
Abstract
Spinal muscular atrophy (SMA) is an autosomal recessive degenerative neuromuscular disorder characterized by loss of spinal motor neurons leading to muscle weakness and atrophy that is caused by survival motor neuron (SMN) protein deficiency resulting from the biallelic loss of the SMN1 gene. The SMN2 gene modulates the SMA phenotype, as a small fraction of its transcripts are alternatively spliced to produce full-length SMN (fSMN) protein. SMN-targeted therapies increase SMN protein; mRNA therapies, nusinersen and risdiplam, increase the amount of fSMN transcripts alternatively spliced from the SMN2 gene, while gene transfer therapy, onasemnogene abeparvovec xioi, increases SMN protein by introducing the hSMN gene into various tissues, including spinal cord via an AAV9 vector. These SMN-targeted therapies have been found effective in improving outcomes and are approved for use in SMA in the US and elsewhere. This article discusses the clinical trial results for SMN-directed therapies with a focus on efficacy, side effects and treatment response predictors. It also discusses preliminary data from muscle-targeted trials, as single agents and in combination with SMN-targeted therapies, as well as other classes of SMA treatments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Medications that increase survival motor neuron (SMN) protein (SMN-targeted therapies), either by upregulating splicing of the SMN2 gene to produce a greater amount of SMN protein (nusinersen and risdiplam) or by gene transfer of a human SMN gene through an AAV9 vector (onasemnogene abeparvovec xioi) were found effective in treating spinal muscular atrophy (SMA). |
Nusinersen and risdiplam are approved to treat SMA in patients of all ages. Onasemnogene abeparvovec xioi is approved in the US for use in SMA infants and children under the age of 2 years. In the EU it is approved for use in patients with SMA type 1 under 21 kg or in presymptomatic SMA neonates with 2 and 3 SMN2 copies. |
No studies have established superior efficacy of any one SMN-targeted therapy. Choice of agent is selected based on age, preference, route of administration, biodistribution and side effects. |
The earlier in the course of the disease any of these treatments are implemented the better the clinical response will be. Presymptomatic treatment results in the best clinical response and offers the best chance of normal development. |
Combination therapy studies of SMN-targeted therapy with myostatin inhibitors are currently underway. Preclinical studies indicate combination therapy targeting different systems offers a chance at a better outcome than SMN-targeted therapies alone. |
1 Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive degenerative neuromuscular disorder caused by mutations in the SMN1 gene [1], which is responsible for producing 90% of the survival motor neuron (SMN) protein in the body [2]. Motor neurons are exquisitely vulnerable to low SMN protein levels, which in turn lead to loss of spinal motor neurons and secondary muscle weakness and atrophy. An understanding of the natural history of SMA phenotypes is needed in symptomatic SMA to fully appreciate the rate of disease progression and treatment response, whereas SMA risk prediction in infants diagnosed and treated presymptomatically relies entirely on genetic burden. This article primarily covers pivotal trials providing information on approved SMA therapies needed for treatment decisions and preliminary data on investigational SMA treatments. For the sake of completeness, it also includes in a nonsystematic fashion, salient clinical studies of non-SMN modifying treatments.
2 Pediatric Phenotypes
Pediatric SMA, which was the focus of clinical trials, varies in age of onset and rate of disease progression. SMA type classifications were developed prior to the advent of treatment based on the maximum motor function achieved: type 1 is characterized by the inability to sit; type 2, by the ability to sit but never walk and type 3, by the ability to walk. Patients with SMA type 0 (also known as type 1a), in which symptoms begin in utero, were excluded from clinical trials.
Historically, SMA was one of the most common genetic causes of infant mortality, with an average carrier rate of about 1:50 [3]. Newborn screening showed population-based birth incidence varies from 1 in 7035 births in Germany [4] to 1 in 28,000 in New York state [5]. SMA type I is the most common SMA type at birth, accounting for over 40% of all cases [6]. Infants are normal at birth but between 2–6 months of age they develop hypotonia, progressive weakness, and poor feeding, which is followed by respiratory impairment. Without intervention few infants survive past 2 years of age. The median age to death or permanent ventilation is between 8 [7] and 13 months [8] of age. Most SMA type 1 patients have two SMN2 copies (Fig. 1). Clinical outcomes in infant clinical trials have been event-free survival (survival or lack of tracheostomy or respiratory support requirements ≥ 16 h), the Children's Hospital of Philadelphia Infant Test for Neuromuscular Disorder scale (CHOP INTEND) [9], Bayley Scales of Infant Development, motor scale [10] or motor milestone scores or response on the Hammersmith Infant Neurological Exam 2 (HINE2) [11] and WHO milestone acquisition [12].

Distribution of SMN2 copy number by SMA type. Percent SMN2 copies by SMA type in 625 Spanish SMA patients: 272 SMA type 1 (44%); 186 SMA type 2 (30%) and 165 SMA type 3 (26%) patients. Adapted from Calucho et al. Neuromuscular Disorders 2018; 28:208 [6]. SMA spinal muscular atrophy
SMA type 2 is an intermediate form that accounts for about a third of all incident cases of SMA. Children develop symptoms typically between 6–18 months of age. SMA type 2 infants can sit independently and some can even stand but they never walk independently. Natural history studies show improvement in motor function between ages 2–5 years [13, 14] that warrants consideration when determining treatment responses, especially in age subgroup analyses. Over time, however, there is ongoing loss of upper body strength and development of musculoskeletal deformities (e.g. contractures and scoliosis) and occasionally dysphagia and hypoventilation during sleep. Most type 2 patients have three SMN2 copies (Fig. 1) [6]. Functional motor outcomes include the Hammersmith Functional Motor Scale Extended (HFMSE) [15], the Motor Function Measure (MFM) [16] and the Revised Upper Limb Module (RULM) [17]. Survival is shortened but patients usually reach adulthood.
SMA type III is the least severe and least common pediatric SMA phenotype (Fig. 1) and is defined by the ability to walk independently. Weakness starts in the pelvic girdle and progresses relatively slowly when compared with infant-onset SMA. Onset under age 3 years (type 3a), however, is associated with a more severe phenotype and earlier loss of ambulation [18]. Diagnosis in SMA type 3 is delayed longer than for other SMA types [19]. Ambulation is assessed typically with the Six Minute Walk Test (6MWT) [20]. The pivotal SMA clinical trials did not include ambulatory SMA patients.
3 Spinal Muscular Atrophy (SMA) Genetics and Molecular Biology
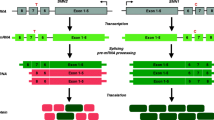
Classic 5q spinal muscular atrophy (SMA) is caused by a biallelic mutation in the Survival Motor Neuron (SMN) 1 gene [1]; 95% of cases are caused by a homozygous deletion, but about 5% of cases are caused by a point mutation in one or both alleles [21]. Located in the telomere of 5.q, the SMN1 gene is responsible for transcribing the majority of functioning full-length SMN protein (see Fig. 2). The ubiquitously expressed SMN protein is vital for survival. Its function is linked to small nuclear ribonuclear protein (snRNP) biogenesis and preMRNA metabolism [22, 23]. SMN protein levels and requirements are highest embryonically and in early postnatal life (< 3 months in human controls) [24,25,26]. High levels coincide with periods of rapid growth and cell differentiation of CNS and muscle [24]. In autopsy spinal tissue of non-SMA controls, basal SMN protein levels were reached after 3 months of age: median SMN protein levels in prenatal controls were 2.3-fold higher than early postnatal controls and 6.5-fold higher than late postnatal controls (> 3 months to 14 years) [25]. Temporal changes in SMN-inducible SMA mouse models note rapid functional decline and developmental abnormalities in the neuromuscular junction (NMJ) when SMN levels are low in early life, but not so when low SMN levels occur in later life [26]. In SMA, the resulting decrease in SMN protein results in degeneration of motor neurons, sensory neurons and NMJ [27]. Both motor neurons [28] and sensory afferents [29, 30] have been postulated to play a neuropathogenic role. The SMN protein requirements in other tissues are only noted in more severe clinical phenotypes [31], presumably because they produce the lowest amount of SMN protein. In infant-onset SMA, especially SMA type 0 (prenatal in onset), reported abnormalities include congenital cardiac malformations [32], vascular defects with necrosis [33, 34], impaired beta oxidation metabolism [35, 36], autonomic dysregulation [37] and gastrointestinal motility and intestinal absorption issues [38]. Affected organs mirror the abnormalities elicited in experimental models with severe SMN protein depletion [39,40,41,42].

