
Abstract
Aducanumab (aducanumab-avwa; Aduhelm™) is a human, immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against aggregated soluble and insoluble forms of amyloid β. It has been co-developed by Biogen and Eisai under license from Neurimmune for the treatment of Alzheimer's disease. In June 2021, aducanumab received its first approval in the USA for the treatment of Alzheimer’s disease. According to the US FDA prescribing information, treatment should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. There are no safety or effectiveness data on initiating treatment at earlier or later stages of the disease than were studied. Aducanumab is under regulatory review in Japan and in Europe. Its long-term safety and tolerability is being evaluated in a multinational phase 3b clinical study in patients with early Alzheimer’s disease (mild cognitive impairment and mild Alzheimer’s disease). This article summarizes the milestones in the development of aducanumab leading to this first approval for Alzheimer’s disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Digital Features for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.14907999. |
A fully human IgG1 recombinant monoclonal antibody that has been co-developed by Biogen and Eisai under license from Neurimmune for the treatment of Alzheimer's disease |
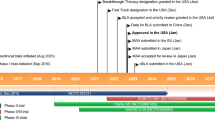
Received its first approval on 7 June 2021 in the USA |
Approved for the treatment of Alzheimer’s disease. Treatment should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. |
1 Introduction
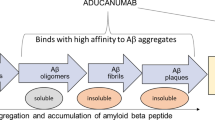
Alzheimer’s disease is a progressive neurodegenerative disorder that accounts for approximately 64% of cases of dementia [1]. In Alzheimer’s disease, the normal soluble amyloid β peptide is converted into oligomeric amyloid β, which is thought to be the most toxic, or converted into fibrillar amyloid β, which is deposited as amyloid plaques and congophilic angiopathy [2]. In addition, abnormally phosphorylated tau accumulates as soluble toxic oligomers and neurofibrillary tangles [2]. According to the ‘amyloid cascade hypothesis’, the main driver of pathogenesis in Alzheimer’s disease is the accumulation of amyloid β because of an imbalance between amyloid β production and clearance in the brain [3]. Preclinical studies in Alzheimer’s disease showed that anti-amyloid β antibodies inhibit amyloid β peptide fibrillization, disaggregate pre-formed fibrils and thus, prevent cell culture based neurotoxicity [2, 4].
Aducanumab (aducanumab-avwa; Aduhelm™) is one such amyloid β-directed antibody that has been co-developed by Biogen and Eisai under license from Neurimmune for the treatment of Alzheimer’s disease. Aducanumab is a human, immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against aggregated soluble and insoluble forms of amyloid β [5]. In patients with Alzheimer’s disease, aducanumab reduces brain amyloid β in a dose- and time-dependent manner [6]. On 7 June 2021 [7], aducanumab received its first approval in the USA for the treatment of Alzheimer’s disease [5]. According to the US FDA prescribing information, treatment should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. There are no safety or effectiveness data on initiating treatment at earlier or later stages of the disease than were studied. This indication is approved under accelerated approval based on reduction in amyloid β plaques observed in patients treated with aducanumab. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trial(s). After initial titration, the recommended dosage of aducanumab is 10 mg/kg. Aducanumab is administered as an intravenous infusion over approximately 1 h every 4 weeks and at least 21 days apart [5]. Aducanumab is under regulatory review in Japan and in Europe.

Key milestones in the development of aducanumab for the treatment of Alzheimer’s disease. EMA European Medicines Agency, MAA marketing authorization application
1.1 Company Agreements
In November 2007, Biogen Idec and Neurimmune Therapeutics AG announced that they had entered into an agreement for the worldwide development and commercialization of novel, fully human antibodies for the treatment of Alzheimer's disease [8]. The alliance was to focus on the development of antibodies that bind to amyloid β. Under the terms of the agreement, Neurimmune was to conduct research to identify potential therapeutic antibodies using the company's Reverse Translational Medicine platform, and Biogen Idec was responsible for the development and commercialization of all products [8]. In December 2010, Biogen Idec and Neurimmune Holding AG announced that Biogen Idec had acquired a subsidiary of Neurimmune, which included the world-wide rights to three pre-clinical immunotherapy programs [9]. These programs focused on the discovery and development of novel human antibodies targeting three central nervous system targets: alpha-synuclein, tau and TDP 43 [9].
