
Abstract
Apalutamide (ERLEADA®), a next-generation androgen receptor (AR) inhibitor, is approved in several countries, including those of the EU and in the USA, for the treatment of non-metastatic castration-resistant prostate cancer (nmCRPC). In the pivotal SPARTAN study in men with nmCRPC who were at high risk of developing metastases despite androgen-deprivation therapy (+ ADT), oral apalutamide (+ ADT) significantly prolonged metastasis-free survival (MFS) compared with placebo (+ ADT) at a median follow-up of 20.3 months, with consistent benefits demonstrated across prespecified subgroups. At this timepoint, apalutamide (+ ADT) also significantly prolonged the time to metastasis and progression-free survival (PFS) and maintained health-related quality of life (HR-QOL) compared with placebo (+ ADT). Apalutamide is generally well tolerated, with most adverse reactions of mild to moderate severity and relatively few patients discontinuing treatment because of these events. Although mature OS results are awaited with interest, given its beneficial effects on MFS and convenient oral once-daily regimen, apalutamide (+ ADT) is an important emerging treatment option for patients with nmCRPC who are at high risk of developing metastatic disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Oral non-steroidal next-generation AR inhibitor that binds directly to the ligand binding domain of the AR |
Significantly prolongs MFS, time to metastasis and PFS |
Overall survival results are immature (at a median follow-up of 41.0 months) |
Maintains health-related quality of life |
Generally well tolerated |
What is the rationale for using apalutamide in non-metastatic castration-resistant prostate cancer?
Prostate cancer (PC), the second most common malignancy in men, may be curable using definitive therapy during early stages; however, biochemical recurrence in some (27–53%) patients is inevitable [1, 2]. Androgen-deprivation therapy (ADT) remains the gold standard treatment for PC and, although ADT is initially effective, disease progression to castration-resistant PC (CRPC) eventually occurs in almost all patients. The risk of progression to metastatic CRPC (mCRPC) and death is highest amongst patients with non-metastatic CRPC (nmCRPC) who have higher PSA levels and a shorter PSA doubling time (PSADT) [2, 3]. Hence, delaying progression to mCRPC is the primary goal of treatment in patients with nmCRPC. Until recently, treatment options for nmCRPC were limited and were not associated with a survival benefit. An improved understanding of the mechanisms underlying ongoing androgen axis signaling in CRPC led to the development of effective targeted treatment strategies to overcome androgen receptor (AR) signaling [1, 4]. One such strategy is apalutamide (ERLEADA®), a next-generation, non-steroidal, selective AR inhibitor.
For whom is apalutamide indicated?
Oral apalutamide is approved in several countries, including those of the EU [5] and in the USA [6], for the treatment of adult men with nmCRPC (featured indication); specific indications may vary between individual countries. Treatment with apalutamide should be initiated and supervised by specialist physicians experienced in the medical treatment of PC. Table 1 provides a summary of the prescribing information for the use of apalutamide in nmCRPC in the EU [5] and USA [6]. Consult local prescribing information for further details.
What are the pharmacological properties of apalutamide?
Apalutamide binds directly to the ligand-binding domain of the AR and, as a consequence, prevents AR nuclear translocation, inhibits DNA binding and impedes AR-mediated transcription (reviewed previously in Drugs [7]) [5, 8]. Apalutamide lacks AR agonist activity. In preclinical studies, apalutamide treatment reduced tumour cell proliferation and increased apoptosis, leading to potent antitumour activity. In vitro, the major metabolite, N-desmethyl apalutamide, exhibited one-third of the activity of the parent drug [5, 8].
Like other antiandrogens, apalutamide exhibited weak binding at the gamma-aminobutyric acid type A (GABAA) receptor (a receptor implicated in seizures), with respective 50% inhibitory constants for apalutamide and enzalutamide of 3.0 µmol/L and 2.7 µmol/L. In vivo data suggest that apalutamide may be associated with lower seizure-inducing potential than enzalutamide [8]; the clinical relevance remains to be established.
Apalutamide exhibits dose-proportional pharmacokinetics after multiple oral once-daily doses (30–480 mg) in patients with mCRPC [9, 10]. At the recommended dose, steady-state concentrations of apalutamide occur after 4 weeks. The median time to maximum plasma concentrations is 2 h [5] and the mean absolute oral bioavailability of apalutamide is ≈ 100% [5, 11]. Apalutamide and its major active metabolite, N-desmethyl apalutamide, are extensively bound (96% and 95%) to plasma proteins (mainly to albumin) [5]. The primary route of apalutamide metabolism is by CYP3A4 and CYP2C8 to N-desmethyl apalutamide [5, 11]. The primary route of elimination of apalutamide is in the urine, with 65% and 24% of a radiolabeled dose recovered in the urine and faeces [5, 11]. The apparent clearance of apalutamide is 1.3 L/h after a single dose (2.0 L/h at steady state) [5].