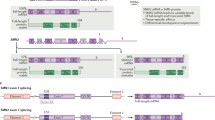
Mechanism of action of SMN-targeted therapies. a In 5q SMA, (center figure), no functional SMN protein is produced by the SMN1 gene due to a bialleic mutation. b Ninety percent of SMN2 gene (left figure) transcripts produce a non-functional truncated SMN protein that is missing exon7 (Δ7 SMN protein). b1 About 10% of SMN2 transcripts are alternatively spliced to include exon 7. c mRNA therapies act by increasing inclusion of exon 7 in SMN2 transcripts (green arrows), thus producing a larger amount of full-length functional (fSMN) protein; c1 nusinersen enhances inclusion of exon 7 by displacing hnRNP (a splicing silencer) from the ISS-N1 site on intron 7; risdiplam is believed to do so by binding to ESE (exon splicing enhancer) and 5′ on exon 7. d Gene transfer therapy onasemnogene abeparvovec uses an AAV9 vector to introduce hSMN (human survival motor neuron) gene into cells. (*) Following viral transduction, hSMN gene reaches the cell nucleus but does not incorporate into host DNA. A beta chicken actin promotor enhances expression of the hSMN gene. SMA spinal muscular atrophy, SMN survival motor neuron
3.1 SMN2 Gene Copy Number and Disease Prediction
The phenotypic expression of the SMN1 biallelic mutation is variable given the presence of a paralogous gene, SMN2, in which a C for T nucleotide substitution disrupts an exonic splicing enhancer (ESE) and results in the exclusion of exon 7. This ∆7 SMN protein is non-functional and rapidly degraded (Fig. 2) [43]. However, about 10–15% of the SMN2 transcript undergoes alternative splicing to include exon 7, and thus produce full-length SMN (fSMN) protein. Humans carry between one and five SMN2 gene copies [6]. The SMN2 gene is a disease modifier; the lower the SMN2 copy number, the earlier the disease onset and more severe the SMA phenotype [44]. Most type I patients have two SMN2 copies, while patients with SMA type 2 usually have three copies and type 3 patients have three and four SMN2 copies [44, 45]; however, there is overlap in the number of SMN2 copy numbers distributed among the different types of SMA patients [6, 45] (Fig. 1).
Not all SMN2 gene copies are equivalent, which may account for divergence between copy number and SMA phenotypic prediction. Several SMN2 variants have been identified that are associated with milder phenotypes. Variant c.859 G>C in exon 7 results in a 20% increase in fSMN protein [46, 47]. Variants A-44G [48, 49], A549G and C-1897 [49] on intron 6 are also linked to milder SMA phenotypes. Less is understood regarding SMN2 variants that result in more severe SMA phenotypes or whether specific SMN2 variants are associated with a more muted response to mRNA therapies.
4 Survival Motor Neuron (SMN)-Targeted Therapies
Currently in the US and elsewhere, three SMN-targeted therapies have been approved for treatment in SMA. Nusinersen (Spinraza™) is an antisense oligonucleotide (ASO) mRNA splicing modulator that was the first to be approved for SMA of all types and ages by the US Food and Drug Administration (FDA) in December 2016 [50] and by the European Medicine Agency (EMA) in April 2017 [51]. Regardless of age, a dose of 12 mg is delivered intrathecally (IT) on days 1, 15, 30 and 60, followed by maintenance every 4 months. In the US, gene transfer therapy (GTT) with onasemnogene abeparvovec xioi (Zolgensma™) was the second treatment approved for use in infant SMA under age 2 years [52]. It is administered as a one-time intravenous infusion at a dose of 1.1 × 1014 vg/kg. The EMA provided onasemnogene with conditional authorization in May 2020 for use in infants up to 21 kg who have a diagnosis of SMA type 1 or up to three copies of the SMN2 gene [53]. A third agent, mRNA splicing modulator risdiplam (Evrysdi™), is an oral liquid preparation administered daily that was approved in the US in August 2020 [54] and in the EU in March 2021 [55] for use in SMA patients 2 months of age or older. Risdiplam was recently approved by the FDA for use in SMA infants under 2 months of age [56]. Dose is weight based: 5-mg dose for ≥ 20 kg; 0.25 mg/kg/day for < 20 kg.
4.1 Antisense Oligonucleotide (ASO) Nusinersen
ASOs are short synthetic strings of nucleotides designed to selectively bind through base pairing hybridization to RNA that encodes the protein of interest. In SMA, the ASO is designed to bind to a specific sequence in the intron of exon 7 in the region occupied by the heterogeneous nuclear ribonucleoprotein (hRNP A1/2 proteins) that masks the intron splicing silencer N1 (ISS-N1) site. By displacing hRNP A1/2 from the ISS-N1 site, the ASO promotes the inclusion of exon 7 (Fig. 2) [57].
ASOs do not cross the blood–brain barrier and must be administered directly into the central nervous system. Application of an SMA-specific ASO to various SMA systems (cell cultures, transgenic mice) showed an increase in expression of SMN protein [58]. Intraventricular injection of ASO in the SMA ∆7 mouse model corrected SMA-related molecular and histological abnormalities (muscle size, motor neuron number, NMJ integrity), and mitigated the severity of the phenotype, improving survival and motor function [59]. These studies set the stage for the clinical trials that followed.
Several nusinersen clinical trials have been performed—a phase II dose escalation infant study (CS3A) and late-onset SMA phase II dose escalation open-label studies (CS2-CS10), which included ambulatory SMA patients. The pivotal phase III trials were sham procedures controlled for both infant-onset (ENDEAR) and late-onset (CHERISH) SMA. Patients from all these trials were invited for follow-up in the SHINE (CS11) study. NURTURE is the presymptomatic nusinersen study.
4.1.1 Symptomatic Infant SMA Trials
4.1.1.1 ENDEAR (CS3B) Sham-Control Study (NCT02193074)
CS3B was a phase III global randomized multicenter sham procedure-controlled double-blind study that included 121 SMA type 1 infants with two SMN2 copies treated 2:1 with nusinersen. Study design, eligibility criteria and dosing regimen with 12 mg intrathecally/sham are listed in Table 1. Entry criteria include disease onset under age 6 months, age < 7 months at screening and being free of respiratory requirements, although preventive non-invasive ventilation was allowed. Significant improvements from baseline compared with control were noted on HINE2 milestones and CHOP INTEND scores by 13 months (Table 2). At study end, 51% of nusinersen-treated compared with 0% of sham control infants were motor milestone responders on the HINE2 (p < 0.001) [60]. Of note, infants with disease duration < 13.1 weeks had a greater chance (77%) of being event free (i.e. alive and with no permanent ventilation needs) than those with ≥ 13 weeks’ disease duration (46%). Only 8% were sitting independently at 13 months when the study closed prematurely due to lack of clinical equipoise. Infants tolerated medication well and no significant safety issues were identified related to the medication or the procedure itself. Most side effects were due to SMA-related respiratory issues, and decreased growth (height/length) was also noted in nusinersen-treated infants compared with controls [61]. Laboratory abnormalities include proteinuria and mild thrombocytopenia.
4.1.1.2 Open-Label Dose Infant SMA Study, CS3A (NCT01839656)
CS3A was a phase II nusinersen study of infants with SMA symptoms between 3 weeks and 6 months of age who were initially treated with either 6 or 12 mg of nusinersen. The high-dose cohort showed beneficial effects in event-free survival and motor functions (CHOP INTEND and HINE2 scores) [62]. Most infants had two SMN2 copies but two infants in the 12-mg group had three SMN2 copies. Infants showed continued gains in developmental milestones at 36 months of follow-up: 42% (8/19) of patients were sitting independently and only two required permanent ventilation [63].
4.1.2 Later Onset SMA Trials
4.1.2.1 CHERISH (CS4) SMA Type 2 Sham-Controlled Study (NCT02292537)
CS4 was a phase III, double-blind, placebo–sham procedure-controlled (2:1 randomization), multinational multicenter study that enrolled 126 children with SMA type 2 between 2–12 years of age (oldest enrollee was 9 years old) and free of significant scoliosis or contractures (Table 2). Dosing of nusinersen 12 mg IT or sham was administered on days 1, 29, 85 and then every 6 months. Primary outcome was change from baseline on the HFMSE assessed at 15 months, with + 4.0 points versus − 1.9 points, respectively, for the treated versus control group for a 5.9-point difference between groups in favor of nusinersen (Table 3) [64]. The importance of age of first dosing in predicting motor response was evident in an interim analysis performed at day 690 in CHERISH patients enrolled in the CS11-Shine extension study. When stratified by age at first dosing into three groups, the largest change from baseline on the HFMSE, an 8.4-point increase, was observed in the youngest age group (2.1 to < 3.7 years), an intermediate increment (+ 3 points) in the 3.7- to < 4.9-year age group, while a slight decline in function was observed in the oldest (4.9–9.0 years) age group (− 2 points) [65]. Nusinersen was safe to administer. There were no differences in adverse events across the initial dose cohorts. Most side effects were related to the lumbar puncture (LP), comprising headache, back pain and post-LP headache. In later onset, longer treated SMA subjects, 41% reported post-LP headache, typically within 5 days of the procedure [50]. Laboratory abnormalities include proteinuria and mild thrombocytopenia.
