
Opinion statement
Spinal muscular atrophy is caused by mutations in the survival motor neuron 1 (SMN1) gene, leading to the reduction of SMN protein. The loss of alpha motor neurons in the ventral horn of the spinal cord results in progressive paralysis and premature death. There is no current treatment other than symptomatic and supportive care, although over the past decade, there has been an outstanding advancement in understanding the genetics and molecular mechanisms underlying the physiopathology of SMA. The most promising approach, from current trials, is the use of antisense oligonucleotide (ASOs) to redirect SMN2 translation and increase exon 7 inclusion in the majority of the RNA transcript, to increase the production of fully functional SMN protein. Recently, ISIS Pharmaceuticals Inc. (2855 Gazelle Court, Carlsbad CA 92010) reported an interim analysis from a multiple dose study in children with SMA between 2 and 14 years of age, using ASO therapy. The results indicated good tolerability at all dose levels, increases in muscle function in children treated with multiple doses of ISIS-SMNRx, and increase in SMN protein levels in cerebrospinal fluid (CSF) from both single and multiple dose studies. Studies in infants are ongoing in a few centers; soon other institutions may begin enrollment. Infants are fragile and their disease process may differ from the older SMA population. It is not known whether effective drug would best be given to SMA infants or older children. Other promising therapies are still in preclinical phases or early clinical phases. Gene therapy appears to be efficient in improving survival in a severe mouse model of SMA, though a better definition of the route of administration and of the safety profile of the viral vectors is needed before clinical administration is possible.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive disease, characterized by progressive muscle weakness resulting from degeneration and loss of the anterior horn cells in the spinal cord and the brain stem nuclei. SMA was described in 1891 by Guido Werdnig, a physician from the University of Vienna, who reported a case of muscular dystrophy with positive spinal cord findings “neurogenic dystrophy” [1]. Shortly thereafter, Professor Johann Hoffmann from Heidelberg University presented two siblings with progressive atrophy, weakness, and death during early childhood [2]. Autopsies revealed severe atrophy of the ventral roots of the spinal cord and histologic evidence of loss of motor neurons in the anterior horn cells of this region. Hoffmann called the syndrome “spinale muskelatrophie (spinal muscular atrophy).” Byers and Banker in 1960 [3] classified SMA into categories based on the severity and age of onset of the symptoms, in an effort to predict prognosis.
SMA is the second most frequent and fatal autosomal recessive disease of childhood after cystic fibrosis. The estimated incidence is one in 6000 to 10,000 live births, and the carrier frequency of SMN1 mutations in general population of 2 % to 3 %[4]. Zaldivar et al. [5] found that the incidence of SMA type I in Cuba was 3.53 per 100,000 live births. When the population was classified according to self-reported ethnicity, the incidence was eight per 100,000 for whites, 0.89 per 100,000 for blacks, and 0.96 per 100,000 for those of mixed ethnicity. Zaldivar et al. (2005) concluded that SMA I may occur less frequently in those of African ancestry [5].
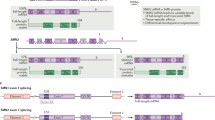
This disorder is caused by a mutation in 5q11.2–q13.3, affecting the survival of motor neuron (SMN) gene with a consequential absence of SMN1 exon 7. The severity of SMA is determined in part by the number of copies of the disease-modifying gene (SMN2 gene). More copies of SMN2 are associated with less severe forms of the disease. SMN2 has a C-to-T transition in an exonic splicing enhancer site of exon 7. The base substitution at this position is translationally silent, but it impairs the ability of the spliceosome to recognize exon 7; subsequently, this exon is excluded in 90 % of SMN2 mRNA transcripts. The aberrant messenger RNA (mRNA) transcripts lacking exon 7 encode a truncated protein (SMNΔ7). Despite this, the spliceosome is still capable of generating a small percentage of SMN2 mRNAs that incorporate exon 7 and, therefore, a small amount of full length SMN protein is generated.
