
Abstract
Elafibranor (IQIRVO®) is a first-in-class peroxisome proliferator-activated receptor (PPAR) agonist being developed by Ipsen, under license from Genfit, for the treatment of primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC). On 10 June 2024, elafibranor received accelerated approval based on reduction of alkaline phosphatase (ALP) in the USA for the treatment of PBC in combination with ursodeoxycholic acid (UDCA) in adults who have an inadequate response to UDCA, or as monotherapy in patients unable to tolerate UDCA. Elafibranor has also received a positive opinion in the EU. This article summarizes the milestones in the development of elafibranor leading to this first approval for PBC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Digital Features for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.26343709 |
PPAR agonist being developed by Ipsen, under license from Genfit, for the treatment of PBC and PSC |
Received its first approval on 10 June 2024 in the USA under accelerated approval based on reduction of ALP |
Approved for the treatment of PBC in combination with UDCA in adults who have an inadequate response to UDCA, or as monotherapy in patients unable to tolerate UDCA |
1 Introduction
Primary biliary cholangitis (PBC; previously known as primary biliary cirrhosis) is a chronic autoimmune disease of the liver that causes inflammation and progressive destruction of the bile ducts [1]. This leads to cholestasis which, if left untreated, progresses to liver fibrosis and cirrhosis. Clinically, PBC is characterized by fatigue, jaundice and pruritus. The disease is typically diagnosed between the ages of 40 and 60 years, with females being at greater risk than males [1].
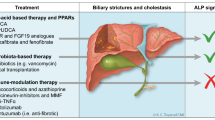
Guidelines for the management of PBC recommend ursodeoxycholic acid (UDCA) as first-line therapy [2]. UDCA has been shown to improve liver biochemistry and slow disease progression in patients with PBC [1, 2]. However, between 30% and 40% of patients have a suboptimal response to UDCA and are at high risk for disease progression [2]. Obeticholic acid has been approved in the USA as a second-line therapy for PBC, but cannot be used in patients with advanced liver cirrhosis [1, 2]. Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear receptor family of ligand-activated transcription factors that heterodimerize with the retinoid X receptor to regulate the expression of genes involved in fatty acid metabolism, inflammation and glucose metabolism [1]. PPAR activation represents a therapeutic target for cholestatic liver diseases [1].
Elafibranor (IQIRVO®) is a first-in-class PPAR agonist that is being developed by Ipsen, under license from Genfit, for the treatment of PBC and primary sclerosing cholangitis (PSC).

Elafibranor was given orphan drug designation in the USA and the EU in July 2019 for the treatment of PBC [3]; prior to this, elafibranor had been granted breakthrough therapy designation [4]. In December 2023, the US FDA granted priority review for elafibranor in PBC [5]. On 10 June 2024, elafibranor received accelerated approval in the USA for the treatment of PBC in combination with UDCA in adults who have an inadequate response to UDCA, or as monotherapy in patients unable to tolerate UDCA [6, 7]. The approval was based on reduction of alkaline phosphatase (ALP) [6] in the phase III ELATIVE trial (Sect. 2.4.1) [7]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s) [6]. The recommended dosage of elafibranor is 80 mg once daily, with or without food. Elafibranor should be administered ≥ 4 h before or 4 h after administration of a bile acid binding sequestrant (or at as great an interval as possible). The use of elafibranor in patients who have or who develop decompensated cirrhosis (e.g. ascites, variceal bleeding, hepatic encephalopathy) is not recommended. The use of elafibranor in patients with complete biliary obstruction should be avoided [6].
Elafibranor received a positive opinion the EU in July 2024 for the treatment of PBC [8]. Elafibranor is under regulatory review in the UK for the treatment of PBC [5]. The drug is in phase III development for PBC in multiple countries worldwide. Elafibranor is also being developed for the treatment of PSC (in phase II). Clinical development of elafibranor in non-alcoholic steatohepatitis was terminated after the phase II trial did not meet the primary endpoint due to lack of efficacy, but not for safety reasons. Clinical development of elafibranor for the treatment of non-alcoholic fatty liver disease, hepatic fibrosis, lipid metabolism disorders and type 2 diabetes mellitus has also been discontinued.
1.1 Company Agreements
In June 2019, Terns Pharmaceuticals acquired rights to develop and commercialize elafibranor in Greater China from Genfit in exchange for upfront, milestone and royalty payments [9, 10].