In March 2014, Eisai and Biogen entered into a collaboration to develop and commercialise Eisai's drug candidates for the treatment of Alzheimer's disease, and provided Eisai with an option to jointly develop and commercialize two of Biogen Idec’s candidates for Alzheimer’s disease, the anti-amyloid β antibody aducanumab and an anti-tau monoclonal antibody [10]. In October 2017, Eisai and Biogen announced that they had expanded their existing agreement to jointly develop and commercialize investigational Alzheimer’s disease treatments [11]. Under the terms of the agreement, Eisai had exercised its option to co-develop and co-promote Biogen’s investigational anti-amyloid β antibody aducanumab. Biogen was to continue to lead the phase 3 development of aducanumab and remained solely responsible for all development costs until April 2018 [11].
2 Scientific Summary
2.1 Pharmacodynamics
Aducanumab was derived from an Alzheimer’s disease patient with an unusual stable clinical course [12]. It was developed by screening of libraries of human memory B cells for reactivity against aggregated amyloid β, leading to the molecular cloning, sequencing and recombinant expression of aducanumab [3, 12]. Aducanumab binds to the N terminus of amyloid β in an extended conformation [13]. Computer modelling suggests that aducanumab interacts weakly with the amyloid β monomer [13]; in an in vitro study, aducanumab was > 10,000-fold selective for aggregated over monomeric amyloid β [6]. Histological staining of autopsy tissue from patients with Alzheimer’s disease confirmed the binding of aducanumab to amyloid β fibrils [6].
A preclinical study in a transgenic mouse model of Alzheimer’s disease showed that aducanumab entered the brain, bound to parenchymal amyloid β and reduced soluble and insoluble amyloid β in a time- and dose-dependent manner [6]. Another study showed that chronic systemic treatment with aducanumab inhibited amyloid β toxicity and increased phagocytosis and cell viability in a transgenic mouse model, suggesting a beneficial effect of aducanumab on the proteome of senile plaques and closely surrounding tissue [14]. Aducanumab was also shown to restore calcium homeostasis disrupted by amyloid β in a transgenic model of Alzheimer’s disease [15].
In substudies of the pivotal phase 3 EMERGE (NCT02484547) and ENGAGE (NCT02477800) studies and in the phase 1 PRIME study in patients with early Alzheimer’s disease (mild cognitive impairment and mild Alzheimer’s disease) (NCT01677572), aducanumab reduced amyloid β plaque levels in a dose- and time-dependent manner [5, 6, 16]. In the EMERGE and ENGAGE substudies, both high- and low-dose aducanumab significantly (p < 0.0001) reduced amyloid β plaque levels in the brain at weeks 26 and 78 [5, 16]. In long-term extensions of EMERGE and ENGAGE, a continued decrease in brain amyloid β plaque levels was observed at week 132 in patients initially randomized to aducanumab [5]. Likewise, in the PRIME study, amyloid β plaque levels were significantly reduced with aducanumab doses of 3, 6 and 10 mg/kg at week 26 and with all aducanumab doses at week 54 (all p < 0.05) [5]. Through week 222 in a long-term extension of PRIME, amyloid β plaque levels in the brain continued to decline in a time- and dose-dependent manner [5].
Statistically significant reductions in markers of tau pathophysiology [cerebrospinal fluid (CSF) phosphorylated-tau (p-tau)] and neurodegeneration [CSF total-tau (t-tau)] were observed with high- and low-dose aducanumab at week 78 in a substudy of EMERGE (all p < 0.05). However, the reductions in p- and t-tau in an ENGAGE substudy were not statistically significant [5].