4.1.2.2 Open-Label (CS2/CS12) SMA Type 2 and 3 Studies
Children in the initial, late-onset SMA, open-label dose-finding study exhibited a 5.8-point increase from baseline in the HFMSE in the 9-mg cohort at 9-month follow-up [66]. All dose cohorts transitioned to receiving 12-mg doses and were followed in the SHINE extension study. At day 1150, SMA type 2 patients showed a + 10.6 mean point increase from baseline on HFMSE score and ambulatory SMA type 3 patients showed a 92-m mean gain from baseline in distance walked on the 6MWT [67].
4.1.3 Presymptomatic SMA Trial
NURTURE (SM201) (NCT023865553), was a presymptomatic SMA nusinersen treatment study that enrolled 25 infants identified prenatally or at the time of birth to < 6 weeks of age. Fifteen infants had two SMN2 copies and 10 infants had three SMN2 copies. Inclusion requirements were a normal neurological exam and an ulnar compound muscle action potential (CMAP) > 1.0 mV. Infants treated with nusinersen (same regimen as ENDEAR) showed 100% survival with no infant requiring permanent ventilation. At median follow-up of 2.9 years, the three SMN2 copies group showed motor development within the norm and all were walking within the WHO window. In the two SMN2 copies group, earlier milestones were achieved more readily than later milestones: median age of sitting of 7.9 months was reached in 15/15 (100%) of infants whereas median age of walking independently of 20.4 months (outside the WHO upper limit) was reached in only 12/15 (80%) of infants, 7/15 (47%) of whom did so within the WHO window. Three infants were not walking by age 36 months [68]. Onset of dysphagia was noted in 4/15 (27%) infants who needed tube feeding, two exclusively and two combined with oral feeds [69]. Plasma heavy-chain neurofilament levels were higher in infants with two SMN2 copies compared with those with three SMN2 copies and levels predicted response to treatment [68].
4.1.4 Adult SMA Trials
No controlled clinical trials with nusinersen have been conducted in adults. However, a large observational German study in SMA patients aged 16–65 years, 40% of whom were ambulatory, reported nusinersen-related improvements over time. The proportion of patients that gained ≥ 3 points from baseline on the HFMSE score was 28% at 6 months, 35% at 10 months and 40% at 14 months. Among ambulatory patients, distance walked from baseline increased 22 m at 6 months, 31 m at 10 months and 46 m at 14 months (only 40% of participants were assessed at 14 months). Of note, the treated cohort was more functional than expected for age (mean baseline HFMSE score of 20 and 6MWT of 360 m) [70]. Another large observational study found that SMA type 3 patients (N = 103) (both sitters and walkers) treated with nusinersen for > 6 months showed significant improvement on HFMSE and RULM. Non-ambulatory patients showed a greater improvement in RULM scores than ambulatory ones. Too few SMA type 2 patients (n = − 13) were included to expect a response [71]. Results from two other smaller observational studies of adult SMA patients treated with nusinersen, one of which included very weak SMA type 2 patients [72] and the other which included both ambulatory and non-ambulatory SMA type 3 patients [73], reported no effect on HFMSE. A modest change was noted, however, in baseline distance walked on 6MWT (+ 8.24 m) in ambulatory patients on day 300 [73].
Laboratory findings reported in the pediatric clinical trials include mild thrombocytopenia, not reported below 50,000 per microliter, and mild proteinuria. Proteinuria was more frequent in later-onset, longer-treated SMA patients (69%) compared with infants (33%). Post-marketing experience identified five cases of hydrocephalus linked to nusinersen (four with SMA type 1 and one in an adult with SMA) [74]. It is unclear if hydrocephalus was related to nusinersen treatment, given the 4.7 times higher incidence of hydrocephalus noted in SMA patients compared with healthy controls [75]. One case of recurrent aseptic meningitis has also been reported following repeated doses of nusinersen [76].
4.2 AAV9 Gene Transfer Therapy (GTT), Onasemnogene Abeparvovec (OAV)
Advances in viral gene delivery, especially identification of AAV serotypes suitable for gene delivery and possessing variable tissue tropism and lack of pathogenicity, facilitated gene transfer studies. AAV9 was found to be an ideal vector for SMA given its CNS tropism and long lasting expression in tissues [77]. Preclinical SMA mice studies showed that sc-AAV9 gene transfer therapy (GTT) injected intravascularly traversed the blood–brain barrier, transfected about 60% of motor neurons and improved motor function, neurophysiology and survival [78]. Phenotypic rescue, however, was contingent on age of treatment: the best response occurred at postnatal day 1 and 2, partial response at postnatal day 5 (15-day survival) and no response at postnatal day 10. The lower response noted with age was attributed possibly to the preferential astrocytic transfection reported following intravascular GTT delivery in animals as they mature [79]. Phenotypic rescue with GTT was also documented in a large animal SMA model [80].
Following intravenous administration in neonatal mice, vector and transgene was distributed widely with highest expression in heart and liver and substantial expression in brain and spinal cord. Transgene expression in humans had a similar tissue biodistribution [81]. The transgene is diluted from cells that replicate (e.g. endothelium) but remains in neurons as these cells do not replicate. In preclinical GTT toxicity studies, the main organs affected were heart and liver. Onasemnogene dose-related findings in the cardiac ventricles were inflammation, edema and fibrosis and in the atria, inflammation, thrombosis, myocardial degeneration/necrosis and fibrosis [52]. Many cardiac effects occurred at the lower dose tested (1.4 times the recommended dose). In the liver, hepatocellular hypertrophy, Kupffer cell activation and scattered hepatocellular necrosis was observed.
4.2.1 Symptomatic Infant SMA Trials
START (NCT02122952), the first human gene transfer trial in infants with SMA, was a phase I, dose-escalating study which administered a single intravenous dose at two concentrations of scAAV9 vector carrying the hSMN transgene. Infants enrolled were symptomatic type 1 SMA patients with two SMN2 copies. Fifteen infants were divided into two cohorts: low and high doses (Table 1). Infants in the higher dose cohort on average were very young and of high function. The mean age at enrollment was 3.4 months with two infants who were 1 month of age or younger; the maximum CHOP INTEND score reported was 50, a score that is not expected to be reached in symptomatic SMA type 1. In the higher dose cohort at age 20 months or older, 12/12 were event free, 11/12 were able to sit for 5 s, 11/12 fed orally and all had reached CHOP INTEND scores over 40 points [82], a threshold not surpassed in SMA type 1 patients (Table 2) [7]. Of note, infants in the high-dose cohort treated early (< 3 months of age) and with a higher baseline motor function on CHOP INTEND (> 20 points) showed the best response, while early dosing with lower baseline scores (< 20 points) had an intermediate response and infants dosed after 3 months had the worst response. By age 24 months, the ability to sit for 30 s was achieved in 100% of each early dosing groups, but this was achieved much earlier in the high function group compared with the low function group (mean age 10 ± 1.9 months vs 21.2 (1.86) months, respectively). Only 50% in the late dosing group achieved this milestone by 24 months and about a third required non-invasive respiratory and nutritional support [83]. Given that both the early dosing groups (high and low function) had the same mean treatment age (1.5 months), the differential impact of baseline motor function on response to treatment underscores the importance of the size of the residual motor neuron pool at the time of GTT.
4.2.1.1 STRIVE-US and STRIVE-EU (Phase III)
Two phase III onasemnogene abeparvovec (OAV) trials (STRIVE-US and STRIVE-EU) have recently been completed. The studies ran in parallel but employed slightly different eligibility criteria. STRIVE-US treated 22 SMA type 1 infants with two SMN2 copies under 6 months of age. To be eligible infants had to have no swallowing issues or respiratory requirements (no hypoxia when sleeping). Infants had a mean age of 3.5 months at treatment. Primary outcome, sitting for 30 s, was achieved in 59% of treated infants (13/22) at 18 months [84]. Event-free survival at 14 months was 91% and decreased to 82% by 18 months. As with the START study, about a quarter of patients developed transaminitis which was severe in 5%. Hydrocephalus occurred in one patient.
STRIVE-EU employed a broader eligibility criteria as it allowed respiratory support < 12 h and need for feeding support, which occurred in 27% (9/32) of enrolled infants [85]. Mean age at treatment was 4.1 months. The primary outcome measure, sitting for 10 s, was achieved in 44% (14/32) of infants at 18 months. Median age of achieving sitting milestone was 15.9 months (range 9.6–18 months).
4.2.2 Presymptomatic SMA Trial
SPRINT (NCT03505099) was a phase III, multicenter, single-arm OAV trial of presymptomatic SMA identified with a biallelic SMN1 gene mutation prenatally or at the time of birth that enrolled 29 infants < 6 weeks of age. Fourteen infants had two SMN2 copies and 15 infants had three SMN2 copies. Inclusion requirements were a normal neurological exam, swallowing thin liquids and an ulnar CMAP ≥ 2 mV. About half of infants with two SMN2 copies were following a developmental trajectory within the norm on the Bayley Scales Infant Development (BSID) III (gross motor) [86]. At the 18-month visit, 100% were sitting unsupported for 30 s, 79% (11/14) had reached the sitting milestone within the normal developmental window; 64% (9/14) were walking independently; walking milestone was reached within the WHO developmental window in 36% (5/14) of infants assessed with the Bayley scale and in 43% (6/14) assessed with WHO milestones [87]. No patients required feeding or respiratory assistance. In the three SMN2 copy group, all patients were following a developmental trajectory within the norm.