SMA is traditionally classified into five subtypes according to the age of onset and highest motor function achieved (Table 1). Type 0 corresponds to very severe forms of antenatal-onset SMA. Type 1 (Werdnig-Hoffmann disease) is the most common type (50 % of patients). The onset of clinical signs is before 6 months of age, and patients never acquire the ability to sit unsupported. Without intervention, patients generally do not survive past the first 2 years of life. Type 2 (Dubowitz disease) is characterized by onset between 7 and 18 months of age. Patients achieve the ability to sit unsupported when placed in a sitting position. Some patients are able to acquire standing position, but they do not acquire the ability to walk independently. Type 3 (Kugelberg–Welander disease) manifests after 18 months of age. Patients are able to walk without support at some time, although many of them lose this ability during adolescent. Type 4 is the adult or late onset form that typically manifests after the second or third decade of life.
Symptoms vary greatly according to the type. In general, there is evidence of generalized profound hypotonia, progressive muscle weakness with sparing of the facial muscles, absent or diminished deep tendon reflexes, fine tremors, and contractures. The involvement of bulbar motor neurons often gives tongue fasciculations, and poor suck and swallow with increasing swallowing and feeding difficulty over time. Respiratory function is progressively affected.
As with any hypotonic patient, the first screening testing is creatine kinase level; it is usually normal or minimally elevated in patients with SMA. If the clinical presentation strongly suggests SMA, SMN gene analysis is indicated. There is no current treatment for SMA. Management should be multidisciplinary and can be divided by systems including pulmonary, gastrointestinal, nutritional, orthopedic care, and rehabilitation (Table 2).
There are several ongoing efforts searching various potential therapeutic agents: neuroprotective drugs to rescue motor neurons, small-molecule drugs that modulate endogenous SMN2, SMN1 gene replacement using viral vector or by stem cell transplantation, antisense oligonucleotide to promote exon 7 inclusion for full length SMN protein.
Treatment
Clinical management
Pulmonary
Pulmonary complaints are the leading cause of morbidity and mortality in patients with SMA type 1, 2, and sometimes 3. Progressive respiratory complications are mainly due to chest muscle weakness, abnormal chest wall shape, and underdeveloped lungs. Poor cough causes difficulties clearing secretions and recurrent infections. Patients also develop sleep disordered breathing and nocturnal hypoventilation.
Patients should be referred to a pediatric pulmonology specialist, the options for respiratory care must be discussed with the family, and they should be educated about the prevention and management of chronic restrictive lung disease. As proposed by the consensus statement for standard care in SMA [6], management can be targeted differently for nonsitter, sitter or walker.
Nonsitters
Most nonsitters are too young for formal pulmonary function testing; therefore, other means of assessment should be used. First, the physical examination including respiratory rate, work of breathing, presence of paradoxical breathing, chest wall shape, and skin color should be evaluated. Assessment of cough effectiveness by observation and monitoring of gas exchange by pulse oximetry and End-tidal carbon dioxide are important components of the evaluation.
Overnight pulse oximetry with continuous CO2 monitoring can be used to screen for nocturnal hypoxemia and polysomnography to identify the presence of sleep-disordered breathing or to titrate noninvasive ventilatory support.
Other screening tests include a baseline chest x-ray and if risk of dysphagia and aspiration, video swallow study is helpful.
Nonsitters present with early and rapidly progressive respiratory insufficiency. Options should be discussed with the family, including not using any ventilatory support (palliative care), non-invasive ventilation in the form of bi-level positive airway pressure ventilation and invasive ventilation (tracheostomy and ventilatory support).
Because of the severity of the disease, patients who are started on part time NIV may progress very quickly to needing 24-hour support. With full time NIV, the airway can get damaged, frequent secretion clearance is required, and recurrent infections are very common. Patients usually are admitted to the pediatric critical care unit needing intubation. At that moment, some families decide to withdraw care or transition to invasive ventilation in the form of tracheostomy and ventilator. Most patients are completely paralyzed by that time, some with minimal finger and facial movements, requiring very frequent secretion clearance. Many of them have repeated episodes of desaturation and hypoxemia, causing hypoxic brain injury.
Beside routine immunization, other recommended vaccines include respiratory syncytial virus (RSV) and Flu vaccine. There should not be any exposure to tobacco use at home or smoke exposure.
Sitters
The recommendations for this group of patients are very similar to the ones described above. Children older than 5 or 6 years of age are able to perform pulmonary function test (PFTs) once a year, which gives an objective evaluation of the respiratory status.