Apalutamide is a potent inducer of many enzymes and transporters and may lead to an increase in elimination of many commonly used medicinal products; therefore, interaction with many common medicinal products that are substrates of enzymes and transporters is expected. The reduction in plasma concentrations of co-administered medicinal products can be substantial and lead to loss of/reduced clinical effects. Table 2 summarizes clinically relevant drug interactions (but not limited to these interactions) that may potentially occur with apalutamide, as outlined in EU [5] and US [6] prescribing information.
What are the potential resistance mechanisms for apalutamide?
Point mutations in the AR ligand-binding site, including F877L and T878A (formerly F876L and T877A), have been associated with resistance to AR-targeted therapy (apalutamide, enzalutamide, and abiraterone acetate plus prednisone), albeit these are not common contributors to de novo or acquired resistance to apalutamide [12, 13]. In addition to altering the specificity of ligand binding, F877L also converts AR antagonists to agonists; it spontaneously arose in cells with prolonged exposure to apalutamide and enzalutamide, and neither drug inhibited tumour growth in F877L-expressing cells [12, 14, 15].
The frequency of AR anomalies common in AR signaling-targeted therapy-resistant mCRPC did not increase with apalutamide (+ ADT) in the pivotal SPARTAN study in patients with nmCRPC who had a high risk of developing metastases [16]. At end of treatment, clinically relevant AR mutations (L702H, W742C, H875Y, F877L, T878A) were detected in 8.4% and 6.5% of patients in the apalutamide (n = 118) and placebo (n = 122) groups, with AR amplification occurring in 15.2% and 14.7%. Ligand-independent AR variant 7 (ARv7) was expressed by 9.4% of apalutamide recipients (n = 96) and 12.5% of placebo recipients (n = 104). AR anomaly positivity was associated with a shorter median second progression-free survival (PFS2) time than AR anomaly negativity. AR anomaly positivity had a minimal impact on PFS2 time in the apalutamide group. PFS2 was defined as time from randomization to progression during the next sequential therapy or death at any time [16]. Available data from a digital droplet-based detection of ARv7 in patients from SPARTAN suggested that a higher threshold of expression may be biologically important for driving treatment resistance [17].
What is the efficacy of apalutamide in non-metastatic castration-resistant prostate cancer?
The efficacy of oral once-daily apalutamide 240 mg (+ ADT) in men with nmCRPC who had a high risk of developing metastases (defined as a PSADT of ≤ 10 months during continuous ADT) was established in the randomized, double-blind, placebo-controlled, multinational, phase III SPARTAN study (Table 3) [18]. Eligibility criteria included age ≥ 18 years, cytologically or histologically confirmed adenocarcinoma of the prostate that was castration resistant, and a high risk of developing metastases with either no nodal disease (N0) or regional malignant pelvic nodes that were located below the aorto-iliac bifurcation (N1) and which measured < 2 cm in the short axis [18]. Patients were excluded if they had distant metastases on conventional imaging. Randomized treatment (apalutamide or placebo) was continued until protocol-defined disease progression, adverse events (AEs) or withdrawal of consent. Baseline demographics and disease characteristics were well-balanced across the two groups [18].
The primary endpoint was metastasis-free survival (MFS) and secondary endpoints were the time to metastasis, progression-free survival (PFS), time to symptomatic progression, overall survival (OS) and time to initiation of cytotoxic chemotherapy. A hierarchical adaptive, group-sequential procedure was used to assess the secondary endpoints; the first interim analysis of OS was done at the time of the primary analysis (i.e. final analysis of MFS). At the time of the clinical data cutoff for the primary analysis (19 May 2017), the median follow-up was 20.3 months. The study was unblinded in July 2017, with patients in the placebo group given the option of receiving apalutamide (hereafter referred to as the placebo → apalutamide group). The final analysis for MFS, the primary endpoint, was performed after distant metastasis or death had occurred in 378 patients (184 and 194 patients in the apalutamide and placebo groups) [18].
