirmed with Sanger sequencing.
3 Results
3.1 Results from the Large Panel Analysis by the Reference Laboratory
A total of 480 reference laboratory solid tumor NGS reports, each belonging to a unique patient, were analyzed. The most common primary tumor sites were CNS, hepatobiliary/pancreas, lung, and colon, in descending order (Fig. 1). A total of 2072 genomic variants were reported (Supplementary Table 4). This included 1008 (48.6%) SNVs, 405 (19.5%) indels, 598 (28.9%) CNVs, and 61 (2.9%) rearrangements (Fig. 2). Variants associated with FDA-approved therapies were reported in 61 (12.7%) patients. No patient had more than one variant associated with FDA-approved therapy. Variants associated with off-label therapy were reported in 89 (18.5%) patients. Finally, 312 (65.0%) patients had variants associated with eligibility for clinical trials (Table 1). In total, the variants with associated FDA-approved therapeutic indications were found in ten genes (ALK, BRAF, BRCA1/2, EGFR, ERBB2, KRAS, NRAS, NTRK1, PIK3CA) with the addition of emerging therapies adding two genes (MET and RET) (Fig. 3).

Tumor site of origin for the 480 cases

Variant subtypes identified by the reference laboratory panel

Number of variants located in each respective gene/gene family associated with an indication for US Food and Drug Administration-approved therapy (blue) or National Comprehensive Cancer Network (NCCN) emerging therapies (orange). RAS includes K-RAS and N-RAS, BRCA includes BRCA1 and BRCA2
3.2 Comparison of the Large and Medium Panels
Both panels had considerable overlap with 134 common genes for SNVs and indels and 15 common genes for select rearrangements (Fig. 4). The genomic coverage of the 161-gene panel encompassed the loci of 1354 (65.3%) of the variants detected by the large panel. All patients with variants associated with FDA (61/61) and off-label (89/89) therapies would have been identified by the 161-gene panel, and all 312 patients would have been identified for genomically matched clinical trials with the 161-gene panel as well.

Genes overlapping (light gray) that cover (a) single nucleotide variants (SNVs)s and indels; (b) select rearrangements in the large- (white) and the medium- (dark gray) sized next-generation sequencing (NGS) panels. The small panels coverage was almost entirely included in both the large- and the medium-sized panel
Within the 55 cases run in parallel with the medium panel, 170/172 (98.8%) SNVs, 37/37 (100%) indels, 47/49 (95.9%) CNVs, and 3/3 (100%) fusions that were covered by the medium panel were detected. One frameshift indel was detected by the medium panel and confirmed by Sanger sequencing but not present on the report from the large panel. Both SNVs detected by the large panel not seen in the medium panel were listed as “sub clonal” on the outside report. They were not confirmed by Sanger sequencing but were likely below the limit of detection of both Sanger and the medium panel. Across all variants reported, concordance was seen in 257/262 (98.1%). All five variants with discordance were not considered to be clinically actionable at this time.
3.3 Comparison of the Large Panel and the Small Panel
The 50-gene panel was not as comprehensive in identifying reportable (or actionable) lesions. Only 737 (35.5%) of the variants were encompassed; however, eligibility for FDA-approved therapy was only minimally affected, with 54 out of 61 (88.5%) patients being correctly identified. Variants eligible for off-label therapy and clinical trials would have been identified in 54 (60.7%) and 256 (82.1%) patients, respectively. The major reason for lack of detection was inability to recognize errors in genes in the homologous recombination (HR) repair pathway (i.e., BRCA1, BRCA2, ATM, etc.).