4.2.3 Immunogenicity
The most common complications following intravenous OAV treatment are liver and cardiac enzyme elevations that occur to a varying degree and result from tissue transfection and its corresponding immune response. Immune response is mitigated by prophylactic treatment with 1 mg/kg of prednisone. Enzyme elevation can occur shortly after OAV infusion and be severe enough to warrant increasing doses of prednisone and rarely intravenous steroids to suppress. Enzyme elevations can also occur during steroid taper and require more prolonged treatment before resolving [88]. Cardiac enzymes elevation, however, has not been found to affect cardiac function [52]. Rare side effects reported with OAV include liver failure [88] and hepatic fibrosis, thrombotic microangiopathy [89] and hydrocephalus [84].
4.2.4 Investigational Intrathecal OAV Trials
STRONG was the initial dose-escalating intrathecal trial for SMA type 2 patients, ages ≥ 2 and < 5 years, that found significant improvement in HFMSE. It was placed on hold by the FDA in 2019 because of concerns of dorsal root ganglia (DRG) toxicity. The hold was lifted in August 2021 following the results of toxicity studies in nonhuman primates that addressed these concerns [90]. Currently underway is STEER (NCT05089656), the new phase III, randomized, double-blind, sham-control trial designed to evaluate the clinical efficacy, safety and tolerability of a one-time intrathecal (IT) dose of OAV-101 in treatment-naïve SMA type 2 patients between 2 and 18 years of age and able to sit.
4.3 mRNA-Based Small Molecule Risdiplam
Risdiplam is an orally bioavailable SMN2 splicing modifier that enhances exon 7 inclusion from SMN2 transcripts. Based on findings from a precursor SMN2 splicing molecule RG7800, risdiplam effects on SMN2 premRNA are thought to occur through action on the SMN2 5′ splice site to enhance the binding affinity of U1 snRNP and help stabilize the transient double-strand RNA structure formed between the SMN2 pre-mRNA and U1 snRNP complex [91].
4.3.1 Preclinical Studies
When administered shortly after birth in the SMN∆7 severe SMA-like mouse model, the small molecule was found to diminish muscle atrophy, prevent motor impairment and extend survival into adulthood [92]. Small molecules, such as risdiplam, have the potential for systemic off-target effects. Toxicity studies performed with the risdiplam precursor, RG7800, found non-reversible histological change in the retina of monkeys that were administered doses higher than those ever reached in the clinical trial [93]. As a result, the small molecule was refined further for greater selectivity and improved physicochemical properties.
FOXM1 and MADD are two genes whose function, respectively, affects cell regulation and apoptosis that may match the preclinical toxicology data. FOXM1 protein is highly expressed in rapidly dividing cells (e.g. gastrointestinal tract, male germ cells, skin and blood cell progenitors in the bone marrow). Preclinical findings are largely reversible following discontinuation of the small molecule [93]. Testicular germ cell degeneration observed in prepubescent rats and monkeys was largely reversible at 4 weeks, when testes were examined. Findings had no effect on subsequent fertility in rats [54]. Toxicity observed in preclinical safety studies—parakeratosis, bone marrow depression and retinal findings [94]—has not been observed in the clinical trials discussed hereafter. Risdiplam showed good distribution throughout the body, achieving drug levels and risdiplam-induced SMN protein elevations in CNS and other tissues that mirror plasma levels [95]. Risdiplam is bound by albumin and metabolized primarily by the liver, but minimally by the CYP3A system.
Risdiplam has completed an infant SMA phase II study (FIREFISH study part 1) and a phase III, late-onset SMA study (SUNFISH study). Currently still underway are the part 2, phase III FIREFISH study; the JEWELFISH clinical trial targeting treatment non-naïve SMA patients and RAINBOWFISH, an international presymptomatic SMA study.
4.3.2 Symptomatic Infant-Onset SMA Trials
FIREFISH (NCT02913482) is a two-part (phase II/III), multi-center, open-label study of the safety and efficacy of risdiplam in infants aged 1–7 months at enrollment with Type 1 SMA and two SMN2 gene copies. Four infants received the low dose and 17 the high dose (Table 1). Part 1 entails the completed dose-finding component that enrolled 21 infants [96]. The median age at first dose was 6.7 months, which is older than the age of infants enrolled in other symptomatic SMA trials. Study design and baseline participant information are depicted in Tables 1 and 2, respectively. Baseline SMN protein levels increased > 2-fold in both therapeutic-dose groups by 4 weeks, and remained elevated throughout the 12 months of treatment. Survival among the entire cohort at 12 months (mean age 17.5 months) was 86%. All surviving infants in the therapeutic dose group were fed orally (15/15), either exclusively or combined with tube feeding, and 41% (7/17) of this group were able to sit for 5 s unsupported. Frequent side effects, seen in about a third or more infants, were pyrexia, upper respiratory tract infection and diarrhea. The most common serious adverse events were respiratory and SMA related. Of note, no patient withdrew or had serious side effects related to risdiplam. Part 2, the pivotal phase III efficacy study, is currently underway and 41 infants with Type 1 SMA have been enrolled. The primary endpoint of FIREFISH Part 2 is sitting unassisted for 5 s.
4.3.3 Later-Onset SMA Trials
SUNFISH (NCT02908685) is also a two-part multicenter study. Part 1 entails the dose-finding component. Part 2 is a phase III, placebo-controlled 2:1 (N = 180) component that included non-ambulant SMA (type 2 and 3) patients 2–24 years of age. Primary outcome was change from baseline on the MFM32. Baseline characteristics are noted in Table 2. The treated cohort was composed mostly of SMA type 2 patients (70%) with a median age at screening of 9 years. SUNFISH patients exhibited more advanced SMA disease than patients enrolled in the other late-onset SMA study, as over 60% of the cohort had scoliosis, which was severe (Cobb angle > 40°) in 28% of the treated participants. Older patients (between 12–24 years) had higher rates of scoliosis (78.6%) than the younger ones. When compared with placebo, significant treatment differences favoring risdiplam were observed at month 12 of treatment on MFM32 scores (+ 1.55 points), especially in the D3 domain that corresponds to distal limb function (+ 2.34 points) and RULM scores (+ 1.59 points). The proportion of patients whose baseline MFM score increased ≥ 3 points was 2.35-fold higher (p = 0.04) in the risdiplam-treated group (see Table 3) [97]. Younger patients tended to show a greater response to risdiplam, while older patients tended towards stabilization. Safety analyses were performed in the intent-to-treat cohort (N = 180). The most common side effects reported were upper respiratory tract infections, pyrexia, headache, diarrhea, and rash. Serious adverse events were comparable across groups and none were related to risdiplam. No risdiplam-related laboratory abnormalities or serious adverse events were noted, specifically none of the preclinical toxicity findings were observed in any subject.
JEWELFISH (NCT03032172) is a multicenter, open-label study of daily risdiplam in non-naïve patients with SMA (N = 174) with a broad inclusion criteria of age (6 months to 60 years) and SMA types (1–3), who previously received therapy with RG7800 (N = 13), nusinersen (N = 76), olesoxime (N = 70) or onasemnogene abeparvovec (N = 16). Risdiplam treatment led to a rapid and sustained > 2-fold increase in SMN protein level from baseline that mirrored findings from the SUNFISH study. No serious adverse events were reported. No drug-related safety findings leading to withdrawal were reported for any patient in JEWELFISH. The safety profile was consistent with the safety profile observed in treatment-naïve patients [98].
4.3.4 Presymptomatic SMA Trial
RAINBOWFISH (NCT03779334) is an ongoing open-label, single-arm, multicenter, global clinical study enrolling infants recruited from birth to 6 weeks of age (at first dose), regardless of SMN2 copy number. Unlike other presymptomatic studies, CMAP amplitude was not an entry criteria. Preliminary data after 12 months of risdiplam treatment in five infants found a favorable development profile and all infants were fed exclusively by mouth. Infants will receive risdiplam for 24 months, followed by a 36-month extension. The primary endpoint is the proportion of infants sitting without support for at least 5 s after 12 months of treatment [99].
4.4 Branaplam
Branaplam is an SMN2 slicing modulator currently being studied in SMA type 1 infants in an open-label, multipart, first-in-human phase I/II study in which the primary outcome is safety and tolerability. Efficacy is a secondary outcome. A preliminary report after 24 months of branaplam treatment found that 28% (6/21) of infants required no ventilation support and 90% (19/21) were fed orally. Motor function at 24 months also improved: CHOP INTEND score increased 25 + points from baseline and 37.5% of infants were sitting independently or better on the HINE2 [100]. Branaplam was well tolerated.