Apalutamide (+ ADT) significantly prolonged MFS compared with placebo (+ ADT), reducing the risk of metastasis or death in apalutamide recipients by 72% at the time of the primary analysis (Table 3) [18]. MFS benefits significantly favoured apalutamide over placebo (+ ADT) [i.e. hazard ratios (HRs) < 1; 95% CIs did not cross 1] for all prespecified subgroups (except the subgroup of Black patients for whom low patient numbers confounded interpretation of data), including based on age, race, region and number of previous hormonal therapies, and baseline ECOG performance status, PSA level and stratification factors at randomization [i.e. PSADT (> 6 vs ≤ 6 months), use of bone-sparing agents (yes vs no) and classification of nodal disease (N0 vs N1)] [18]. In post hoc analyses, the beneficial effects of apalutamide (+ ADT) over placebo (+ ADT) on MFS were similar in Asian and non-Asian subpopulations (HRs 0.29 and 0.28; both p < 0.001) [19] and in Japanese patients (vs overall population) [20], and evident (all HRs < 1) regardless of prior local therapy (i.e. ± prior radical prostatectomy and/or external radiotherapy; p < 0.05) [21], DECIPHER genomic classifier score (i.e. low to average score ≤ 0.6 or high score > 0.6; p < 0.05) [22], baseline comorbidities (any comorbidity, diabetes/hyperglycaemia, cardiovascular disease, hypertension and renal insufficiency; significant based on the 95% CIs) [23], whether post-treatment PSA levels did or did not decline to < 0.2 ng/mL (p < 0.0001) [24] and age (< 65, 65–74 or ≥ 75 years) [25]. In a multivariate analysis, independent predictors of MFS (p < 0.05) included treatment with apalutamide, PSADT > 6 months, N0 classification, Gleason score ≤ 7 at diagnosis and baseline PSA ≤ 7.8 ng/mL; a continuous relationship between shorter PSADT and faster time to metastasis or death was also observed [26].
At the time of the final MFS analysis (median follow-up of 20.3 months), secondary outcomes also generally favoured apalutamide (+ ADT) over placebo (+ ADT), with the median time to metastasis, PFS and time to symptomatic progression significantly (p < 0.001) prolonged in the apalutamide group (Table 3) [18]. Symptomatic progression events had occurred in 8% and 16% of apalutamide and placebo recipients [5]. The median time to initiation of cytotoxic chemotherapy was not yet reached in either treatment group (Table 3) [18].
At the time of the final MFS analysis, results for OS were immature [104 of the 427 events (24%) required for the planned final OS analysis had occurred], with the median OS not yet reached in the apalutamide group (Table 3) [18]. At the time of the second prespecified analysis [median follow-up of 41 months; 285 OS events (67%) had occurred], median OS was not reached in either treatment group, with the risk of death reduced by 25% in the apalutamide (+ ADT) versus the placebo group (HR 0.75; 95% CI 0.59–0.96; p = 0.0197); the p-value did not cross the prespecified O’Brien-Fleming boundary [27]. In the apalutamide and placebo groups, respective 4-year OS rates were 72% and 65% and, after adjustment for patients who crossed over from placebo to apalutamide, 4-year OS rates were 72% vs 61%. Results for OS were robust despite crossover of 19% of placebo recipients to apalutamide treatment, as determined in sensitivity analysis using naïve censoring and inverse probability of censoring weighted analyses [27].
In a landmark analysis of SPARTAN, patients who developed metastases at 6 (n = 103), 9 (n = 167) or 12 (n = 214) months had significantly shorter OS than patients without metastases (n = 1099, 1026 and 967 respectively) [HR 4.12; 95% CI 2.60–6.54; p < 0.0001], with the association between metastases development and OS remaining after adjustments for baseline covariates. A positive correlation between MFS and OS was demonstrated by the Spearman’s correlation statistics (co-efficient: 0.62; p < 0.0001) and confirmed using Fleischer’s statistical model (co-efficient: 0.69), with ≈ 50% of the variability in OS explained by MFS [28].
In terms of exploratory endpoints at the time of final MFS analysis (median follow-up of 20.3 months), the median time to PSA progression was not reached in the apalutamide group and was 3.7 months in the placebo group (HR 0.06; 95% CI 0.05–0.08), with a PSA response achieved by 89.7% and 2.2% of patients (relative risk 40; 95% CI 21–77) [18]. At a median follow-up of 41.0 months (i.e. second interim analysis), the median PFS2 was 55.6 months in the apalutamide group versus 43.8 months in the placebo group, with a 45% reduction in the risk of disease progression or death (from any cause) following second-line therapy in the apalutamide group (HR 0.55; 95% CI 0.45–0.68; p < 0.0001). At this timepoint, apalutamide prolonged the time to initiation of cytotoxic chemotherapy compared with placebo (median time not reached in either group; HR 0.60; 95% CI 0.45–0.80) [27].