4 Discussion
NGS has revolutionized cancer genomics, allowing detailed assessment of the mutational landscape of a tumor to be completed in a relatively short time. Since NGS technology was developed, the number of available therapies linked to specific mutations has increased significantly, with more than 80 currently approved by the FDA (four in the last year alone) [35]. Most academic laboratories struggle to decide which test or tests to employ for the molecular diagnosis of solid tumors. Single-gene tests and small NGS panels containing 5–50 genes identify some clinically actionable lesions and can be performed rapidly and at low cost. However, these small panels do not provide comprehensive coverage of clinically actionable mutations or more extended coverage that may lead to the discovery of novel actionable variants. Large panels of hundreds to thousands of genes, WES or WGS are not currently economically justified, although it is apparent that this may change in the future as the cost and reporting times are reduced and variant annotations become more standardized. In the meantime, the line between the clinical applicability of a panel and its research value has begun to blur as clinical panels have grown to number in the hundreds of genes [29, 36]. This, together with heavy marketing by large reference labs, has led to an erroneous impression of “bigger is better” by clinicians and patients alike. In addition, the larger panel results pose an extensive interpretive burden, especially in the evaluation of variants that are supported by anecdotal or low-level data. The question of what constitutes the appropriate size for a cancer gene panel based on a specific goal—clinical actionability—is relatively unaddressed in the literature.
To test the hypothesis that a large panel is necessarily superior to a small- or medium-sized panel for the detection of actionable variants, we directly compared the results of nearly 500 large (300 + gene) panel analyses of solid tumor specimens with those that would have been obtained using a medium-sized panel developed specifically to detect actionable mutations [34], and a small 50-gene hotspot panel. While the larger panel detected more overall variants, the additional variants beyond those contained in the medium panel had no impact on patient management. Every variant identified by the larger panel that was associated with an FDA-approved therapy, both on (61/61) and off label (89/89), would have been identified by the medium panel. Further, almost all (88.5%) of the patients genomically matched to FDA-approved therapies would have been detected by the 50-gene panel as well. Finally, while most of the comparison in this study was in silico, parallel testing of 55 cases with the medium-sized panel showed an overall concordance of 98.1%, with no clinically actionable variants showing discordance.
Fortunately, the issue of appropriate testing panels has begun to be addressed by a joint guideline of the College of American Pathologists (CAP), Association of Molecular Pathology (AMP), and the American Society of Clinical Oncology (ASCO) [37]. These groups recommend classification of variants into four tiers, with tier I having strong clinical significance and each subsequent level having less. Most importantly for tier I variants, the therapeutic evidence should be either level A or level B, meaning that the therapy is FDA approved for that specific variant or that there is strong evidence for the efficacy of the therapy based on well-powered clinical studies with consensus from experts in the field. Importantly, the use of these guidelines is designed to allow for clinicians to better synthesize progressively larger amounts of genomic data.
An important limitation of our study is that it is limited to the time span (2013–2017) when the collected cases were initially reported, so that consideration of panel size on large genomic biomarker such as microsatellite instability (MSI), tumor mutational burden (TMB), loss of heterozygosity (LOH), and chromosomal aneuploidy was not addressed. With recent advances in biomarker-driven response to immunotherapy [38,39,40] and the pan-cancer designation of MSI as an indication for pembrolizumab [41], TMB and MSI are especially important. While WGS/WES with matched normal samples is the gold standard for determination of TMB [42,43,44], multiple studies have shown TMB derived from large panels (~ 300 genes), with and without matched normal samples, to be highly concordant and predictive of response to therapy [45,46,47]. Additionally, similarly sized and smaller panels have been shown to be able to reliably assess MSI status [48, 49]. Currently, a panel size of at least one megabase is considered ideal for the detection of TMB, with one study finding significant variance from WES with any panel below 500 kilobases [50]. While development of TMB calculation with the OCAv3 was attempted, the panel is approximately 350 kilobases and currently does not support TMB or MSI calculation capabilities. However, a panel with similar gene content, the TruSight™ Tumor 170 (Illumina, San Diego, CA, USA) panel, has been shown to be able to calculate both TMB and MSI reliably [51, 52]. These shortcomings can also be compensated for by the use of additional assays specifically designed for TMB and/or MSI evaluation. These can be highly automated, accurate, run in parallel from the same extracted genomic material, reported concurrently, and only require an additional 20–30 ng of DNA [53, 54]. Of note, recent data show that TMB is currently not a mature biomarker for therapy selection [55,56,57] and even MSI has high amounts of variability across tumor types [58]. Revaluation of both of these markers may be necessary regardless of expert guidelines or even FDA approval.