4.5 Prediction of Response to Treatment
Across the various SMA types, disease evolution varies in age of onset and slope of decline, but all types share a similar pattern of decline, namely a subclinical phase of motor neuron loss (presymptomatic), followed by a relatively more rapid progression of symptoms (early acute phase) that in time is followed by a pseudo plateau phase (late chronic phase), in which disease progression continues but at a much slower pace [101]. Response to treatment is highly dependent on how early in this curve the patient is treated, that is, before or after the occurrence of substantial loss of motor neurons. Given the variability of disease progression for the same SMN2 copy number, both age and baseline motor function should be taken into account when reviewing clinical trials results. During the late chronic phase, not only is there a smaller pool of motor neurons available to recover, but also musculoskeletal changes (contractures and scoliosis) interfere with treatment response. The finding of improvement in CMAP and motor unit potentials, but not in functional motor outcome measures in nusinersen-treated very weak adult SMA patients [72], would tend to support this notion. Hence, during the SMA late chronic phase, stability of function (i.e. lack of disease progression) is a reasonable treatment response. Further, given the differences in ages, baseline function and study design of the various SMN-targeted treatment trials performed (Tables 1, 2, 3), superior efficacy of any one agent cannot be established. Aside from preference, other factors that inform treatment decisions include age, route of drug administration, distribution and potential side effects discussed in the following sections. It should be noted that more has been published on nusinersen treatment in SMA than for the other two approved agents because of its earlier date of approval and widespread use (11,000 + SMA patients treated thus far worldwide).
4.6 Treatment Considerations
The SMA treatment considerations in selecting approved SMN-targeted therapies are summarized in Table 4 and detailed below.
4.6.1 Nusinersen
4.6.1.1 Advantages
Nusinersen is relatively inert, non-immunogenic and safe to administer. Routine laboratory tests have a rapid turn-around time that facilitates early treatment. With over 7 years of commercial treatment experience, the feasibility and tolerability of repeated IT nusinersen injections are well documented.
4.6.1.2 Disadvantages
Nusinersen requires repeated intrathecal injections for life, which poses a cumulative risk of post-lumbar puncture headaches that can be mitigated with the use of smaller caliber spinal needles. Additional treatment risks arise from repeated anesthesia (necessary in young children), for patients with spinal hardware, and from radiation due to repeated fluoroscopy. About half of adult, treated SMA patients report a wearing-off effect of nusinersen during the month prior to dosing [102]. Other potential drawbacks are nusinersen’s restricted tissue distribution (mostly to CNS) and its spinal cord distribution gradient. Compared with lumbar spinal cord, nusinersen concentrations are lower cephalad: levels are 2-fold lower in the cervical spine [25] and brainstem levels are only about 30% of that achieved in the lumbar spine [62]. The lower nusinersen concentration cephalad could explain persistent bulbar symptoms in treated type 1 patients or the onset of swallowing symptoms with preservation of motor function reported in a few older NURTURE patients [69]. Because the same dose is administered to neonates and adults, concerns have arisen regarding under-dosing older patients, an issue that is being currently addressed in the DEVOTE study, which is designed to assess safety, pharmacokinetics and efficacy of higher doses of nusinersen [103].
4.6.2 Onasemnogene Abeparvovec Xioi
4.6.2.1 Advantages
OAV is administered as a single intravenous injection. Theoretically, transfected neurons should retain functioning transgene indefinitely, however, long term durability (past 6.2 years) of OAV treatment is unknown [104].
4.6.2.2 Disadvantages
Often there are delays in administration related to normalization of pre-dose labs and, in some settings, in the time needed to obtain insurance approval. It is a novel biological agent with inflammatory and immune-mediated effects on liver and heart (enzyme elevations) that often requires prolonged immune suppression of young infants, up to 220 days [88], raising the risk of serious viral co-infections. Long-term safety and durability is unknown. OAV has been administered in a very small, restricted population, insufficient in number to show rare complications.
4.6.3 Risdiplam
4.6.3.1 Advantages
Risdiplam is a daily orally administered medication which has excellent CNS and tissue distribution [95] and a favorable safety profile [54].
4.6.3.2 Disadvantages
Risdiplam has potential effects on male fertility; although degeneration of testicular germ cells is largely reversible in preclinical models, assessment at 4 weeks was insufficient to assess full recovery [54]. Treatment is contraindicated during pregnancy, and contraception must be maintained in women of child-bearing age during treatment and up to 1 month after risdiplam is discontinued. Care must be taken for interaction with drugs that are MATE (multidrug and toxin extrusion) substrates.
5 Non-SMN-Targeted Therapies
5.1 Muscle-Targeted Therapies
Myostatin inhibitors and skeletal troponin activator reldesemtiv are the most recent agents under study in SMA. Efficacy of myostatin inhibitors is being evaluated individually or as an addition to SMN-targeted therapy—combined with nusinersen in the TOPAZ study and planned for study combined with risdiplam in the MANATEE study.
5.1.1 Apitegromab
Myostatin is a muscle cell regulator. Myostatin inhibitors block this effect, which enables greater muscle growth. In SMA mouse models, myostatin inhibition increased muscle mass and bone trabeculations [105]. TOPAZ (NCT03921528) is an open-label, phase II study to assess the safety, tolerability and efficacy of apitegromab, a myostatin monoclonal blocking antibody that is administered monthly via intravenous infusion. In the non-ambulatory nusinersen-treated group who received apitegromab 20 mg/kg for 12 months, the mean increase from baseline in HFMSE was + 7.1 points in the < 5 years age group (mean age 3.8 years) and + 0.6 points in the ≥ 5 years (mean age 11.7 years) age group, analysed by intention to treat [106]. Seven of 35 non-ambulatory patients treated with apitegromab and nusinersen gained WHO Motor Development Milestones, including three patients in the ≥ 5 years age group. SAPPHIRE (NCT05156320) is a phase III, multicenter, double-blind, placebo-control apitegromab study targeting non-ambulatory SMA type 2 and 3 patients treated with risdiplam or nusinersen that is currently underway.
5.1.2 Reldesemtiv
Reldesemtiv is a second-generation, fast skeletal muscle troponin activator that increases force production at submaximal stimulation. In a phase II, placebo-controlled, dose-escalation reldesemtiv study of SMA patients types 2 to 4 and over age 12 years not on SMN-targeted therapy, the higher reldesemtiv dose of 450 mg twice daily resulted in a 35.6-m (p = 0.004) increase from baseline in distance walked (6MWT) at 4 weeks and a 24.9-m increase (p = 0.58) at 8 weeks [107]. Mean expiratory pressure at 150-mg and 450-mg doses also showed a significant increase from baseline with reldesemtiv. Both dose levels of reldesemtiv were well tolerated.
5.2 Histone Deacetylase Inhibitors (HDAi)
Some of the earliest SMA clinical trials focused on histone deacetylase inhibitors (HDAi), which were found to increase the expression of the SMN2 gene and ameliorate SMN2 splicing defects in SMA patient fibroblasts, purportedly by up-regulating positive regulators of exon 7 splicing [108]. Subsequently, several clinical trials were conducted involving different HDAis, including valproic acid in CARNI-VAL I [109] and CARNIVAL II [110], phenylbutyrate [111] and hydroxyurea [112], all of which failed to find any significant effects on SMA outcome.
A novel HDACi (LBH589), reported to have greater (10 ×) potency than valproic acid on SMN protein upregulation [113], was studied in SMA cells in combination with an mRNA splicing enhancer antisense oligonucleotide and found to have a possible synergistic response on SMN protein levels that may potentiate effects [114]. Further SMA preclinical studies are needed to assess whether improvement in motor outcome with such combination therapy exceeds that of isolated ASO treatment.
5.3 Neuromuscular Junction (NMJ)-Targeted Therapies
Fatigue is an important finding in SMA [115], which has been attributed to impairments in maturation and degeneration of NMJ structure identified in animal models [116] and in SMA1 neuropathology specimens [27]. Electrophysiological studies in SMA patients report an electrodecremental response to repetitive nerve stimulation [117] and electrodecremental responses that correlated with fatigue on 6MWT [118] in some studies, but not others [115, 119]. SMA-controlled clinical trials to evaluate the efficacy of agents that increase NMJ transmission are few. The SPACE study (NCT02941328) is a phase II pyridostigmine/placebo control crossover trial in SMA types 2–4 [120] that is underway. The other clinical trial (NCT 01645787) tested 4-aminopyridine (4-AP, dalfampridine, Ampyra™), a K+ voltage gated inhibitor that enhances synaptic transmission centrally, as well as at the NMJ. Ambulatory adult SMA patients were assessed with 4-AP in a two-part, randomized, placebo-controlled, crossover pilot study composed of a short-term (2-week) and a long-term (6-week) trial. No effect on distance walked or percent fatigue (difference in distance walked between 1 vs 6 min) on 6MWT was noted with 4-AP in either the short-term or long-term trial [119].