Apalutamide treatment (+ ADT) maintained health-related quality of life (HR-QOL), with both Functional Assessment of Cancer Therapy-Prostate (FACT-P) total and subscale scores and European Quality of Life-5 dimension three-level (EQ-5D-3L) scores generally maintained from baseline until cycle 29 (prespecified exploratory analysis) [29]. HR-QOL also generally remained stable in the placebo (+ ADT) group. In the apalutamide and placebo groups, mean FACT-P total and subscale scores and FACT-General (FACT-G) total scores were similar from baseline up to 12 months after metastasis in patients with distant metastases, with both groups showing similar decreases in these scores after symptomatic progression events [29].
What is the tolerability profile of apalutamide?
Oral apalutamide (+ ADT) was generally well tolerated in the pivotal SPARTAN study in men with nmCRPC who were at high risk of developing metastases [18]. In clinical studies the following adverse reactions were reported during apalutamide treatment [5]:
Very common (frequency ≥ 10%) Skin rash (as per discussion below), fracture (as per discussion below), arthralgia, fatigue, decreased weight, fall
Common (frequency ≥ 1% to < 10%) Hypothyroidism (includes hypothyroidism, blood thyroid-stimulating hormone increased, thyroxine decreased, autoimmune thyroiditis, thyroxine free decreased, tri-iodothyronine decreased), hypercholesterolaemia, hypertriglyceridaemia, pruritus
Uncommon (frequency ≥ 0.001% to < 1%) seizure
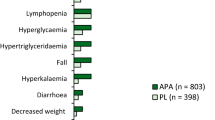
At the time of the final analysis of MFS (median follow-up of 20.3 months), the median duration of exposure in the safety population in the apalutamide (n = 803) and placebo (n = 398) groups was 16.9 and 11.2 months [6]. Treatment-emergent AEs (TEAEs) of any grade occurred in 96.5% and 93.2% of patients in the apalutamide and placebo groups, with 10.6% and 7.0% of patients discontinuing study drug because of these events [18]. With longer-term treatment (i.e. at the time of the second prespecified analysis; respective median treatment durations in the apalutamide, placebo and placebo → apalutamide groups were 31.4, 11.5 and 15.0 months), TEAEs of any grade had occurred in 97%, 94% and 86% of patients in the apalutamide, placebo and placebo → apalutamide groups, respectively (n = 803, 398 and 76), with 13.6%, 7.3% and 10.5% of patients discontinuing study drug because of these TEAEs [27]. The most common TEAEs of any grade occurring in the apalutamide, placebo and placebo → apalutamide group were fatigue (31.9, 21.4 and 14.5%, respectively), hypertension (27.6, 20.9 and 9.2%), diarrhoea (22.2, 15.3 and 11.8%) and fall (20.9, 9.5 and 6.6%) [27].
No new safety signals were observed during longer-term treatment with apalutamide [27]. In apalutamide recipients, the incident rate of grade 3 or 4 events after adjusting for exposure did not differ significantly between the time of the first and second interim analysis, including for grade 3 or 4 rash (4.2 vs 2.7 events/100 patient-years’ exposure (PYE)], falls (1.2 vs 1.2 events/100 PYE) and fractures (2.1 vs 2.0 events/100 PYE) [27].
In the apalutamide and placebo groups, serious AEs occurred in 24.8% and 23.1% of patients during the first prespecified analysis period [18]. AEs leading to death occurred in ten apalutamide recipients (one case each of acute myocardial infarction, cardiorespiratory arrest, cerebral haemorrhage, myocardial infarction, multiple organ dysfunction and pneumonia, and two cases each of sepsis and prostate cancer) and one placebo recipient (cardiorespiratory arrest) [18]. At the time of the second interim analysis, AEs leading to death had occurred in 17, 2 and 2 patients in the apalutamide, placebo and placebo → apalutamide groups, respectively (individual causes of death not specified) [27].
Selected adverse reactions of interest
See Table 1 regarding potential risks and management of selected adverse events, including those discussed below.