Our results suggest that a gene panel large enough to identify cancer gene sequence variants for which an FDA-approved or off-label drug is available, or for which a clinical trial is available, is prudent when clinical actionability is the major criterion. Reflecting this, a retrospective study at University of Michigan has shown that even with the additional information provided by a large panel, ~ 90% of patients received the standard of care [59]. Very few patients received off-label therapy (< 10%) or were entered into a clinical trial (< 5%) based on the molecular results received, a trend that has been seen in large studies looking at community oncologists in NSCLC [60, 61]. Their conclusion mirrors our own, which is that most, if not all, variants that affect patient management are detectable using smaller, readily available oncology panels that can be implemented in a small- to medium-sized clinical laboratory.
One prudent approach to the care of cancer patients would be to follow a scaled molecular testing approach. Either an independent smaller panel tailored to the tumor types typically seen by the laboratory could be run first or smaller “actionable” panels could be informatically carved out of larger assays. If the initial panel is negative, then the full panel can be run or unmasked. While cheaper to run a smaller assay, when the case mix is significantly heterogenous for tumor type it becomes simpler from a workflow perspective to run informatically created sub panels from a larger assay. Assuming that an actionable variant is detected in a subpanel and the panel is not unmasked, all detected variants should still be stored in a centralized database and be available at the request of the clinician. Guidelines regarding reporting of NGS results have been addressed by numerous organizations. A particularly relevant one is from the New York State Department of Health stating that inclusion of a phrase such as “This test is designed to detect x, y and z…in genes a, b and c… However, variants other than the ones listed above may also have been detected. If interested, these can be released upon request.” Additionally, the panel should cover all currently actionable variants and those that have a high probability of reaching actionability in a clinically relevant time frame and should be subject to regular periodic review to ensure newly relevant genes and variants were not missed. If no actionable information is obtained, the sample can then be analyzed, in the setting of a clinical trial, by WES or WGS since gene discovery efforts may reveal a new drug or clinical trial that is not currently available. Actionable variants beyond those that have been identified to date will only be discovered through analysis of tens to hundreds of thousands of sequences from tumor specimens, plus basic science research on individual oncogenes.
In summary, our comparative analysis of the clinical utility of large (315 genes), medium (161 genes), and small (50 gene) NGS panels demonstrates that, when the primary clinical goal is the identification of clinically actionable mutations, an optimized medium-size panel is equally as informative as a larger panel. Thus, in this context, we conclude that bigger is not better.
References
Benson AB 3rd, Venook AP, Cederquist L, Chan E, Chen YJ, Cooper HS, et al. Colon cancer, version 1.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw. 2017;15(3):370–98.
Ettinger DS, Wood DE, Aisner DL, Akerley W, Bauman J, Chirieac LR, et al. Non-small cell lung cancer, version 5.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw. 2017;15(4):504–35.
Coit DG, Thompson JA, Algazi A, Andtbacka R, Bichakjian CK, Carson WE 3rd, et al. Melanoma, version 2.2016, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2016;14(4):450–73.
Dagogo-Jack I, Shaw AT. Screening for ALK rearrangements in lung cancer: time for a new generation of diagnostics? Oncologist. 2016;21(6):662–3. https://doi.org/10.1634/theoncologist.2016-0179.
Bubendorf L, Büttner R, Al-Dayel F, Dietel M, Elmberger G, Kerr K, et al. Testing for ROS1 in non-small cell lung cancer: a review with recommendations. Virchows Arch. 2016;469(5):489–503. https://doi.org/10.1007/s00428-016-2000-3.