5.4 Salbutamol (Albuterol)
Finally, in a class of its own, β-agonist salbutamol (albuterol), has been found to increase SMN protein in leukocytes [121] and fibroblasts [122] from SMA patients, perhaps by increasing inclusion of exon 7 in SMN2 transcripts, although the precise mechanism is not known. Uncontrolled pilot studies with salbutamol have suggested a beneficial effect of treatment on functional motor function in SMA type 2 patients. Findings were evident in children under age 5 years [123, 124], but not in children over age 5 years [124], raising questions as to whether the age of the younger cohort could have influenced the treatment response. However, findings from a placebo-controlled clinical trial in adult SMA type 3 patients that documented increases in SMN2 transcripts and fSMN protein, as well as improvements in 6MWT and, in a subset of responders, improvements in motor function [125], suggests it is SMA disease severity rather than patient age that influences treatment response.
6 Conclusion
The future of treatment in SMA looks bright with the availability of three approved effective therapies and more under study, as well as with the dissemination of SMA newborn screening. Decisions regarding treatment are based on patient’s age, preference, method of delivery and side effects, given that no difference in efficacy between treatments has been established. Regardless of SMA type and treatment agent, the single most important factor predicting response is early treatment, before motor neuron loss is unrecoverable. Novel therapies such as OAV raise questions as to the long-term durability of effects and of possible late unforeseen side effects that may take decades to properly assess. In infant-onset SMA, the benefits of early treatment clearly justify any potential risks from OAV treatment. Newer oral medication risdiplam offers ease of administration and has shown benefit in adults with more advanced SMA. The main concern raised with risdiplam treatment is with regards to human reproduction: in males, vis a vis the theoretical risk that effects on testicular germ cells would not fully reverse and could affect fertility; in women, it is not risk to fertility but to pregnancy that is of concern, given risdiplam’s embryofetal effects.
Combination therapy in symptomatic SMA offers the opportunity of improving upon the benefit afforded by any single SMN-targeted therapy. Discerning the effect of the agent under study from both the effect of young age (or shorter duration of symptoms) and the response to the background SMN-targeted treatment may prove difficult; to properly do so will require placebo-controlled studies, matched not only for age but also for duration of the background SMN-targeted treatment. Finally, the best outcome is seen with presymptomatic treatment enabled by dissemination of newborn screening programs. With three or more SMN2 copies, normal development of motor milestones is expected; while with two SMN2 copies, regardless of treatment chosen, more than half of infants treated will exhibit delays in motor development while continuing to gain milestones. No doubt approved SMA therapies have altered the natural history of SMA for the better and presymptomatic treatment has transformed two SMN2 copy SMA (expected type 1) from a life-threatening disorder to a disorder of motor development. Only time will tell if the enormous benefits of presymptomatic SMA treatment are sustained through adolescence and adulthood.
References
Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–65.
Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16(3):265–9.
Sugarman EA, Nagan N, Zhu H, Akmaev VR, Zhou Z, Rohlfs EM, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. 2012;20(1):27–32.
Vill K, Kölbel H, Schwartz O, Blaschek A, Olgemöller B, Harms E, et al. One year of newborn screening for SMA—results of a German Pilot Project. J Neuromuscul Dis. 2019;6(4):503–15.
Kay DM, Stevens CF, Parker A, Saavedra-Matiz CA, Sack V, Chung WK, et al. Implementation of population-based newborn screening reveals low incidence of spinal muscular atrophy. Genet Med. 2020;22(8):1296–302.
Calucho M, Bernal S, Alías L, March F, Venceslá A, Rodríguez-Álvarez FJ, et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord. 2018;28(3):208–15.
Kolb SJ, Coffey CS, Yankey JW, Krosschell K, Arnold WD, Rutkove SB, et al. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol. 2017;82(6):883–91.
Finkel RS, McDermott MP, Kaufmann P, Darras BT, Chung WK, Sproule DM, et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014;83(9):810–7.
Glanzman AM, McDermott MP, Montes J, Martens WB, Flickinger J, Riley S, et al. Validation of the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND). Pediatr Phys Ther. 2011;23(4):322–6.
Bayley N. Bayley Scales of Infant Development. 3rd ed. Coushatta: Pearson Assessments; 2005.
Haataja L, Mercuri E, Regev R, Cowan F, Rutherford M, Dubowitz V, et al. Optimality score for the neurologic examination of the infant at 12 and 18 months of age. J Pediatr. 1999;135(2 Pt 1):153–61.
WHO. WHO Motor Development Study: windows of achievement for six gross motor development milestones. Acta Paediatr Suppl. 2006;450:86–95.
Kaufmann P, McDermott MP, Darras BT, Finkel R, Kang P, Oskoui M, et al. Observational study of spinal muscular atrophy type 2 and 3: functional outcomes over 1 year. Arch Neurol. 2011;68(6):779–86.
Mercuri E, Lucibello S, Pera MC, Carnicella S, Coratti G, de Sanctis R, et al. Long-term progression in type II spinal muscular atrophy: a retrospective observational study. Neurology. 2019;93(13):e1241–7.
O’Hagen JM, Glanzman AM, McDermott MP, Ryan PA, Flickinger J, Quigley J, et al. An expanded version of the Hammersmith Functional Motor Scale for SMA II and III patients. Neuromuscul Disord. 2007;17(9–10):693–7.
Vuillerot C, Payan C, Girardot F, Fermanian J, Iwaz J, Berard C, et al. Responsiveness of the motor function measure in neuromuscular diseases. Arch Phys Med Rehabil. 2012;93(12):2251-2256 e1.
Mazzone ES, Mayhew A, Montes J, Ramsey D, Fanelli L, Young SD, et al. Revised upper limb module for spinal muscular atrophy: development of a new module. Muscle Nerve. 2017;55(6):869–74.
Zerres K, Rudnik-Schoneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-Petrusewicz I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci. 1997;146(1):67–72.
Lin CW, Kalb SJ, Yeh WS. Delay in diagnosis of spinal muscular atrophy: a systematic literature review. Pediatr Neurol. 2015;53(4):293–300.
Montes J, McDermott MP, Martens WB, Dunaway S, Glanzman AM, Riley S, et al. Six-Minute Walk Test demonstrates motor fatigue in spinal muscular atrophy. Neurology. 2010;74(10):833–8.
Wirth B, Herz M, Wetter A, Moskau S, Hahnen E, Rudnik-Schoneborn S, et al. Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet. 1999;64(5):1340–56.
Burghes AH, Beattie CE, Burghes AHM, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10(8):597–609.
Pellizzoni L, Yong J, Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298(5599):1775–9.
Gabanella F, Carissimi C, Usiello A, Pellizzoni L. The activity of the spinal muscular atrophy protein is regulated during development and cellular differentiation. Hum Mol Genet. 2005;14(23):3629–42.
Ramos DM, d’Ydewalle C, Gabbeta V, Dakka A, Klein SK, Norris DA, et al. Age-dependent SMN expression in disease-relevant tissue and implications for SMA treatment. J Clin Investig. 2019;129(11):4817–31.
Le TT, McGovern VL, Alwine IE, Wang X, Massoni-Laporte A, Rich MM, et al. Temporal requirement for high SMN expression in SMA mice. Hum Mol Genet. 2011;20(18):3578–91.
Harding BN, Kariya S, Monani UR, Chung WK, Benton M, Yum SW, et al. Spectrum of neuropathophysiology in spinal muscular atrophy type I. J Neuropathol Exp Neurol. 2015;74(1):15–24.
McGovern VLIC, Arnold WD, Gombash SE, Zaworski PG, Blatnik AJ 3rd, et al. SMN expression is required in motor neurons to rescue electrophysiological deficits in the SMNΔ7 mouse model of SMA. Hum Mol Genet. 2015;24(19):27.
Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, et al. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011;69(3):453–67.
Imlach WL, Beck ES, Choi BJ, Lotti F, Pellizzoni L, McCabe BD. SMN is required for sensory-motor circuit function in Drosophila. Cell. 2012;151(2):427–39.
Singh RN, Howell MD, Ottesen EW, Singh NN. Diverse role of survival motor neuron protein. Biochim Biophys Acta. 2017;1860(3):299–315.
Rudnik-Schöneborn S, Heller R, Berg C, Betzler C, Grimm T, Eggermann T, et al. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J Med Genet. 2008;45(10):635–8.
Rudnik-Schöneborn S, Vogelgesang S, Armbrust S, Graul-Neumann L, Fusch C, Zerres K. Digital necroses and vascular thrombosis in severe spinal muscular atrophy. Muscle Nerve. 2010;42(1):144–7.
Araujo A, Araujo M, Swoboda KJ. Vascular perfusion abnormalities in infants with spinal muscular atrophy. J Pediatr. 2009;155(2):292–4.
Tein I, Sloane AE, Donner EJ, Lehotay DC, Millington DS, Kelley RI. Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy: primary or secondary defect(s)? Pediatr Neurol. 1995;12(1):21–30.
Crawford TO, Sladky JT, Hurko O, Besner-Johnston A, Kelley RI. Abnormal fatty acid metabolism in childhood spinal muscular atrophy. Ann Neurol. 1999;45(3):337–43.