Patients with vital capacity <65 % predicted or with signs of nocturnal hypoventilation should have overnight pulse oximetry. Nocturnal hypoventilation should be treated with NIV. Airway clearance with chest physio-therapy and cough assist is important to prevent infections and to reduce the duration of respiratory sickness. Furthermore, sitters should be evaluated for presence and severity of scoliosis. Beside routine immunization, other recommended vaccines include Pneumovax and influenza.
Walkers
Walkers have a delayed presentation of respiratory symptoms or never develop them. Nevertheless, the same assessment described above should be performed, including overnight pulse oximetry, PFTs, chest x-ray and polysomnography. PFTs may remain normal through adulthood.
Gastrointestinal
The main gastrointestinal problems identified by the International Standard of Care Committee for SMA [6] were feeding and swallowing problems, gastrointestinal dysmotility, growth and under nutrition/over nutrition problems. Growth and under nutrition/over nutrition problems will be discussed under nutrition and diet.
Nonsitters
In this group of patients, the first sign of disease progression is increased feeding time, oral pooling, cough, fatigue, aspiration, and recurrent infections. Initially, weight gain is appropriate but around 3 to 4 months of age, a plateau in weight gain is evident. If an appropriate intervention is not initiated, weight loss occurs.
There is no consensus on patients who are recently diagnosed and asymptomatic; but if the patient has inadequate oral intake, a proactive approach is recommended. Feedings can be initially administered by nasogastric or nasojejunal tube before the patient is taken for gastrostomy tube (GT) placement. GT feeding is the ideal method of feeding when inadequate caloric intake or unsafe oral feeding is of concern. Some families will decide against GT placement. In these cases, the morbidities associated with long-term nasogastric (NG) and nasojejunal (NJ) tube should be discussed. These include local skin irritation because of the tape, increased secretions, and nasopharyngeal discomfort.
Nissen fundoplication for type 1 patients at the time of gastrostomy placement is strongly recommended in cases of severe gastro-esophageal reflux (GERD). Nissen can be recommended in patients without GERD given that a high percentage will developed reflux at some point. A routine upper gastrointestinal series may be recommended for presurgical evaluation for gastrostomy tube placement to rule out anatomic anomalies and to document GERD.
Sitters
The main symptoms of feeding problems include prolonged mealtime, fatigue and coughing during or after swallowing. Other difficulties in this group of patients can be sub-divided according to the specific feeding phase [3–5].
Difficulties in the pre-oral phase include poor head control, limited mouth opening, and difficulties conveying food to the mouth. In the oral phase, difficulties chewing, oral hypersensitivity, tongue movement abnormalities, fasciculations and atrophy of the tongue, reduced range of mandibular motion limiting mouth opening, and increased fatigue of masticatory muscles. The pharyngeal phase is usually affected because of poor coordination of swallowing with airway closure, which can lead to penetration and aspirations.
In a multidisciplinary clinic setting, the speech therapist should perform a baseline evaluation, and a video swallow study should be obtained when indicated. Most of the time, mild to moderate dysphagia is managed with dietary modifications. A semisolid diet can be used to compensate for poor chewing and reduce length of mealtimes. Thickened liquids may protect against aspiration of thin fluids [8, 9]. Positioning and seating may improve self-feeding ability as well as swallow safety and efficiency.
Gastro-esophageal reflux can be managed with acid neutralizers and/or inhibitors of acid secretion. Most commonly, patients are started on histamine blockers such as ranitidine and famotidine or proton pump inhibitors like esomeprazole or omeprazole. Proton pump inhibitors have been associated with an increased risk of vitamin and mineral deficiencies impacting vitamin B12, vitamin C, calcium, iron, and magnesium metabolism. While these risks are considered to be relatively low in the general population, they may be notable in malnourished patients [10].
When delayed gastric emptying or diminished motility is present, prokinetic agents may be useful like metoclopramide or erythromycin.
Constipation can be another complication related to slow motility. Dietary modifications like increased water intake and fiber can be used initially. If these measurements do not correct the problem, stool softeners like Docusate or laxatives like Miralax, can be safely used.