Komai Y, Fujiwara M, Fujii Y, Mukai H, Yonese J, Kawakami S, et al. Adult Xp11 translocation renal cell carcinoma diagnosed by cytogenetics and immunohistochemistry. Clin Cancer Res. 2009;15(4):1170.
Wolff AC, Hammond MEH, Hicks DG, Dowsett M, McShane LM, Allison KH, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31(31):3997–4013. https://doi.org/10.1200/JCO.2013.50.9984.
Abrahao-Machado LF, Scapulatempo-Neto C. HER2 testing in gastric cancer: an update. World J Gastroenterol. 2016;22(19):4619–25. https://doi.org/10.3748/wjg.v22.i19.4619.
Kawakami H, Okamoto I, Okamoto W, Tanizaki J, Nakagawa K, Nishio K. Targeting MET amplification as a new oncogenic driver. Cancers. 2014;6(3):1540–52. https://doi.org/10.3390/cancers6031540.
Ellison G, Zhu G, Moulis A, Dearden S, Speake G, McCormack R. EGFR mutation testing in lung cancer: a review of available methods and their use for analysis of tumour tissue and cytology samples. J Clin Pathol. 2013;66(2):79–89. https://doi.org/10.1136/jclinpath-2012-201194.
Benlloch S, Paya A, Alenda C, Bessa X, Andreu M, Jover R, et al. Detection of BRAF V600E mutation in colorectal cancer: comparison of automatic sequencing and real-time chemistry methodology. J Mol Diagn. 2006;8(5):540–3. https://doi.org/10.2353/jmoldx.2006.060070.
De Castro DG, Angulo B, Gomez B, Mair D, Martinez R, Suarez-Gauthier A, et al. A comparison of three methods for detecting KRAS mutations in formalin-fixed colorectal cancer specimens. Br J Cancer. 2012;107(2):345–51.
Lindeman NI, Cagle PT, Aisner DL, Arcila ME, Beasley MB, Bernicker E, et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors. J Thorac Oncol. 2018. https://doi.org/10.1016/j.jtho.2017.12.001.
Shao D, Lin Y, Liu J, Wan L, Liu Z, Cheng S, et al. A targeted next-generation sequencing method for identifying clinically relevant mutation profiles in lung adenocarcinoma. Sc Rep. 2016;6:22338. https://doi.org/10.1038/srep22338.
Tuononen K, Mäki-Nevala S, Sarhadi Virinder K, Wirtanen A, Rönty M, Salmenkivi K, et al. Comparison of targeted next-generation sequencing (NGS) and real-time PCR in the detection of EGFR, KRAS, and BRAF mutations on formalin-fixed, paraffin-embedded tumor material of non-small cell lung carcinoma—superiority of NGS. Genes Chromosom Cancer. 2013;52(5):503–11. https://doi.org/10.1002/gcc.22047.
Angulo B, Conde E, Suarez-Gauthier A, Plaza C, Martinez R, Redondo P, et al. A comparison of EGFR mutation testing methods in lung carcinoma: direct sequencing, real-time PCR and immunohistochemistry. PLoS ONE. 2012;7(8):e43842. https://doi.org/10.1371/journal.pone.0043842.
Endris V, Penzel R, Warth A, Muckenhuber A, Schirmacher P, Stenzinger A, et al. Molecular diagnostic profiling of lung cancer specimens with a semiconductor-based massive parallel sequencing approach: feasibility, costs, and performance compared with conventional sequencing. J Mol Diagn. 2013;15(6):765–75. https://doi.org/10.1016/j.jmoldx.2013.06.002.
Hadd AG, Houghton J, Choudhary A, Sah S, Chen L, Marko AC, et al. Targeted, high-depth, next-generation sequencing of cancer genes in formalin-fixed, paraffin-embedded and fine-needle aspiration tumor specimens. J Mol Diagn. 2013;15(2):234–47. https://doi.org/10.1016/j.jmoldx.2012.11.006.