Skin rashes with apalutamide (+ ADT) treatment were typically macular or maculopapular rashes, had a median time to onset of 82 days and resolved within a median of 60 days; recurrence of rash was reported in ≈ 50% of patients re-challenged with apalutamide [18]. In the apalutamide and placebo group, 23.8% and 5.5% of patients experienced skin rash, with grade 3 rashes (i.e. covering > 30% of the body surface area) occurring in 5.2% and 0.3% of patients. There were no occurrences of toxic epidermal necrolysis or Stevens-Johnson syndrome. In the apalutamide group, skin rash AEs led to treatment discontinuation, dose interruption and dose reduction in 2.4%, 6.8% and 2.7% of patients, respectively, with corresponding rates in the placebo group of 0%, 1.3% and 0.3% [18].
Hypothyroidism AEs were reported in 8.1% and 2.0% of patients in the apalutamide and placebo groups, all of which were of grade 1 or 2 severity [18]. The median time to onset of hypothyroidism, based on assessments of thyroid-stimulating hormone every 4 months, was 113 days [5, 18]. In patients not receiving thyroid replacement therapy, hypothyroidism was reported in 5.7% and 0.8% of patients in the apalutamide and placebo groups. Hypothyroidism was manageable with increases in or initiation of thyroid replacement therapy [18]. When clinically indicated, thyroid replacement therapy should be initiated or adjusted [5, 6].
At the time of the first interim analysis, falls were reported in 11.7% and 6.5% of patients in the apalutamide and placebo groups, of which 2.7% an 0.8% were grade 3 or 4 [18]. Of patients with a fracture, ≈ 50% of patients experienced a fall during the 7 days before a fracture event [5]. The median time to onset of fracture was 314 days in the apalutamide group; falls were not associated with a loss of consciousness or seizures [6]. In a multivariate analysis, older age, poor ECOG performance status, history of neuropathy and pre-study use of α-blockers were independently associated with time to fall, whereas older age and low serum albumin levels were independently associated with time to fracture (p < 0.05 for all) [30].
What is the current clinical position of apalutamide in non-metastatic castration-resistant prostate cancer?
Although mature OS results are awaited with interest, given its beneficial effects on MFS and convenient once-daily regimen, apalutamide (+ ADT) is an important emerging option for the treatment of patients with nmCRPC who are at high risk of developing metastatic disease, as reflected in recent EU [31] and US [32] guidelines. Prolonging MFS is recognised by the FDA as a clinically relevant measure in PC trials (i.e. MFS represents a landmark in PC care) [33, 34]. In the pivotal, multinational SPARTAN study in this patient population, relative to placebo (+ ADT), apalutamide (+ ADT) significantly prolonged MFS and was more effective than placebo for secondary outcomes of the median time to first metastasis, PFS and time to symptomatic progression. The beneficial effects of apalutamide (+ ADT) in prolonging MFS were observed in all evaluable prespecified subgroups. The p-values for OS did not cross the prespecified efficacy boundary at the time of the first and second interim analysis. Thus, the final OS analysis is planned after the specified 427 OS events have occurred. A landmark analysis of SPARTAN showed a positive correlation between MFS and OS [28], with the correlation between MFS and OS in nmCRPC supported by a retrospective analysis of a Japanese claims database [35].
Reflecting results of clinical trials, the recent approvals of AR inhibitors has revolutionized the treatment paradigm for patients with nmCRPC at high risk of developing metastases [4, 31, 32]. EU [31] and NCCN guidelines [32] strongly recommend the use of apalutamide or enzalutamide in men with nmCRPC at high risk of developing metastasis to prolong the time to metastases. Continuation of ADT to maintain castrate serum levels of testosterone < 50 ng/dL is recommended, with monitoring recommended in patients whose PSA levels do not increase. In those in whom PSA levels are increasing, give consideration to changing or maintaining treatment (with monitoring) in non-metastatic disease (i.e. M0) or to changing to systemic therapy for metastatic disease (i.e. M1).