Nutritional
Children with spinal muscular atrophy are at risk for growth failure or weight gain. Nonsitters tend to develop growth failure, whereas obesity is a problem of sitters. Walkers tend to be thin. To evaluate growth progression, the routine history, physical examination, and growth velocity (weight, height to length, weight to height) measures should be combined. Body mass index by itself should not be used as a measure of ideal weight. Children with SMA have decreased lean body mass but usually acceptable fat mass. Body mass index may plot patients as underweight due to decrease in lean body mass and underestimation of body fat. The goal is to maintain each child on his or her own growth velocity and meet the daily-recommended nutrient intake for age.
A dietician should make a baseline assessment of the nutritional intake and formulate a plan accordingly with the patient needs. Currently, there is no indication for increasing or decreasing specific nutrients (ie, protein, fat, carbs, vitamins, or minerals). Although some families report benefits with the use of elemental or semi-elemental formulas, including, satisfactory growth, decreased reflux, gas and decreased secretions. If an elemental formula is used, a dietitian should be involved to help ensure the child does not receive insufficient or excessive amounts of nutrients, to perform laboratory assessments as needed, and to monitor adequate growth [11–13].
Orthopedic care and rehabilitation
The main problems are related to progressive muscle weakness, limited mobility, contracture formation, spinal deformity, increased risk of pain, osteopenia, and fractures.
A multidisciplinary team that includes physical and occupational therapists should evaluate all patients with SMA. The main goals are to provide stretching exercise, adequate equipment to support function, and to prevent or ameliorate contractures without limiting function.
Nonsitters present with severe weakness and only limited upper extremity and head movements. Upper extremity orthotics like mobile arm supports or slings can increase active range of motion and functional abilities. Hand orthotics should be carefully used because attempts to correct postural deviations, such as ulnar deviation, with an orthotic may result in reduced function. Lower extremity orthotics includes supra malleolar orthotics, ankle foot orthotics, knee orthotic and hip stabilizers. Medical strollers and custom manual wheelchair are important to provide good support of the neck and trunk, as well to allow tilting.
Sitters suffer from progressive weakness, contractures, and scoliosis. Contractures can be managed with a regular stretching and bracing program to preserve flexibility. Serial casting can be used to improve standing and tolerance of bracing. The use of ankle foot orthotics is recommended in some cases, for standing (independently or with a standing frame) or assisted ambulation. Upper extremity orthotics with mobile arm supports or slings increase functional capacity.
Regular exercise should be encouraged to maintain fitness and endurance and may include supervised swimming, aquatic therapy, horseback riding, and adaptive sports.
The degree and progression of scoliosis is evaluated through x-rays. Surgical correction of scoliosis should be considered based on the patient’s curve progression, pulmonary function, and bony maturity. Some evidence suggests that earlier surgery results in better outcome. Beneficial effects on pulmonary function remain controversial, but the rate of pulmonary decline may be slowed [14].
Equipment evaluation includes seating and mobility, positioning, and equipment for self-care. Evaluations for manual and power mobility may be conducted as early as 12 to 18 months of age.
For walkers, musculoskeletal deformities, scoliosis, and leg/foot pain are the most common problems reported. Patients may present with fractures or other musculoskeletal injuries like sprains. The goal for these patients is to provide appropriate mobility aids, adaptive equipment, assistive technology, and environmental access to maintain independence and mobility. Orthotics support functional walking. Wheelchair mobility for distance transportation helps with independence. Contracture management and education to maximize joint protection and school/home health evaluation by occupational therapy allow for safe accessibility and optimal independence.
Emerging therapies
Antisense oligonucleotides (ASO)
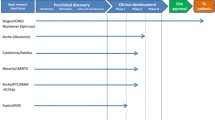
This therapy uses nucleotides that bind to the mRNA sequence of SMN2 and change the splicing so that exon 7 is included instead of repressed. This then allows for a full length and more functional SMN protein to be created [15•, 16••]. Evidence has shown success of this therapy in the mouse model of SMA causing increased survival and reduced tail necrosis in the mice [17]. Currently, ISIS Pharmaceuticals is conducting phase II clinical trials of intrathecal injections of their ASO, ISIS-SMNRx, at various doses and intervals.