Genfit and Ipsen entered into a long-term strategic global partnership in December 2021 [11]. Under the terms of the licensing agreement, Ipsen acquired exclusive worldwide rights to develop, manufacture and commercialize elafibranor from Genfit in exchange for upfront, milestone and royalty payments [11].
2 Scientific Summary
2.1 Pharmacodynamics
Elafibranor and its main active metabolite GFT1007 are PPAR agonists that activate PPAR-α, PPAR-γ and PPAR-δ in vitro [6]. Uncertainty exists regarding the exact mechanism by which elafibranor exerts its therapeutic effects in PBC. However, it is thought to involve inhibition of bile acid synthesis via activation of PPAR-α and PPAR-δ.

The key enzyme involved in bile acid synthesis from cholesterol, CYP7A1, is thought to be downregulated by fibroblast growth factor 21 as part of the PPAR-δ signaling pathway [6].
Elafibranor and GFT1007 produced activation of PPAR-α in vitro, with half-maximal effective concentration (EC50) values of 46 nM and 14 nM, respectively, and maximum effect (Emax) values of 56% and 61%, respectively, compared with reference PPAR agonists [6]. Elafibranor and GFT1007 were ≈ 3- to 8-fold more potent for PPAR-α than for PPAR-γ and PPAR-δ. Although the in vitro pharmacology studies detected PPAR–γ activation by elafibranor and GFT1007, animal toxicology studies demonstrated no adverse effects associated with activation of PPAR-γ [6].
In a phase IIa trial in patients with PBC (Sect. 2.4.2), elafibranor was associated with significant (p < 0.05 vs placebo) reductions from baseline in various disease and inflammatory markers, including γ-glutamyltransferase, immunoglobulin M, 5’-nucleotidase, high-sensitivity C-reactive protein and haptoglobin [12].
No clinically significant QTc interval prolongation was observed after administration of an elafibranor dose 3.75 times the recommended dose [6].
2.2 Pharmacokinetics
Elafibranor demonstrated time-dependent pharmacokinetics following 16 days of repeated once-daily administration [6]. Steady-state plasma concentrations of GFT1007 and elafibranor were achieved after 7 and 14 days, respectively. The area under the concentration-time curve (AUC) over the last 24-h dosing interval increased dose-proportionally across the dose range of 40–300 mg. Following repeated administration of elafibranor 80 mg once daily in patients with PBC, peak plasma concentrations (Cmax) of elafibranor were reached after a median of 1.25 h (Tmax). When a single dose of elafibranor was administered with a high-fat and -calorie meal, the Tmax of elafibranor increased by 30 min and elafibranor Cmax and AUC decreased by 50% and 15%, respectively, compared with fasted conditions. Elafibranor is highly (≈ 99.7%) bound to plasma proteins (mainly serum albumin). Elafibranor has a mean apparent volume of distribution of 4731 L following a single dose of 80 mg in healthy fasted volunteers [6].
Elafibranor is extensively metabolized by the cytosolic enzyme 15-ketoprostaglandin 13-∆ reductase to form the major active metabolite GFT1007 [6]. GFT3351 is a major inactive metabolite. The metabolism of elafibranor is also mediated by CYP2J2, UGT1A3, UGT1A4 and UGT2B7. GFT1007 is further metabolized by CYP2C8, UGT1A3 and UGT2B7. Following a single radiolabeled oral dose of elafibranor 120 mg (1.5 times the recommended dose) in healthy volunteers, ≈ 77% of the administered dose was recovered in faeces, primarily as unchanged drug (57%) and GFT1007 (6%). Approximately 19% of the administered dose was recovered in urine, primarily as GFT3351 (12%). Following a single dose of 80 mg under fasted conditions, the mean apparent total clearance of elafibranor is 50 L/h and the mean elimination half-life is 70.2 h. GFT1007 has a mean elimination half-life of 15.4 h [6].
Sex, body weight (43 kg to 120 kg), body mass index (14.5 kg/m2 to 53.5 kg/m2), renal impairment and hepatic impairment have no clinically relevant effects on the pharmacokinetics of elafibranor [6]. Following a single dose of 120 mg (1.5 times the recommended dose), the exposure of elafibranor was 23% higher in healthy elderly volunteers aged 75–80 years than in healthy younger volunteers aged 26–42 years [6].