Hachiya Y, Arai H, Hayashi M, Kumada S, Furushima W, Ohtsuka E, et al. Autonomic dysfunction in cases of spinal muscular atrophy type 1 with long survival. Brain Dev. 2005;27(8):574–8.
Mercuri E, Finkel RS, Muntoni F, Wirth B, Montes J, Main M, et al. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28(2):103–15.
Shababi M, Habibi J, Yang HT, Vale SM, Sewell WA, Lorson CL. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Mol Genet. 2010;19(20):4059–71.
Gombash SE, Cowley CJ, Fitzgerald JA, Iyer CC, Fried D, McGovern VL, et al. SMN deficiency disrupts gastrointestinal and enteric nervous system function in mice. Hum Mol Genet. 2015;24(19):5665.
Bowerman M, Swoboda KJ, Michalski JP, Wang GS, Reeks C, Beauvais A, et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann Neurol. 2012;72(2):256–68.
Mutsaers CA, Wishart TM, Lamont DJ, Riessland M, Schreml J, Comley LH, et al. Reversible molecular pathology of skeletal muscle in spinal muscular atrophy. Hum Mol Genet. 2011;20(22):4334–44.
Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96(11):6307–11.
Mailman MD, Heinz JW, Papp AC, et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med. 2002;4(1):20–6.
Crawford TO, Paushkin SV, Kobayashi DT, et al. Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS One. 2012;7(4): e33572.
Prior TW, Krainer AR, Hua Y, Swoboda KJ, Snyder PC, Bridgeman SJ, et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. 2009;85(3):408–13.
Vezain M, Saugier-Veber P, Goina E, Touraine R, Manel V, Toutain A, et al. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum Mutat. 2010;31(1):E1110–25.
Wu X, Wang SH, Sun J, Krainer AR, Hua Y, Prior TW. A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Hum Mol Genet. 2017;26(14):2768–80.
Ruhno C, McGovern VL, Avenarius MR, Snyder PJ, Prior TW, Nery FC, et al. Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Hum Genet. 2019;138(3):241–56.
FDA. Spinraza (nusinersen) product label: Reference ID: 4625921—December 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/209531lbl.pdf (Accessed 15 Feb 2022)
EMA. Summary of opinion (initial authorization) Spinraza nusinersen 2017 April 21. https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-spinraza_en.pdf.
FDA. BLA Clinical Review Memorandum (ZOLGENSMA, onasemnogene abeparvovec xioi); 2019 May 23. https://www.fda.gov/vaccines-blood-biologics/zolgensma (Accessed 15 Feb 2022).
EMA. Zolgensma (onasemnogene abeparvovec) [updated 6/12/2021]; 2020 March 26. https://www.ema.europa.eu/en/medicines/human/EPAR/zolgensma (Accessed Oct 2020).
FDA. Center for Drug Evaluation and Research. Application number: 213535Orig1s00. Clinical Reviews. EVRYSDI (risdiplam). 2020 June 30. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213535Orig1s000MedR.pdf (Accessed 15 Feb 2022).
EMA. Orphan Maintenance Assessment report-Evrysdi (risdiplam). 2021 March 26. https://www.ema.europa.eu/en/documents/orphan-maintenance-report/evrysdi-orphan-maintenance-assessment-report-initial-authorisation_en.pdf (Accessed 20 June 2022).
Genentech. FDA approves Genentech’s Evrysdi (risdiplam) for use in babies under two months with spinal muscular atrophy (SMA); 2022 May 30. https://www.gene.com/media/press-releases/14955/2022-05-30/fda-approves-genentechs-evrysdi-risdiplam (Accessed 6 July 2022).
Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26(4):1333–46.
Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–48.
Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;2(3):11.
Finkel RSME, Darras B, Connolly A, Kunz N, Kirschner J, et al. For the ENDEAR Study Group efficacy and safety of nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377:1723–32.
FDA. Center for Drug Evaluation and Research. Application number: 209531Orig1s000. Medical Reviews. Spinraza (nusinersen); December 15, 2016. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/209531Orig1s000MedR.pdf (Accessed March 2017).
Finkel RS, Chiriboga CA, Vajsar J, Day J, Montes J, De Vivo DC, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388:10063.
Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: final report of a phase 2, open-label, multicentre, dose-escalation study. Lancet Child Adolesc Health. 2021;5(7):491–500.
Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378(7):625–35.
Chiriboga CA, Darras BT, Farrar MA, Mercuri E, Kirschner J, Kuntz NL, et al. Interim report on the safety and efficacy of longer-term treatment with nusinersen in later-onset spinal muscular atrophy (SMA): results from the SHINE study. 48th Child Neurology Society Meeting 2019 October 23–29, Charlotte.
Chiriboga CA, Swoboda KJ, Darras BT, Iannaccone ST, Montes J, De Vivo DC, et al. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology. 2016;86(10):890–7.
Darras BT, Chiriboga CA, Iannaccone ST, Swoboda KJ, Montes J, Mignon L, et al. Nusinersen in later-onset spinal muscular atrophy: long-term results from the phase 1/2 studies. Neurology. 2019;92(21):e2492–506.
De Vivo DC, Bertini E, Swoboda KJ, Hwu WL, Crawford TO, Finkel RS, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord. 2019;29(11):842–56.
Sansone V SK, DeVivo DC, Bertini E, Hwu WL, Makepeace C, et al. Preserved swallowing function in Infants who Initiated nusinersen treatment in the presyptomatic stage of SMA: results from the NURTURE study World Muscle Society 26th International Congress; Virtual 2021 Sep 20–24.
Hagenacker T, Wurster CD, Günther R, Schreiber-Katz O, Osmanovic A, Petri S, et al. Nusinersen in adults with 5q spinal muscular atrophy: a non-interventional, multicentre, observational cohort study. Lancet Neurol. 2020;19(4):317–25.
Maggi L, Bello L, Bonanno S, Govoni A, Caponnetto C, Passamano L, et al. Nusinersen safety and effects on motor function in adult spinal muscular atrophy type 2 and 3. J Neurol Neurosurg Psychiatry. 2020;91(11):1166–74.
Elsheikh B, Severyn S, Zhao S, Kline D, Linsenmayer M, Kelly K, et al. Safety, tolerability, and effect of nusinersen treatment in ambulatory adults with 5q-SMA. Front Neurol. 2021;12: 650535.
Walter MC, Wenninger S, Thiele S, Stauber J, Hiebeler M, Greckl E, et al. Safety and treatment effects of nusinersen in longstanding adult 5q-SMA type 3—a prospective observational study. J Neuromuscul Dis. 2019;6(4):453–65.
Biogen. Spinraza (nusinersen): reports of communicating hydrocephalus not related to meningitis or bleeding; 2018 July 31. https://assets.publishing.service.gov.uk/media/5b7edf83ed915d14f4404bf6/Spinraza_UK_DHPC_SPZ_GBR_0020.pdf (Accessed 15 Feb 2022).
Viscidi E, Wang N, Juneja M, Bhan I, Prada C, James D, et al. The incidence of hydrocephalus among patients with and without spinal muscular atrophy (SMA): results from a US electronic health records study. Orphanet J Rare Dis. 2021;16(1):207.
Moshe-Lilie O, Riccelli LP, Karam C. Possible recurrent aseptic meningitis associated with nusinersen therapy. Muscle Nerve. 2020;62(5):E79–80.
Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16(6):1073–80.
Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010;28(3):271–4.
Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27(1):59–65.
Duque SI, Arnold WD, Odermatt P, Li X, Porensky PN, Schmelzer L, et al. A large animal model of spinal muscular atrophy and correction of phenotype. Ann Neurol. 2015;77(3):399–414.
Thomsen G, Burghes AHM, Hsieh C, Do J, Chu BTT, Perry S, et al. Biodistribution of onasemnogene abeparvovec DNA, mRNA and SMN protein in human tissue. Nat Med. 2021;27(10):1701–11.
Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713–22.
Lowes LP, Alfano LN, Arnold WD, Shell R, Prior TW, McColly M, et al. Impact of age and motor function in a phase 1/2A study of infants with SMA type 1 receiving single-dose gene replacement therapy. Pediatr Neurol. 2019;98:39–45.
Day JW, Finkel RS, Chiriboga CA, Connolly AM, Crawford TO, Darras BT, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021;20(4):284–93.
Mercuri E, Muntoni F, Baranello G, Masson R, Boespflug-Tanguy O, Bruno C, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (STR1VE-EU): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021;20(10):832–41.
Strauss KA FM, Swoboda KJ, Saito K, Chiriboga CA, Finkel RS, et al. Onasemnogene abeparvovec in presymptomatic spinal muscular atrophy: SPR1NT study update as of 31 Dec 2019. Muscular Dystrophy Association (MDA) Clinical and Scientific Meeting Virtual; 2020 March 21–24.
Strauss KA MF, Farra MA, Saito K, Mendell J, Servais L, et al. Abeparvovec in presymptomatic spinal muscular atrophy: SPR1NT study update in children with 2 copies of SMN2. American Academy of Neurology 73rd Annual Meeting; 2021 April 17.