To date, no randomized controlled trials (RCTs) have directly compared the efficacy of apalutamide with that of enzalutamide (+ ADT) or darolutamide (+ ADT). A recent review of phase 3 RCTs of apalutamide (approved in EU and USA), enzalutamide (approved in the EU and USA), and darolutamide (approved in USA [36]) indicated that all three drugs (+ ADT) delayed the time to MFS or death in men with high-risk nmCRPC [37]. In a matching-adjusted indirect comparison (MAIC) of pivotal trials evaluating apalutamide (+ ADT; SPARTAN study) and enzalutamide (+ ADT; PROSPER study) in patients with nmCRPC, along with Bayesian probability data, apalutamide recipients had more favourable MFS and OS outcomes than enzalutamide recipients [38]. However, a network meta-analysis (NMA) suggested that there were no significant differences between apalutamide and enzalutamide for MFS or any secondary outcomes (including efficacy and safety endpoints) [39]. The equivocal results between these MAIC [38] and NMA [39] analyses may, at least in part, reflect the different methodological approaches taken in each of these analyses (i.e. a Bayesian anchored MAIC approach and a Bucher technique/approach, respectively), with several potential limitations associated with the Bucher approach (see Chowdhury et al. [38] for an in-depth discussion of these limitations). In another Bayesian anchored MAIC of SPARTAN and PROSPER, apalutamide (+ ADT) recipients had a higher probability of an improvement in HR-QOL than enzalutamide (+ ADT) recipients, based on FACT-P total and subscale scores (probabilities for more favourable improvement with apalutamide ranged from 57–73%) and FACT-G total and subscale scores (80–97%) [40]. Ultimately, the choice of treatment for nmCRPC will rely upon physician familiarity and pharmaceutical coverage, as well as other patient and health economic considerations. Results of a systematic review and NMA of RCTs suggest that cost and availability should drive clinical decision making in patients with nmCRPC [41].
Apalutamide treatment (+ ADT) is generally well tolerated in patients with nmCRPC, with most adverse reactions of mild to moderate severity and relatively few patients discontinuing treatment because of these events. Albeit ongoing clinical experience is required to fully determine the long-term safety profile of apalutamide (+ ADT), no new safety signals were identified at a median follow-up of 41 months. In a Bayesian anchored MAIC of SPARTAN and PROSPER, apalutamide recipients had a higher probability of an improved tolerability profile than enzalutamide recipients, with a reduced probability (all odds ratios < 1) of the occurrence of any AEs (66.9% probability in favour of apalutamide + ADT vs enzalutamide + ADT), serious AEs (90.9%), fatigue (99.5%), hypertension (99.2%), decreased appetite (98.3%), fall (90.3%), headaches (86.7%) and nausea (80.0%) [40].
Apalutamide (+ ADT) is associated with a low number needed-to-treat (NNT) and a high number needed-to-harm (NNH) in patients with nmCRPC, based on clinical event rates from survival curves and grade 3/4 AE rates in SPARTAN [42]. In terms of NNTs, compared with ADT alone, one additional patient is free of developing metastases or death at 12, 24 and 36 months for every 3.6, 2.7 and 2.3 patients treated with apalutamide (+ ADT), respectively; the NNH with apalutamide treatment (+ ADT) for grade 3/4 AEs is ≥ 20 [42].
References
El-Amm J, Aragon-Ching JB. The current landscape of treatment in non-metastatic castration-resistant prostate cancer. Clin Med Insights Oncol. 2019. https://doi.org/10.1177/1179554919833927.
Esther J, Maughan BL, Anderson N, et al. Management of nonmetastatic castration-resistant prostate cancer: recent advances and future direction. Curr Treat Options Oncol. 2019. https://doi.org/10.1007/s11864-019-0611-z.
Freedland SJ, Ramaswamy K, Lechpammer S, et al. Association of prostate-specific antigen trajectories with mortality in veterans with non-metastatic castration-resistant prostate cancer [abstract no. MP34-07]. J Urol. 2019;201(4S Suppl):e497–8.
Crawford ED, Schellhammer PF, McLeod DG, et al. Androgen receptor targeted treatments of prostate cancer: 35 years of progress with antiandrogens. J Urol. 2018;200(5):956–66.
ERLEADA (apalutamide) film-coated tablets: EU summary of product characteristics. Beerse: Janssen-Cilag International NV; 2019.
ERLEADA® (apalutamide tablet, film coated): US prescribing information. Horsham (PA): Janssen Products, LP; 2019.
Al-Salama ZT. Apalutamide: a review in non-metastatic castration-resistant prostate cancer. Drugs. 2019;79(14):1591–8.
Clegg NJ, Wongvipat J, Joseph JD, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72(6):1494–503.
Rathkopf DE, Morris MJ, Fox JJ, et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2013;31(28):3525–30.
Peréz-Ruixo C, Peréz-Blanco JS, Chien C, et al. Population pharmacokinetics of apalutamide and its active metabolite N-desmethyl-apalutamide in healthy and castration-resistant prostate cancer subjects. Clin Pharmacokinet. 2019. https://doi.org/10.1007/s40262-019-00808-7.
de Vries R, Jacobs F, Mannens G, et al. Apalutamide absorption, metabolism, and excretion in healthy men, and enzyme reaction in human hepatocytes. Drug Metab Dispos. 2019;47:453–64.