Features and properties of elafibranor
Alternative names | GFT505; IPN60190; IQIRVO |
Class | Anti-inflammatories; antifibrotics; antihyperglycaemics; antihyperlipidaemics; hepatoprotectants; organic sulfur compounds; phenyl ethers; propionic acids; small molecules |
Mechanism of action | PPAR-α agonists; PPAR-δ agonists |
Route of administration | Oral |
Pharmacodynamics | Mechanism by which elafibranor exerts its therapeutic effects in PBC appears to involve inhibition of bile acid synthesis via activation of PPAR-α and PPAR-δ |
Pharmacokinetics | Time-dependent pharmacokinetics; median Tmax 1.25 h; ≈ 99.7% bound to plasma proteins; mean apparent volume of distribution 4731 L; mean apparent total clearance 50 L/h; mean elimination half-life 70.2 h |
Most frequent adverse events | COVID-19, pruritus, abnormal weight gain, abdominal pain, diarrhoea, nausea, UTI, vomiting |
WHO ATC codes | A05AX06 (Other Drugs for Bile Therapy) |
Chemical name | 2-[2,6-dimethyl-4-[(E)-3-(4-methylsulfanylphenyl)-3-oxoprop-1-enyl]phenoxy]-2-methylpropanoic acid |
2.3 Drug Interactions
Elafibranor is a substrate of CYP2J2, UGT1A3, UGT1A4, UGT2B7, PTGR1, MRP2 and BCRP, a weak inducer of CYP3A4 and an inhibitor of BSEP and BCRP [6]. Therefore, clinically significant drug interactions may occur when elafibranor is coadministered with other drugs. Coadministration of elafibranor with hormonal contraceptives may reduce the systemic exposure of ethinyl estradiol and progestin (both CYP3A4 substrates) and lead to contraceptive failure and/or increased breakthrough bleeding. The risk of myopathy may be increased when elafibranor is coadministered with HMG-CoA reductase inhibitors. Coadministration of elafibranor with rifampin (an enzyme inducer) may reduce the systemic exposure of elafibranor and lead to a delayed or suboptimal biochemical response. Coadministration of elafibranor with bile acid binding sequestrants (Sect. 1) may reduce the absorption and systemic exposure of elafibranor and lead to reduced efficacy. Consult local prescribing information for specific recommendations [6].
Key clinical trials of elafibranor
Drug(s) | Indication | Phase | Status | Location(s) | Identifier | Company |
|---|---|---|---|---|---|---|
Elafibranor | PBC | IIa | Completed | Multinational | NCT03124108 | Genfit |
Elafibranor | PBC | III | Active, no longer recruiting | Multinational | NCT04526665; ELATIVE | Genfit, Ipsen |
Elafibranor | PBC | III | Recruiting | Multinational | NCT06016842; ELFIDENCE | Ipsen |
Elafibranor | PBC | III | Not yet recruiting | Multinational | NCT06383403; ELSPIRE | Ipsen |
Elafibranor | PBC | IV | Not yet recruiting | Multinational | NCT06447168; ELFINITY | Ipsen |
Elafibranor | PSC | II | Recruiting | Multinational | NCT05627362; ELMWOOD | Ipsen |
2.4 Therapeutic Trials
2.4.1 Phase III ELATIVE Trial
Elafibranor significantly improved biochemical indicators of cholestasis in patients with PBC participating in the randomized, double-blind, multinational, phase III ELATIVE trial (NCT04526665) [13]. The proportion of patients achieving a biochemical response at week 52, defined as an ALP level < 1.67 x upper limit of normal (ULN), with a 15% reduction from baseline, and a total bilirubin level ≤ ULN, was 51% in the elafibranor group and 4% in the placebo group (primary endpoint; p < 0.0001). The proportion of patients with normalization of ALP at week 52 was 15% with elafibranor and 0% with placebo (p = 0.0019). Among patients with moderate to severe pruritus at baseline (n = 66), the least squares mean change in score on the Worst Itch Numeric Rating Scale [WI-NRS; scores range from 0 (no itch) to 10 (worst itch imaginable)] was not significantly different between the elafibranor and placebo groups from baseline through week 52 (– 1.93 vs – 1.15) and from baseline through week 24 (– 1.60 vs – 1.26). However, the change from baseline to week 52 on the itch domain of the PBC-40 quality of life (QOL) questionnaire (scores range from 0/1 to 5, with higher scores indicating worse QOL) and the 5-D itch scale total score (scores range from 5 to 25, with higher scores indicating worse itch-related QOL) appeared to favour elafibranor over placebo [13].