Substudies of EMERGE and ENGAGE also evaluated the effect of aducanumab on neurofibrillary tangles composed of tau protein using positron emission tomography (PET) imaging (tau PET; another marker of tau pathophysiology) [5]. The PET signal was quantified using the Standard Uptake Value Ratio (SUVR) method to estimate levels of tau in brain regions expected to be affected by Alzheimer’s disease pathology (medial temporal, temporal, frontal, cingulate, parietal and occipital cortices) versus levels in a brain region expected to be spared of such pathology (cerebellum). In pooled data from 37 patients with longitudinal follow-up in the two substudies, the adjusted mean changes from baseline in tau PET SUVR favoured high-dose aducanumab over placebo in the medial temporal (p < 0.001), temporal (p < 0.05) and frontal (p < 0.05) brain regions. No statistically significant between-group differences were observed for the cingulate, parietal or occipital cortices [5].
In pooled data from EMERGE and ENGAGE, up to 0.6% (15/2689) of patients receiving aducanumab once monthly for up to 41 months developed anti-aducanumab antibodies [5]. Because of limited data, no definitive conclusions can be made regarding a potential effect of neutralizing activity of anti-aducanumab antibodies on pharmacokinetics, safety or efficacy of aducanumab.
2.2 Pharmacokinetics
The pharmacokinetics of aducanumab were based on data from 2961 patients with Alzheimer’s disease who received single- or multiple-dose aducanumab [5]. Steady-state concentrations of aducanumab were reached by week 16 after repeat administration of aducanumab every 4 weeks. Systemic accumulation of aducanumab after repeat dosing was 1.7-fold [5]. A dose proportional increase in aducanumab peak concentration, trough concentration and area under the plasma concentration-time curve at steady state was observed over a dose range of 1–10 mg/kg every 4 weeks [3, 5]. At steady state, the mean volume of distribution of aducanumab was 9.63 L. Aducanumab, like endogenous IgG, is expected to be degraded into small peptides and amino acids via catabolic pathways. Aducanumab clearance was 0.0159 L/h and the terminal half-life was 24.8 days [5].
Bodyweight, age, sex and race did not affect the pharmacokinetics of aducanumab to a clinically significant extent [5]. No studies have assessed the effect of renal or hepatic impairment on the pharmacokinetics of aducanumab. However, aducanumab is not expected to undergo renal elimination or metabolism by hepatic enzymes [5].
Features and properties of aducanumab
Alternative names | Aducanumab—Biogen; aducanumab-avwa; Aduhelm; Anti-beta amyloid monoclonal antibody—Biogen Idec; BART; BIIB 037; NI-10 |
Class | Antibodies; antidementias; monoclonal antibodies |
Mechanism of action | Reduces amyloid β plaques in the brain |
Route of administration | Intravenous |
Pharmacodynamics | > 10,000-fold selective for aggregated over monomeric amyloid β in vitro |
Bound to amyloid β fibrils in autopsy tissue from patients with Alzheimer’s disease | |
Reduced amyloid β plaque levels in a dose- and time-dependent manner in Alzheimer’s disease patients | |
Pharmacokinetics | Cmax, Cmin and AUCss increased dose-proportionally over a dose-range of 1–10 mg/kg every 4 weeks |
Mean volume of distribution at steady state 9.63 L | |
Clearance 0.0159 L/h; terminal half-life 24.8 days | |
Most frequent adverse reactions | ARIA-E, headache, ARIA-H microhaemorrhage, ARIA-H superficial siderosis, fall, diarrhoea, confusion/delirium/altered mental status/disorientation |
ATC codes | |
WHO ATC code | N06D (anti-dementia drugs) |
EphMRA ATC code | N7D9 (all other anti-Alzheimer products) |
Chemical name | Immunoglobulin G1-kappa, anti-[Homo sapiens amyloid beta (Abeta, Aβ) peptide], Homo sapiens monoclonal antibody |
2.3 Therapeutic Trials
2.3.1 Phase 3 Studies