Chand D, Mohr F, McMillan H, Tukov FF, Montgomery K, Kleyn A, et al. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J Hepatol. 2021;74(3):560–6.
Chand DH, Zaidman C, Arya K, Millner R, Farrar MA, Mackie FE, et al. Thrombotic microangiopathy following onasemnogene abeparvovec for spinal muscular atrophy: a case series. J Pediatr. 2021;231:265–8.
Novartis. Novartis announces lift of partial clinical trial hold and plans to initiate a new, pivotal Phase 3 study of intrathecal OAV-101 in older patients with SMA. News release. August 3, 2021. https://www.novartis.com/news/media-releases/novartis-announces-lift-partial-clinical-trial-hold-and-plans-initiate-new-pivotal-phase-3-study-intrathecal-oav-101-older-patients-sma (Accessed 15 Feb 2022).
Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang X, et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat Chem Biol. 2015;11(7):511–7.
Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345(6197):688–93.
Ratni H, Ebeling M, Baird J, Bendels S, Bylund J, Chen KS, et al. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem. 2018;61(15):6501–17.
Sergott RC, Amorelli GM, Baranello G, Barreau E, Beres S, Kane S, et al. Risdiplam treatment has not led to retinal toxicity in patients with spinal muscular atrophy. Ann Clin Transl Neurol. 2021;8(1):54–65.
Poirier A, Weetall M, Heinig K, Bucheli F, Schoenlein K, Alsenz J, et al. Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol Res Perspect. 2018;6(6): e00447.
Baranello G, Darras BT, Day JW, Deconinck N, Klein A, Masson R, et al. Risdiplam in type 1 spinal muscular atrophy. N Engl J Med. 2021;384(10):915–23.
Mercuri E, Deconinck N, Mazzone ES, Nascimento A, Oskoui M, Saito K, et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2022;21(1):42–52.
Chiriboga CA, Bruno CDT, Fischer D, Kirschner J, Mercuri E, et al. JEWELFISH: safety, pharmacodynamic and exploratory efficacy data in non-naïve patients with SMA receiving treatment with risdiplam. MDA Clinical & Scientific Conference, Nashville, 13–16 March 2022.
Servais L, Al-Muhaizea M, Farrar M, Nelson L, Prufer A, Finkel R, et al. RAINBOWFISH: a study of risdiplam in infants with presymptomatic spinal muscular atrophy (SMA). Neuromuscul Disord J. 2021;31:S48.
Svetlana J CD, Dobrzycka-Ambrozewicz A, Kotulscka-Jozwiak K, Lvova O, Pervinina Y, et al. Branaplam in type 1 spinal muscular atrophy: second and third parts of a phase I/II study 26th World Muscle Society Virtual Annual Congress, 20–24 September 2021.
Sumner CJ, Crawford TO. Two breakthrough gene-targeted treatments for spinal muscular atrophy: challenges remain. J Clin Investig. 2018;128(8):3219–27.
Osmanovic A, Schreiber-Katz O, Petri S. Nusinersen wearing-off in adult 5q-spinal muscular atrophy patients. Brain Sci. 2021;11(3):367.
Mercuri EFR, Day JW, Pascual Pascual PI, Ryan MM, DeVivo DC, et al. editors. Part A result from ongoing DEVOTE study to explore higher doses of nusinersen in SMA. World Muscle Society 26th International Virtual Congress, 20–24 Sep 2021.
Mendell JR, Al-Zaidy SA, Lehman KJ, McColly M, Lowes LP, Alfano LN, et al. Five-year extension results of the phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol. 2021;78(7):834–41.
Long KK, O’Shea KM, Khairallah RJ, Howell K, Paushkin S, Chen KS, et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum Mol Genet. 2019;28(7):1076–89.
Cure SMA. Scholar rock presents TOPAZ phase 2 data showing the transformative potential of apitegromab in patients with type 2 and 3 SMA; 2021 June 11. https://www.curesma.org/scholar-rock-topaz-data-for-apitegromab-researcher-meeting-2021/ (Accessed 15 Feb 2022).
Rudnicki SA, Andrews JA, Duong T, Cockroft BM, Malik FI, Meng L, et al. Reldesemtiv in patients with spinal muscular atrophy: a phase 2 hypothesis-generating study. Neurotherapeutics. 2021;18(2):1127–36.
Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, et al. Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003;12(19):2481–9.
Swoboda KJ, Scott CB, Crawford TO, Simard LR, Reyna SP, Krosschell KJ, et al. SMA CARNI-VAL trial part I: double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy. PLoS One. 2010;5(8): e12140.
Kissel JT, Scott CB, Reyna SP, Crawford TO, Simard LR, Krosschell KJ, et al. SMA CARNIVAL TRIAL PART II: a prospective, single-armed trial of l-carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PLoS One. 2011;6(7): e21296.
Mercuri E, Bertini E, Messina S, Solari A, D’Amico A, Angelozzi C, et al. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology. 2007;68(1):51–5.
Chen TH, Chang JG, Yang YH, Mai HH, Liang WC, Wu YC, et al. Randomized, double-blind, placebo-controlled trial of hydroxyurea in spinal muscular atrophy. Neurology. 2010;75(24):2190–7.
Garbes L, Riessland M, Hölker I, Heller R, Hauke J, Tränkle C, et al. LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate. Hum Mol Genet. 2009;18(19):3645–58.
Pagliarini V, Guerra M, Di Rosa V, Compagnucci C, Sette C. Combined treatment with the histone deacetylase inhibitor LBH589 and a splice-switch antisense oligonucleotide enhances SMN2 splicing and SMN expression in spinal muscular atrophy cells. J Neurochem. 2020;153(2):264–75.
Bartels B, de Groot JF, Habets LE, Wadman RI, Asselman FL, Nieuwenhuis EES, et al. Correlates of fatigability in patients with spinal muscular atrophy. Neurology. 2021;96(6):e845–52.
Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, et al. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17(16):2552–69.
Wadman RI, Vrancken AF, van den Berg LH, van der Pol WL. Dysfunction of the neuromuscular junction in spinal muscular atrophy types 2 and 3. Neurology. 2012;79(20):2050–5.
Pera MC, Luigetti M, Pane M, Coratti G, Forcina N, Fanelli L, et al. 6MWT can identify type 3 SMA patients with neuromuscular junction dysfunction. Neuromuscul Disord. 2017;27(10):879–82.
Chiriboga CA, Marra J, LaMarca NM, Young SD, Weimer LH, Levin B, et al. Lack of effect on ambulation of dalfampridine-ER (4-AP) treatment in adult SMA patients. Neuromuscul Disord. 2020;30(8):693–700.
Stam M, Wadman RI, Wijngaarde CA, Bartels B, Asselman FL, Otto LAM, et al. Protocol for a phase II, monocentre, double-blind, placebo-controlled, cross-over trial to assess efficacy of pyridostigmine in patients with spinal muscular atrophy types 2–4 (SPACE trial). BMJ Open. 2018;8(7): e019932.
Tiziano FD, Lomastro R, Pinto AM, Messina S, D’Amico A, Fiori S, et al. Salbutamol increases survival motor neuron (SMN) transcript levels in leucocytes of spinal muscular atrophy (SMA) patients: relevance for clinical trial design. J Med Genet. 2010;47(12):856–8.
Angelozzi C, Borgo F, Tiziano FD, Martella A, Neri G, Brahe C. Salbutamol increases SMN mRNA and protein levels in spinal muscular atrophy cells. J Med Genet. 2008;45(1):29–31.
Pane M, Staccioli S, Messina S, D’Amico A, Pelliccioni M, Mazzone ES, et al. Daily salbutamol in young patients with SMA type II. Neuromuscul Disord. 2008;18(7):536–40.
Frongia AL, Natera-de Benito D, Ortez C, Alarcón M, Borrás A, Medina J, et al. Salbutamol tolerability and efficacy in patients with spinal muscular atrophy type II. Neuromuscul Disord. 2019;29(7):517–24.
Tiziano FD, Lomastro R, Abiusi E, Pasanisi MB, Di Pietro L, Fiori S, et al. Longitudinal evaluation of SMN levels as biomarker for spinal muscular atrophy: results of a phase IIb double-blind study of salbutamol. J Med Genet. 2019;56(5):293–300.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was provided for the writing of this manuscript.
Conflict of interest
Dr. Chiriboga receives grant funding from Avexis/Novartis, Biogen and Roche. She serves/served as consultant for Roche, Genentech, Avexis/Novartis and PTC and has served as Speaker in unlabeled educational talks for Biogen, Roche and Genentech.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
Dr. Chiriboga was the sole author of this article.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chiriboga, C.A. Pharmacotherapy for Spinal Muscular Atrophy in Babies and Children: A Review of Approved and Experimental Therapies. Pediatr Drugs 24, 585–602 (2022). https://doi.org/10.1007/s40272-022-00529-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-022-00529-8