ELATIVE enrolled patients aged 18–75 years with a diagnosis of PBC, an inadequate response to or unacceptable adverse events (AEs) with UDCA, an ALP level ≥1.67 x ULN and a total bilirubin level ≤ 2 x ULN [13]. Patients were randomized to receive oral elafibranor 80 mg (n = 108) or placebo (n = 53) once daily. Randomization was stratified by ALP > 3 x ULN or total bilirubin > ULN (yes or no) and a WI-NRS score of ≥ 4 (yes or no). At the end of the double-blind treatment period, patients were eligible to enter an open-label extension during which they received elafibranor for up to 5 additional years [13].
2.4.2 Phase IIa Trial
Elafibranor significantly improved markers of liver injury in patients with PBC and an incomplete response to UDCA participating in a randomized, double-blind, multicentre, phase IIa trial (NCT03124108) [12]. The relative reduction in ALP levels from baseline to week 12 (primary endpoint) was – 48.3% in the elafibranor 80 mg group and – 40.6% in the elafibranor 120 mg group, compared with a 3.2% increase in the placebo group (both p < 0.001). Significantly (p < 0.001) more elafibranor than placebo recipients achieved the composite secondary endpoint of ALP ≤ 1.67 x ULN, total bilirubin < ULN and > 15% reduction in ALP (67% with elafibranor 80 mg and 79% with elafibranor 120 mg vs 7% with placebo). Similar results were seen for the more stringent composite endpoint of ALP < 1.5 x ULN, total bilirubin < ULN and > 40% reduction in ALP (53% and 36% vs 0%; p < 0.001) [12].
This study included patients aged 18–75 years with PBC, defined as the presence of at least two of the following: ≥ 6-month history of elevated ALP levels; positive anti-mitochondrial antibody titre or positive PBC specific anti-nuclear antibodies; liver biopsy consistent with PBC [12]. All patients had been treated with UDCA for ≥ 12 months (on a stable dose for ≥ 6 months) and had ALP levels ≥ 1.67 x ULN (ULN = 104 U/L for females and 129 U/L for males). Patients were randomized to receive elafibranor 80 mg (n = 15), elafibranor 120 mg (n = 15) or placebo (n = 15) once daily for 12 weeks. All patients continued to receive UDCA during the study [12].
2.5 Adverse Events
Elafibranor was generally well tolerated in patients with PBC [13]. In the phase III ELATIVE trial, treatment-related adverse events (AEs) occurred in 39% of patients receiving elafibranor and 40% of patients receiving placebo. The most common treatment-emergent AEs (TEAEs) occurring in ≥ 10% of patients in either treatment group were COVID-19 (29% with elafibranor vs 38% with placebo), pruritus (20% vs 26%), abnormal weight gain (19% vs 19%), abdominal pain (11% vs 6%), diarrhoea (11% vs 9%), nausea (11% vs 6%), urinary tract infection (11% vs 19%), vomiting (11% vs 2%), fatigue (9% vs 13%), headache (8% vs 11%) and back pain (4% vs 11%). Most TEAEs were of mild or moderate severity; severe AEs occurred in 11% of patients in both treatment groups. TEAEs led to discontinuation of treatment in 10% of elafibranor recipients and 9% of placebo recipients. Fatal AEs occurred in two elafibranor recipients, neither of which was considered to be treatment-related [13].
Elafibranor has been associated with cases of myalgia, myopathy and rhabdomyolysis [6]. Elevated creatine phosphokinase (CPK) levels and muscle injury were more common with elafibranor than with placebo in ELATIVE [13]. Four elafibranor recipients (vs no placebo recipients) discontinued treatment due to CPK levels > 5 x ULN with or without associated symptoms or > 3 x ULN with symptoms; of these patients, two had myalgia and one had rhabdomyolysis [13].
There have been reports of drug-induced liver injury with elafibranor [6]. In ELATIVE, one elafibranor recipient and two placebo recipients had elevated aminotransferase levels (> 3 x baseline value if baseline value was elevated or 5 x ULN if baseline value was normal), elevated bilirubin levels (> 2 x ULN) or both that met protocol-defined thresholds for consideration of potential drug-induced liver injury [13]. The event in the elafibranor group was adjudicated as a possible drug-induced liver injury and the events in the placebo group were adjudicated as probable drug-induced liver injuries. One patient in each group discontinued treatment due to elevated aminotransferase levels. All cases of elevated aminotransferase levels were reversible, with levels returning toward baseline after treatment discontinuation [13].