Two identically designed randomized, double-blind, placebo-controlled, multicentre studies EMERGE (NCT02484547) and ENGAGE (NCT02477800) assessed the efficacy of intravenous aducanumab in patients with early Alzheimer’s disease [5, 16]. Eligible patients had confirmed presence of amyloid pathology and mild cognitive impairment or mild dementia, consistent with Stage 3 or Stage 4 Alzheimer’s disease, stratified to include 80% Stage 3 patients and 20% Stage 4 patients [5]. Key inclusion criteria included a positive amyloid PET scan, Clinical Dementia Rating (CDR) global score of 0.5, a Repeatable Battery for Assessment of Neuropsychological Status (RBANS) delayed memory index score ≤ 85, and a Mini-Mental State Examination (MMSE) score of 24–30 [5, 17]. Patients were randomized to receive a target dose of aducanumab 3 mg/kg for ApoE ε4 carriers or 6 mg/kg for noncarriers (referred to as the low-dose group), aducanumab 10 mg/kg [high-dose group (ApoE ε4 carriers were initially titrated to a 6 mg/kg dose; dosage was later adjusted to 10 mg/kg)], or placebo every 4 weeks for 18 months [5]. The double-blind period was followed by an optional, dose-blind, long-term extension period. Both studies included an initial titration period of ≤ 6 months [5].
The primary endpoint was the change from baseline in the Clinical Dementia Rating-Sum of Boxes (CDR-SB) at week 78 [17]. The effect of aducanumab on amyloid β plaque levels in the brain was evaluated in substudies of EMERGE and ENGAGE using PET imaging [5]. The PET signal was quantified using the SUVR method to estimate brain levels of amyloid β plaque in composites of brain areas expected to be widely affected by Alzheimer’s disease pathology (frontal, parietal, lateral temporal, sensorimotor, and anterior and posterior cingulate cortices) versus levels in a brain region expected to be spared of such pathology (cerebellum). The SUVR was also expressed on the Centiloid scale [5].
Both studies were halted in March 2019 based on the results of a prespecified interim analyses for futility, which were conducted, per protocol, after approximately 50% of the participants had the opportunity to complete Week 78 [16, 18]. Following the futility analysis, additional blinded data from these studies became available, which were analysed after final database lock according to the prespecified statistical analysis plan [16, 19].
High-dose aducanumab (10 mg/kg) reduced clinical decline in patients with Alzheimer’s disease who were participating in the EMERGE study (NCT02484547) [16, 18]. Eligible patients (n = 1638; mean age 71 years) were randomized 1:1:1 to receive low-dose or high-dose aducanumab or placebo [intent-to-treat (ITT) n = 543, 547 and 548 in the respective groups] [5, 16]. In the ITT population at week 78, there was a significantly greater decrease from baseline in CDR-SB scores (primary endpoint) with high-dose aducanumab than with placebo [difference from placebo − 0.39 (− 22%); p = 0.012] [5, 16]. In the subgroup of patients who received at least 14 doses of aducanumab 10 mg/kg, the weighted mean difference in the decrease from baseline in CDR-SB scores versus placebo was − 23% [16, 19]. High-dose aducanumab relative to placebo also significantly reduced the MMSE [0.6 (− 18%); p = 0.0493], Alzheimer’s Disease Assessment Scale-Cognitive Subscale [ADAS-Cog 13; − 1.4 (− 27%); p = 0.0097] and Alzheimer’s Disease Cooperative Study – Activities of Daily Living Inventory (Mild Cognitive Impairment version) [ADCS-ADL-MCI; 1.7 (− 40%); p = 0.0006] and Neuropsychiatric Inventory [NPI-10; − 1.3 (− 87%); p = 0.0215] scores from baseline to week 78 [5, 16]. The low-dose aducanumab group did not differ significantly from placebo in terms of the decline in CDR-SB scores [difference from placebo − 0.26 (− 15%); p = 0.0901] and no significant differences were seen between the low-dose aducanumab and placebo groups for the change from baseline in the MMSE, ADAS-Cog 13, ADA-ADL-MCI and NPI-10 scores [16].