Joseph JD, Lu N, Qian J, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3(9):1020–9.
Rathkopf DE, Smith MR, Ryan CJ, et al. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Ann Oncol. 2017;28(9):2264–71.
Balbas MD, Evans MJ, Hosfield DJ, et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife. 2013. https://doi.org/10.7554/eLife.00499.
Korpal M, Korn JM, Gao X, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013;3(9):1030–43.
Smith MR, Thomas S, Chowdhury S, et al. Androgen receptor (AR) anomalies and efficacy of apalutamide (APA) in patients (pts) with nonmetastatic castration-resistant prostate cancer (nmCRPC) from the phase 3 SPARTAN study [abstract no. 2605]. Cancer Res. 2018;78(13 Suppl).
Espaillat MP, Rajpurohit Y, Gormley M, et al. Digital droplet PCR (ddPCR)-based detection of androgen receptor splice variant 7 (AR-v7) in non-metastatic castration resistant prostate [abstract no. 1335]. Cancer Res. 2019;79(13 Suppl).
Smith MR, Saad F, Chowdhury S, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. 2018;378(15):1408–18.
Mainwaring P, Small E, Uemura H, et al. Efficacy and safety of apalutamide (APA) in patients (pts) with nonmetastatic castration-resistant prostate cancer (nmCRPC) from SPARTAN: Asian subpopulation [abstract no. 2130]. Ann Oncol. 2018;29(Suppl 9).
Sato T, Imanaka K, Noguchi H, et al. The efficacy and safety of apalutamide in Japanese patients with non-metastatic castration-resistant prostate cancer (NM-CRPC): sub-analysis of randomized, multicentre, double-blind, phase 3 study [oral presentation]. In: 107th Annual Meeting of the Japanese Urological Association; 2019.
Hadaschik BA, Saad F, Graff JN, et al. Efficacy and safety of apalutamide (APA) in nonmetastatic castration-resistant prostate cancer (nmCRPC) patients (pts) with or without prior radical prostatectomy (RP) and/or external radiotherapy (XRT): post hoc analysis of SPARTAN [abstract no. MP34-19]. J Urol. 2019;201(Suppl 4):e502–3.
Saad F, Thomas S, Feng FY, et al. Response to apalutamide (APA) among patients (pts) with nonmetastatic castration-resistant prostate cancer (nmCRPC) from SPARTAN by Decipher Genomic Classifier (GC) score [abstract no. PD11-03]. J Urol. 2019;201(4S Suppl):e216.
Small EJ, Saad F, Chowdhury S, et al. Efficacy of apalutamide (APA) plus ongoing androgen deprivation therapy (ADT) in patients (pts) with nonmetastatic castration-resistant prostate cancer (nmCRPC) and baseline (BL) comorbidities (CM) [abstract no. 5023 ]. J Clin Oncol. 2019;37(15 Suppl).
Graff J, Saad F, Hadaschik BA, et al. Metastasis-free survival (MFS) in nonmetastatic castration-resistant prostate cancer (NMCRPC) patients (pts) with prostate-specific antigen (PSA) decline to < 0.2 ng/ml following apalutamide (APA) treatment: post hoc results from the phase 3 SPARTAN study [abstract no. MP34-20]. J Urol. 2019;201(4S Suppl):e503.
Graff JN, Smith MR, Saad F, et al. Age-related efficacy and safety of apalutamide (APA) plus ongoing androgen deprivation therapy (ADT) in subgroups of patients (pts) with nonmetastatic castration-resistant prostate cancer (nmCRPC): post hoc analysis of SPARTAN [abstract no. 5024]. J Clin Oncol. 2019;37(15 Suppl).
Small EJ, Saad F, Rathkopf DE, et al. Predicting disease progression in patients (pts) with nonmetastatic castration-resistant prostate cancer (nmCRPC): an analysis from the phase 3 SPARTAN trial [abstract no. 5034]. J Clin Oncol. 2018;36(15 Suppl).
Small EJ, Saad F, Chowdhury S, et al. Apalutamide and overall survival in non-metastatic castration-resistant prostate cancer. Ann Oncol. 2019;30(11):1813–20.
Smith MR, Mehra M, Nair S, et al. Relationship between metastasis-free survival and overall survival in patients with nonmetastatic castration-resistant prostate cancer. Clin Genitourin Cancer. 2019. https://doi.org/10.1016/j.clgc.2019.10.030.