Spencer DH, Sehn JK, Abel HJ, Watson MA, Pfeifer JD, Duncavage EJ. Comparison of clinical targeted next-generation sequence data from formalin-fixed and fresh-frozen tissue specimens. J Mol Diagn. 2013;15(5):623–33. https://doi.org/10.1016/j.jmoldx.2013.05.004.
Dacic S, Villaruz LC, Abberbock S, Mahaffey A, Incharoen P, Nikiforova MN. ALK FISH patterns and the detection of ALK fusions by next generation sequencing in lung adenocarcinoma. Oncotarget. 2016;7(50):82943–52. https://doi.org/10.18632/oncotarget.12705.
Liu X, Jia Y, Stoopler MB, Shen Y, Cheng H, Chen J, et al. Next-Generation sequencing of pulmonary sarcomatoid carcinoma reveals high frequency of actionable MET gene mutations. J Clin Oncol. 2016;34(8):794–802. https://doi.org/10.1200/jco.2015.62.0674.
Pfarr N, Penzel R, Endris V, Lier C, Flechtenmacher C, Volckmar AL, et al. Targeted next-generation sequencing enables reliable detection of HER2 (ERBB2) status in breast cancer and provides ancillary information of clinical relevance. Genes Chromosom Cancer. 2017;56(4):255–65. https://doi.org/10.1002/gcc.22431.
Hagemann IS, Devarakonda S, Lockwood CM, Spencer DH, Guebert K, Bredemeyer AJ, et al. Clinical next-generation sequencing in patients with non-small cell lung cancer. Cancer. 2015;121(4):631–9. https://doi.org/10.1002/cncr.29089.
Van Allen EM, Wagle N, Stojanov P, Perrin DL, Cibulskis K, Marlow S, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med. 2014;20(6):682–8. https://doi.org/10.1038/nm.3559.
Robbe P, Popitsch N, Knight SJL, Antoniou P, Becq J, He M, et al. Clinical whole-genome sequencing from routine formalin-fixed, paraffin-embedded specimens: pilot study for the 100,000 Genomes Project. Genet Med. 2018;20(10):1196–205. https://doi.org/10.1038/gim.2017.241.
Shimoda Y, Nagashima T, Urakami K, Tanabe T, Saito J, Naruoka A, et al. Integrated next-generation sequencing analysis of whole exome and 409 cancer-related genes. Biomed Res. 2016;37(6):367–79. https://doi.org/10.2220/biomedres.37.367.
Schwarze K, Buchanan J, Taylor JC, Wordsworth S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet Med. 2018;20(10):1122–30. https://doi.org/10.1038/gim.2017.247.
Schwarze K, Buchanan J, Fermont JM, Dreau H, Tilley MW, Taylor JM, et al. The complete costs of genome sequencing: a microcosting study in cancer and rare diseases from a single center in the United Kingdom. Genet Med. 2020;22(1):85–94. https://doi.org/10.1038/s41436-019-0618-7.
Kamps R, Brandão RD, van den Bosch BJ, Paulussen ADC, Xanthoulea S, Blok MJ, et al. Next-generation sequencing in oncology: genetic diagnosis, risk prediction and cancer classification. Int J Mol Sci. 2017;18(2):308. https://doi.org/10.3390/ijms18020308.
Miller EM, Patterson NE, Zechmeister JM, Bejerano-Sagie M, Delio M, Patel K, et al. Development and validation of a targeted next generation DNA sequencing panel outperforming whole exome sequencing for the identification of clinically relevant genetic variants. Oncotarget. 2017;8(60):102033–45. https://doi.org/10.18632/oncotarget.22116.
Senft D, Leiserson MDM, Ruppin E, Ronai ZA. Precision oncology: the road ahead. Trends Mol Med. 2017;23(10):874–98. https://doi.org/10.1016/j.molmed.2017.08.003.
Hyman DM, Solit DB, Arcila ME, Cheng DT, Sabbatini P, Baselga J, et al. Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov Today. 2015;20(12):1422–8. https://doi.org/10.1016/j.drudis.2015.08.005.