2.6 Ongoing Clinical Trials
In addition to the ongoing ELATIVE open-label extension (NCT04526665) described in Sect. 2.4.1, a number of other clinical trials are currently underway. The randomized, double-blind, multinational, phase III ELFIDENCE trial (NCT06016842) is currently recruiting patients and plans to evaluate the long-term efficacy of elafibranor in adults with PBC. The randomized, double-blind, phase III ELSPIRE trial (NCT06383403) will evaluate the effect of elafibranor on normalization of ALP in adults with PBC and an inadequate response or intolerance to UDCA. The prospective, multicentre, non-interventional, phase IV ELFINITY trial (NCT06447168) plans to assess the effectiveness, safety and tolerability of elafibranor in patients with PBC receiving treatment in a real-world setting.
A randomized, double-blind, multinational, phase II trial (ELMWOOD; NCT05627362) is also underway to evaluate the safety and efficacy of elafibranor in adults with PSC.
3 Current Status
Elafibranor received its first approval on 10 June 2024 (accelerated approval based on reduction of ALP) in the USA for the treatment of PBC in combination with UDCA in adults who have an inadequate response to UDCA, or as monotherapy in patients unable to tolerate UDCA [7]. Elafibranor received a positive opinion in the EU in July 2024 for the treatment of PBC [8].
Change history
06 September 2024
A Correction to this paper has been published: https://doi.org/10.1007/s40265-024-02088-3
References
Schnabl B. PPAR agonists in primary biliary cholangitis. N Engl J Med. 2024;390(9):855–8.
You H, Duan W, Li S, et al. Guidelines on the diagnosis and management of primary biliary cholangitis (2021). J Clin Transl Hepatol. 2023;11(3):736–46.
Genfit. FDA and EMA grant Genfit’s elafibranor orphan drug designation for primary biliary cholangitis (PBC) [media release]. 29 Jul 2019. http://www.genfit.com.
Genfit. Genfit announces FDA grant of breakthrough therapy designation to elafibranor for the treatment of PBC [media release]. 18 Apr 2019. http://www.genfit.com.
Genfit. Genfit upates 2024 outlook following acceptance of elafibranor filings in primary biliary cholangitis (PBC) [media release]. 8 Dec 2023. http://www.genfit.com.
Ipsen Biopharmaceuticals Inc. IQIRVO (elafibranor) tablets, for oral use: US prescribing information. 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/218860s000lbl.pdf. Accessed 4 Jul 2024.
Ipsen Pharma. Ipsen’s Iqirvo® receives US FDA accelerated approval as a first-in-class PPAR treatment for primary biliary cholangitis [media release]. 10 Jun 2024. https://www.ipsen.com/press-releases/ipsens-iqirvo-receives-u-s-fda-accelerated-approval-as-a-first-in-class-ppar-treatment-for-primary-biliary-cholangitis/.
European Medicines Agency. CHMP summary of positive opinion for Iqirvo (elafibranor). 2024. https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-iqirvo_en.pdf. Accessed 29 Jul 2024.
Terns P. Terns Pharmaceuticals announces exclusive licensing and collaboration agreement with Genfit to develop and commercialize elafibranor in the Greater China region [media release]. 24 Jun 2019. http://www.ternspharma.com.
Genfit. Genfit and Terns Pharmaceuticals Announce $228MM strategic partnership to develop and commercialize elafibranor in Greater China [media release]. 24 Jun 2019. http://www.genfit.com.
Ipsen, Genfit. Ipsen and Genfit enter into exclusive licensing agreement for elafibranor, a phase III asset evaluated in primary biliary cholangitis, as part of a long-term global partnership [media release]. 17 Dec 2021. http://www.ipsen.com.
Schattenberg JM, Pares A, Kowdley KV, et al. A randomized placebo-controlled trial of elafibranor in patients with primary biliary cholangitis and incomplete response to UDCA. J Hepatol. 2021;74(6):1344–54.
Kowdley KV, Bowlus CL, Levy C, et al. Efficacy and safety of elafibranor in primary biliary cholangitis. N Engl J Med. 2023;390(9):795–805.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of Interest
During the peer review process, the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Hannah Blair is a salaried employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to this article and are responsible for its content.
Ethical Approval, Consent to Participate, Consent to Publish, Availability of Data and Material, Code Availability
Not applicable.
Additional information
The original online version of this article was revised to update the missing correction and they have been highlighted in the correction article.
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Blair, H.A. Elafibranor: First Approval. Drugs (2024). https://doi.org/10.1007/s40265-024-02075-8
Accepted:
Published:
DOI: https://doi.org/10.1007/s40265-024-02075-8