Amyloid PET in a subset of patients (n = 488) showed a dose- and time-dependent reduction in amyloid β pathology with high- and low-dose aducanumab at weeks 26 and 78 (all p < 0.0001) [16] In the high-dose group at week 78, the difference from placebo in the mean change from baseline in amyloid β PET composite SUVR was − 0.278 and in PET Centiloid was − 64.2 (both p < 0.0001) [5]. Additionally, in a subset of patients with both baseline and post baseline CSF assessments (n = 78), CSF biomarkers of Alzheimer’s disease, p-tau and t-tau, were significantly reduced both with high-dose (both p < 0.01) and low-dose (both p < 0.05) aducanumab relative to placebo [16]. In the high-dose group at week 78, the difference from placebo in the mean change from baseline in p-tau was − 22.44 pg/mL and in t-tau was − 112.05 pg/mL (both p < 0.01) [5].
The ENGAGE study (NCT02477800) did not meet its primary endpoint [16]. Eligible patients (n = 1647; mean age 71 years) were randomized 1:1:1 to receive low-dose or high-dose aducanumab or placebo [intent-to-treat (ITT) n = 547, 555 and 545 in the respective groups] [5, 16]. At week 78, there was no significant difference between the high- [difference from placebo + 0.03 (+ 2%)] or low-dose [difference from placebo − 0.18 (− 12%)] aducanumab group and placebo in terms of the change from baseline in CDR-SB scores (primary endpoint) [5, 16]. In an analysis of data from the subgroup of patients (n = 298) in ENGAGE who received at least 14 doses of aducanumab 10 mg/kg, the weighted mean difference in the decrease from baseline in CDR-SB scores versus placebo was − 23%, consistent with the outcome seen in a similar patient subgroup in the EMERGE study [16, 19]. There were no significant differences between the high- and low-dose aducanumab groups and placebo for the secondary endpoints of change from baseline in MMSE, ADAS-Cog 13 and ADCS-ADL-MCI scores and for the tertiary endpoint of change from baseline in the NPI-10 score [5, 16].
As in the EMERGE study, in an amyloid β PET substudy of ENGAGE (n = 585), a dose- and time-dependent reduction in amyloid β pathology was observed with high- and low-dose aducanumab at weeks 26 and 78 (all p < 0.0001; Sect. 2.1). In the high-dose group at week 78, the difference from placebo in the mean change from baseline in amyloid β PET composite SUVR was − 0.232 and in PET Centiloid was − 53.5 (both p < 0.0001) [5, 16]. However, in terms of CSF biomarkers of Alzheimer’s disease assessed in a subset of patients (n = 53), no significant differences between the high- or low-dose aducanumab and placebo groups were observed for p-tau or t-tau [5, 16]. In the high-dose group at week 78, the difference from placebo in the mean change from baseline in p-tau was − 10.95 pg/mL and in t-tau was − 69.25 pg/mL [5].