Kopetz S, Mills Shaw KR, Lee JJ, Zhang J, Litzenburger B, Holla V, et al. Use of a targeted exome next-generation sequencing panel offers therapeutic opportunity and clinical benefit in a subset of patients with advanced cancers. JCO Precis Oncol. 2019;3:1–14. https://doi.org/10.1200/PO.18.00213.
Conley BA, Chen AP, O'Dwyer PJ, Arteaga CL, Hamilton SR, Williams PM, et al. NCI-MATCH (Molecular Analysis for Therapy Choice)—a national signal finding trial. J Clin Oncol. 2016;34(15_suppl):TPS2606. https://doi.org/10.1200/JCO.2016.34.15_suppl.TPS2606.
Abramson R. Overview of targeted therapies for cancer. My Cancer Genome. 2017. https://www.mycancergenome.org/content/molecular-medicine/overview-of-targeted-therapies-for-cancer/. Accessed Feb 2020
Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251–64. https://doi.org/10.1016/j.jmoldx.2014.12.006.
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer. J Mol Diagn. 2017;19(1):4–23. https://doi.org/10.1016/j.jmoldx.2016.10.002.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20. https://doi.org/10.1056/NEJMoa1500596.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–13. https://doi.org/10.1126/science.aan6733.
Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377(25):2500–1. https://doi.org/10.1056/NEJMc1713444.
Lemery S, Keegan P, Pazdur R. First FDA approval agnostic of cancer site—when a biomarker defines the indication. N Engl J Med. 2017;377(15):1409–12. https://doi.org/10.1056/NEJMp1709968.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21. https://doi.org/10.1038/nature12477.
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–8. https://doi.org/10.1038/nature12213.
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99. https://doi.org/10.1056/NEJMoa1406498.
Garofalo A, Sholl L, Reardon B, Taylor-Weiner A, Amin-Mansour A, Miao D, et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med. 2016;8(1):79. https://doi.org/10.1186/s13073-016-0333-9.
Johnson DB, Frampton GM, Rioth MJ, Yusko E, Xu Y, Guo X, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res. 2016;4(11):959–67. https://doi.org/10.1158/2326-6066.Cir-16-0143.
Campesato LF, Barroso-Sousa R, Jimenez L, Correa BR, Sabbaga J, Hoff PM, et al. Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to PD-1 blockade in clinical practice. Oncotarget. 2015;6(33):34221–7. https://doi.org/10.18632/oncotarget.5950.
Salipante SJ, Scroggins SM, Hampel HL, Turner EH, Pritchard CC. Microsatellite instability detection by next generation sequencing. Clin Chem. 2014;60(9):1192–9. https://doi.org/10.1373/clinchem.2014.223677.
Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018;7(3):746–56. https://doi.org/10.1002/cam4.1372.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34. https://doi.org/10.1186/s13073-017-0424-2.
Zhang S, So AS, Kaplan S, Kruglyak KM. Abstract 5358: Comprehensive evaluation of Illumina’s TruSight® tumor 170 panel to estimate tumor mutational burden. Can Res. 2017;77(13 Supplement):5358. https://doi.org/10.1158/1538-7445.Am2017-5358.
So A, Zhang S, Kaplan S, Yao J, Le P, Glidewell-Kenney C, et al. Abstract 3414: Determining microsatellite instability (MSI) status of colorectal cancers through next-generation sequencing (NGS). Can Res. 2018;78(13 Supplement):3414. https://doi.org/10.1158/1538-7445.Am2018-3414.
Fang P, Yan Z, Vu Q, Smith D, Galderisi C, Spittle CS, et al. Abstract 3614: Evaluation of a commercial targeted NGS panel for tumor mutation burden assessment in FFPE tissue. Can Res. 2018;78(13 Supplement):3614. https://doi.org/10.1158/1538-7445.Am2018-3614.