Saad F, Cella D, Basch E, et al. Effect of apalutamide on health-related quality of life in patients with non-metastatic castration-resistant prostate cancer: an analysis of the SPARTAN randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2018;19(10):1404–16.
Pollock YY, Smith MR, Saad F, et al. Predictors of falls and fractures in patients (pts) with nonmetastatic castration-resistant prostate cancer (nmCRPC) treated with apalutamide (APA) plus ongoing androgen deprivation therapy (ADT). [abstract no. 5025]. J Clin Oncol. 2019;37(Suppl).
European Association of Urology. EAU oncology guidelines: prostate cancer; 2019. http://uroweb.org/guideline/prostate-cancer/#1. Accessed 10 Jan 2020.
National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: prostate cancer; 2019. http://www.nccn.org/. Accessed 10 Jan 2020.
Beaver JA, Kluetz PG, Pazdur R. Metastasis-free survival: a new end point in prostate cancer trials. N Engl J Med. 2018;378(26):2458–60.
Chandler R, De Bono J. Second-generation antiandrogens in nonmetastatic CRPC. Nat Rev Urol. 2018;15(6):342–4.
Mori A, Hashimoto K, Koroki Y, et al. The correlation between metastasis-free survival and overall survival in non-metastatic castration-resistant prostate cancer patients from the Medical Data Division claims database in Japan. Curr Med Res Opin. 2019;35(10):1745–50.
NUBEQA® (darolutamide tablets): US prescribing information. Whippany: Bayer HealthCare Pharmaceuticals Inc.; 2019.
Oudard S, Soto AJ. A multi-disciplinary review of the evidence supporting metastasis-free survival (MFS) and the benefit of delaying metastasis in high-risk non-metastatic castration-resistant prostate cancer. EMJ Urol. 2019;6(Suppl 2):2–11.
Chowdhury S, Oudard S, Uemura H, et al. Matching-adjusted indirect comparison of the efficacy of apalutamide and enzalutamide in the treatment of non-metastatic castration-resistant prostate cancer. Adv Ther. 2019. https://doi.org/10.1007/s12325-019-01156-5.
Wallis CJD, Chandrasekar T, Goldberg H, et al. Advanced androgen blockage in nonmetastatic castration-resistant prostate cancer: an indirect comparison of apalutamide and enzalutamide. Eur Urol Oncol. 2018;1(3):238–41.
Chowdhury S, Oudard S, Uemura H, et al. Matching-adjusted indirect comparison of health-related quality of life and adverse events of apalutamide versus enzalutamide in non-metastatic castration-resistant prostate cancer. Adv Ther. 2019. https://doi.org/10.1007/s12325-019-01157-4.
Riaz IB, Bhattacharjee S, Malik S, et al. Apalutamide (APA) or enzalutamide (ENZA) in men with nonmetastatic castration-resistant prostate cancer (CRPC): a systematic review and network meta-analysis of randomized clinical trials (RCTs) [abstract no. 263]. J Clin Oncol. 2019;37(7 Suppl).
Schultz NM, Sugarman R, Ramaswamy K, et al. Clinical benefits and risk of enzalutamide and apalutamide: number needed to treat and number needed to harm analysis for patients with non-metastatic castration-resistant prostate cancer [abstract no. MP34-18]. J Urol. 2019;201(4S Suppl):e502.
Acknowledgements
The manuscript was updated from Drugs 2019;79(14):1591–8 [7], and was reviewed by: U. Emmenegger, Sunnybrook Odette Cancer Centre, Medical Oncology, Toronto, ON, Canada; A. Pinto, University Hospital La Paz, Medical Oncology, Madrid, Spain. During the peer review process, Janssen-Cilag Ltd., the marketing-authorization holder of apalutamide, was offered an opportunity to provide a scientific accuracy review of their data. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflict of interest
Lesley Scott is a salaried employee of Adis International Ltd./Springer Nature, is responsible for the article content and declares no conflicts of interest.
Additional information
Enhanced material for this Adis Drug Q&A can be found at https://doi.org/10.6084/m9.figshare.11301911.
Rights and permissions
About this article

Cite this article
Scott, L.J. Apalutamide in non-metastatic castration-resistant prostate cancer: a profile of its use. Drugs Ther Perspect 36, 97–105 (2020). https://doi.org/10.1007/s40267-020-00707-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40267-020-00707-z