Standard procedure
Patients with SMA types II and III are enrolled in the clinical trial and have the ASO injected into their intrathecal space, CSF, and plasma drug levels are checked at intervals after the injection, and outcome measures, including the Hammersmith Functional Motor Scale-Expanded and compound muscle action potential-motor unit number estimate), are followed. (ISIS Pharmaceuticals, http://clinicaltrials.gov/)
Contraindications
Patients are excluded if they are dependent on gastrostomy feeding, have respiratory failure, ventriculostomy shunt, or have had spinal surgery or significant scoliosis, which would interfere with the injections, or are taking experimental medications aimed to treat SMA [16••].
Complications
Possible complications after intrathecal injections include pain at the injection site and postlumbar puncture headache.
Special points
Intrathecal injection is necessary to bypass the blood brain barrier and to ensure the drug has maximal effect in the central nervous system.
Cost
Unknown at this time.
Gene therapy
Standard procedure
The goal of gene therapy is to replace permanently the missing SMN1 gene in the genome of SMA patients [15•]. This can be done using an adenovirus vector, AAV9, which is able to cross the blood brain barrier after intravenous injection [15•]. A phase I clinical trial for SMA I infants birth to 9 months of age started in early 2014 at Nationwide Children’s Hospital in Columbus, Ohio. The optimal route of delivery for the vector is still being determined. Another study is planned that will use intrathecal administration of the AAV9 vector [16••].
Contraindications
Patients will be excluded from the trial if they require ventilator support or if there are high levels of antibodies to the AAV9 vector.
Complications
The AAV9 vector is not inserted into the chromosome so it could be lost, and not effective, if the cells it is inserted into are rapidly dividing. Therefore, tissues with low cell turnover, such as brain, spinal cord, or muscle, are good options. The immune system could react against the vector. In people who previously were exposed to the same virus, the immune system could neutralize the vector. Furthermore, there is the possibility of the reactivation of the virus from which the vector is made [18•].
Special points
The efficacy of this treatment may be related to how early in life it is administered. In studies, when the vector was given to SMN deficient mice on day one of life, the gene was incorporated into 40 % of motor neurons, but if given at day 10, it has no effect [18•].
Cost
Significant, depending on the frequency treatment is needed.
Stem cell therapy
This therapy, being studied in academic centers, uses pluripotent stem cells to direct either neural stem cells or motor neuron progenitors to the spinal cord or ventral horns [15•]. The stem cells must be injected intrathecally [19]. Currently, this work has only been done in SMA mice, but results have shown increased survival, body weight, grip strength, and decreased motor neuron and muscle loss.
Neuroprotection
Olesoxime is a cholesterol-like molecule, studied by the French pharmaceutical company Trophos, which may provide protection for the survival of motor neurons. The phase II study was conducted in Europe and was completed in September 2013. However, no results are available. The medication was given orally to patients with SMA II or III and outcome measures included Hammersmith Functional Motor Scale, compound muscle action potential, and motor unit number estimate [16••].
Small molecules
Quinazolines are molecules that inhibit the breakdown of RNA, including the SMN2 RNA. Theoretically, they could increase the amount of SMN protein available if less of the SMN2 RNA is degraded. The drug company Pfizer (234 East 42nd st, New York NY 10017) is conducting phase I studies.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Werdnig G. Zwei frühinfantile hereditäre Fälle von progressive Muskelatrophie unter dem Bilde der Dystrophie, aber auf neurotischer Grundlage [Two early infantile hereditary cases of progressive muscular atrophy simulating dystrophy, but on a neural basis; in German]. Arch Psychiatr Nervenkr. 1891;22:437–80.
Hoffmann J. U” ber chronische spinale Muskelatrophie im Kindesalter, auf familiärer Basis [On chronic spinal muscular atrophy in childhood, with a familial basis; in German]. Dtsch Z Nervenheilkd. 1893;3:427–70.
Byers RK, Banker BQ. Infantile muscular atrophy: en eleven-year experience. Trans Am Neurol Assoc. 1960;85:10–4.
Ogino S, Leonard DG, Rennert H, Ewens WJ, Wilson RB. Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet. 2002;110:301–7.