Bacher JW, Flanagan LA, Smalley RL, Nassif NA, Burgart LJ, Halberg RB, et al. Development of a fluorescent multiplex assay for detection of MSI-high tumors. Dis Mark. 2004. https://doi.org/10.1155/2004/136734.
Bazhenova L, Redman M, Gettinger S, Hirsch FR, Mack P, Schwartz L, et al. OA04.01 A Phase III randomized study of nivolumab/ipilimumab vs nivolumab for previously treated stage IV squamous cell lung cancer. J Thorac Oncol. 2019;14(10):S214. https://doi.org/10.1016/j.jtho.2019.08.423.
Garassino M, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, Speranza G, et al. OA04.06 Evaluation of TMB in KEYNOTE-189: pembrolizumab plus chemotherapy vs placebo plus chemotherapy for nonsquamous NSCLC. J Thorac Oncol. 2019;14(10):S216–S217217. https://doi.org/10.1016/j.jtho.2019.08.427.
Langer C, Gadgeel S, Borghaei H, Patnaik A, Powell S, Gentzler R, et al. OA04.05 KEYNOTE-021: TMB and outcomes for carboplatin and pemetrexed with or without pembrolizumab for nonsquamous NSCLC. J Thorac Oncol. 2019;14(10):S216. https://doi.org/10.1016/j.jtho.2019.08.426.
Marabelle A, Le DT, Ascierto PA, Giacomo AMD, Jesus-Acosta AD, Delord J-P, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. 2020. https://doi.org/10.1200/jco.19.02105.
Brown N, Betz B, editors. Clinical Utility of large scale genomic sequencing of solid tumors at a large Academic Medical Center Association of molecular pathology annual meeting—AMP 2017; 2017 Nov 16–18; Salt Lake City, UT: Elsevier Science Inc. 360 Park Ave. South, New York, NY 10010-1710 USA.
Presley CJ, Tang D, Soulos PR, Chiang AC, Longtine JA, Adelson KB, et al. Association of broad-based genomic sequencing with survival among patients with advanced non-small cell lung cancer in the community oncology setting. JAMA. 2018;320(5):469–77. https://doi.org/10.1001/jama.2018.9824.
Singal G, Miller PG, Agarwala V, Li G, Kaushik G, Backenroth D, et al. Association of patient characteristics and tumor genomics with clinical outcomes among patients with non-small cell lung cancer using a clinicogenomic database. JAMA. 2019;321(14):1391–9. https://doi.org/10.1001/jama.2019.3241.
Acknowledgements
We thank Dr. Mark Ewalt, Mary Horwath, Kathy Porpora, Jennifer Price, and Jorge Sincuir Martinez for initiating and curating the database that we used for the project; Lawrence Vail for his indispensable advice; and Sean Kohlmeier for his help with the essential administrative tasks involved with the project.
Author information
Authors and Affiliations
Contributions
EV, SA, JS, DME, and JL designed the experiment. EV, JS, JX, JSF, JTK, JL, RAF, AM, MM, AP, and AA collected and analyzed the data. EV wrote and JS, JX, JSF, JTK, AP, RS, AA, WZ, SA, RAF, AM, MM, DME, and JL edited the manuscript.
Corresponding authors
Ethics declarations
Funding
Department of Pathology and Laboratory Medicine, Cedars-Sinai Medical Center.
Conflict of interest
EV has consulted for and/or received honoraria from Thermo Fisher, Illumina and PierianDx. The remaining authors report no relevant conflicts of interest.
Ethics approval
Appropriate regulatory review for this study was completed by the Cedars Sinai Office of Research Compliance and Quality Improvement (IRB # Pro00051862).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Provided as supplementary appendixes.
Code availability
Not applicable.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Vail, E., Song, J., Xu, J. et al. Comparison of Large, Medium, and Small Solid Tumor Gene Panels for Detection of Clinically Actionable Mutations in Cancer. Targ Oncol 15, 523–530 (2020). https://doi.org/10.1007/s11523-020-00743-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-020-00743-9