2.3.2 PRIME Phase 1 Study
Aducanumab reduced brain amyloid β plaques in patients (mean age 73 years) with prodromal or mild Alzheimer’s disease and positive amyloid β PET scans who were participating in the 12-month, randomized, double-blind, placebo-controlled phase 1b PRIME study (NCT01677572) and its 48-month, optional dose-blinded, long-term extension [5, 6, 20]. In the placebo-controlled period of PRIME, patients received monthly fixed-dose aducanumab 1 mg/kg (n = 31), 3 (n = 32), 6 mg/kg (n = 30) or 10 mg/kg (n = 32), aducanumab titrated over 44 weeks to 10 mg/kg (n = 23), or placebo (n = 48) for 12 months [5, 20]. The mean amyloid β PET SUVR composite and PET Centiloid scores were significantly reduced from baseline with aducanumab 3 mg/kg, 6 mg/kg and 10 mg/kg at week 26 (all p < 0.01) and with all aducanumab doses at week 54 (all p < 0.05) [5]. In the aducanumab 10 mg/kg fixed-dose group at week 54, the difference from placebo in the mean change from baseline in amyloid β PET composite SUVR was − 0.277 and in PET Centiloid was − 61.1 (both p < 0.0001) [5]. In addition, a dose-dependent (p < 0.05) slowing of clinical progression was observed with aducanumab relative to placebo, as assessed by the change from baseline in the CDR-SB scores at week 54 (significant slowing in the aducanumab 10 mg/kg group; p < 0.05) and in MMSE scores at week 52 (significant slowing in the aducanumab 3 and 10 mg/kg groups; p < 0.05) [6]. In the aducanumab 10 mg/kg fixed-dose group at week 52, the difference from placebo in the mean change from baseline in CDR-SB score was − 1.26 (95% CI − 2.356 to − 0.163) and in MMSE score was 1.9 (95% 0.06–3.75).
Benefit with aducanumab was sustained in patients who continued aducanumab therapy in the long-term extension of PRIME, with continued decrease in amyloid β plaque levels and continued benefit in terms if CDR–SB and MMSE scores (quantitative data not available) [20].
Key clinical trials of aducanumab in Alzheimer’s disease sponsored by Biogen
Drug(s) | Phase | Status | Location(s) | Identifier |
|---|---|---|---|---|
Aducanumab, placebo | 3 | Terminated | Multinational | NCT02484547; EMERGE |
Aducanumab, placebo | 3 | Terminated | Multinational | NCT02477800; ENGAGE |
Aducanumab | 3b | Enrolling by invitation | Multinational | NCT04241068; EMBARK |
Aducanumab, placebo | 2 | Terminated | USA | NCT03639987; EVOLVE |
Aducanumab, placebo | 1 | Terminated | USA | NCT01677572; PRIME |
Aducanumab, placebo | 1 | Completed | USA | NCT01397539 |
Aducanumab, placebo | 1 | Completed | Japan | NCT02434718; PROPEL |
Aducanumab | 1 | Completed | USA | NCT02782975 |
2.4 Adverse Events
Intravenous aducanumab 10 mg/kg was generally well tolerated in patients with Alzheimer’s disease in pooled data from the phase 3 EMERGE (NCT02484547) and ENGAGE (NCT02477800) studies and their extensions [5]. Data were pooled from 1105 patients who received aducanumab 10 mg/kg (834 who received ≥ 1 dose once monthly for at least 6 months, 551 for ≥ 12 months and 309 for 18 months) and 1087 patients who received placebo in the placebo-controlled periods. The most common adverse reactions occurring in ≥ 2% of aducanumab recipients and at an incidence ≥ 2% higher than in placebo recipients were amyloid-related imaging abnormalities-oedema (ARIA-E; 35% vs 3%), headache (21% vs 16%), amyloid-related imaging abnormalities hemosiderin deposition (ARIA-H) microhaemorrhage (19% vs 7%), ARIA-H superficial siderosis (15% vs 2%), fall (15% vs 12%), diarrhoea (9% vs 7%) and confusion/delirium/altered mental status/disorientation (8% vs 4%). Adverse reactions led to treatment discontinuation in 5% (66 of 1386) patients in the placebo-controlled and extension periods, with ARIA-H superficial siderosis being the most common adverse reaction leading to treatment withdrawal [5].
2.5 Ongoing Clinical Trials
The 24-month open-label, single-arm, phase 3b EMBARK study (NCT04241068) is enrolling an estimated 2400 patients with early Alzheimer’s disease (mild cognitive impairment and mild Alzheimer’s disease) who had previously participated in aducanumab studies, including the EMERGE, ENGAGE, long-term extension of PRIME and the EVOLVE (NCT03639987) studies [21]. The primary objective of the study is to evaluate the long-term safety and tolerability of a monthly 10 mg/kg dose of aducanumab after a gap period imposed by the discontinuation of feeder studies [21].