Zaldivar T, Montejo Y, Acevedo AM, Guerra R, Vargas J, Garofalo N, et al. Evidence of reduced frequency of spinal muscular atrophy type I in the Cuban population. Neurology. 2005;65:636–8.
Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B, et al. Consensus Statement for Standard of Care in Spinal Muscular Atrophy. J Child Neurol. 2007;22:1027.
Mercuri E, Bertini E, Iannaccone ST. Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurol. 2012;11:443–52. A brief communication bringing the reader up to date for standard of care for children with SMA. The authors who are well known for their expertise in the care of SMA patients include a discussion of new and emerging therapies in pre-clinical or clnical testing.
Willig TN, Paulus J, Lacau-Saint-Guily J, et al. Swallowing problems in neuromuscular disorders. Arch Phys Med Rehabil. 1994;75:1175–81.
Tilton AH, Miller MD, Khoshoo V. Nutrition and swallowing in pediatric neuromuscular patients. Semin Pediatr Neurol. 1998;5:106–15.
Ito T, Jensen RT. Association of long-term proton pump inhibitor therapy with bone fractures and effects on absorption of calcium, vitamin B12, iron, and magnesium. Curr Gastroenterol Rep. 2010;12:448–57.
Jones M, Campbell KA, Duggan C, et al. Multiple micronutrient deficiencies in a child fed an elemental formula. J Pediatr Gastroenterol Nutr. 2001;33:602–5.
Van den Engel-Hoek L, Erasmus C. Dysphagia in spinal muscular atrophy type II. Neurology. 2009;73:1787–91.
Messina S, Pane M, et al. Feeding problems and malnutrition in spinal muscular atrophy type I. Neuromuscul Disord. 2008;18:389–93.
Chng S, Wong Y, Hui J, et al. Pulmonary function and scoliosis in children with spinal muscular atrophy types II and III. J Paediatr Child Health. 2003;39:673–6.
Zanetta C, Riboldi G, Nizzardo M, Simone C, Faravelli I, Bresolin N, et al. Molecular, genetic and stem cell-mediated therapeutic strategies for spinal muscular atrophy (SMA). J Cell Mol Med. 2014;18:187–96. This is an in depth review of new therapeutic strategies for childhood SMA. The authors are prominent Italian clinician scientists and they provide and excellent summary of the mechanism of action for each of the approaches with many references.
Zanetta C, Nizzardo M, Simone C, Monguzzi E, Bresolin N, Comi GP, et al. Molecular therapeutic strategies for spinal muscular atrophies: current and future clinical trials. Clin Ther. 2014;36:128–40. These authors are the same as for #15, but this article presents an up to date summary of actual clinical trials either recently completed or still in process for SMA. there is description of mechanism of action for the investigational product as well as summary of the protocol for each study.
Yee J-K, Lin R-J. Antisense Oligonucleotides Shed new light on the pathogenesis and treatment of Spinal Muscular Atrophy. Mol Ther. 2012;20:8–10.
Braun S. Gene-based therapies of neuromuscular disorders: an update and the pivotal role of patient organizations in their discovery and implementation. J Gene Med. 2013;15:397–413. This is an interesting review/comment on the role that patient/volunteer disease-specific organizations have played in drug development for neuromuscular disorders. Such organizations have become increasingly important for the field in the past 15 years. Changes in the way clinical research is done for neuromuscular patients have been historic, very much like the influence the AIDS community had on clinical drug development in the 1980s.
Singh P, Liew WKM, Darras BT. Current advances in drug development in spinal muscular atrophy. Curr Opin Pediatr. 2013;25:682–8.
Compliance with Ethics Guidelines
Conflict of Interest
Susan T. Iannaccone declares that she is a principle investigator for ISIS Pharmaceutical studies in SMA patients. Diana Castro declares that she is a co-principle investigator for ISIS Pharmaceutical studies in SMA patients.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Pediatric Neurology
Rights and permissions
About this article

Cite this article
Castro, D., Iannaccone, S.T. Spinal Muscular Atrophy: Therapeutic Strategies. Curr Treat Options Neurol 16, 316 (2014). https://doi.org/10.1007/s11940-014-0316-3
Published:
DOI: https://doi.org/10.1007/s11940-014-0316-3