Change history
15 September 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40265-021-01590-2
References
Conway ME. Alzheimer’s disease: targeting the glutamatergic system. Biogerontology. 2020;21(3):257–74.
Wisniewski T, Goni F. Immunotherapy for Alzheimer’s disease. Biochem Pharmacol. 2014;88(4):499–507.
Ferrero J, Williams L, Stella H, et al. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimers Dement. 2016;2(3):169–76.
Wisniewski T, Goni F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron. 2015;85(6):1162–76.
Biogen Inc. ADUHELM™ (aducanumab-avwa): US prescribing information 2021. https://www.accessdata.fda.gov/scripts/cder/daf/. Accessed 5 Jul 2021.
Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537(7618):50–6.
US Food & Drug Administration. FDA grants accelerated approval for Alzheimer’s drug [media release]. 7 Jun 2021. https://www.fda.gov/.
Biogen Idec, Neurimmune Therapeutics AG. Biogen Idec and Neurimmune Therapeutics announce alliance to develop treatments for Alzheimer's disease [media release]. 20 Nov 2017. http://www.biogenidec.com.
Biogen Idec, Neurimmune Holding AG. Biogen Idec and Neurimmune announce agreement on three neurodegenerative disease programs [media release]. 21 Dec 2010. http://www.biogenidec.com.
Eisai Co Ltd, Biogen Idec. Eisai and Biogen Idec enter collaboration to develop and commercialize Alzheimers disease treatments [media release]. 5 Mar 2014. http://www.eisai.com.
Eisai Co Ltd, Biogen Inc. Biogen and Eisai expand existing collaboration agreement to develop and commercialize investigational Alzheimer's disease treatments including phase 3 aducanumab [media release]. 23 Oct 2017. http://www.eisai.com.
Dunstan R, Bussiere T, Rhodes K, et al. Molecular characterization and preclinical efficacy [abstract no. P2–449]. Alzheimers Dement. 2011;7(4 Suppl):S457.
Arndt JW, Qian F, Smith BA, et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci Rep. 2018;8(1):6412.
Bastrup J, Hansen KH, Poulsen TBG, et al. Anti-Aβ antibody aducanumab regulates the proteome of senile plaques and closely surrounding tissue in a transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis. 2021;79(1):249–65.
Kastanenka KV, Bussiere T, Shakerdge N, et al. Immunotherapy with aducanumab restores calcium homeostasis in Tg2576 mice. J Neurosci. 2016;36(50):12549–58.
Haeberlein SB, Salloway S, Aisen P, et al. Evaluation of aducanumab efficacy in early Alzheimer’s disease [oral presentation]. AD/PD 2021, Virtual Conference. 2021.
Von Hehn C, Von Rosenstiel P, Tian Y, et al. Baseline characteristics from ENGAGE and EMERGE: two phase 3 studies to evaluate aducanumab in patients with early Alzheimer's disease [abstract no. P4.1-00]. Neurology. 2019;92(15 Suppl).
Haeberlein SB, von Hehn C, Tian Y, et al. EMERGE and ENGAGE topline results: phase 3 studies of aducanumab in early Alzheimer's disease [abstract no. SO3-02-03 + oral presentation]. Alzheimers Dement. 2020;16(Suppl 9).
Cummings J, Aisen P, Lemere C, et al. Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimers Res Ther. 2021;13(1):98.
Castrillo-Viguera C, Budd Haeberlein S, Von Rosenstiel P, et al. Interim analyses of fixed-dose and titration cohorts from PRIME: a randomized, double-blind, placebo-controlled phase 1b study of aducanumab [abstract no. S9.006]. Neurology. 2019;92(15 Suppl).
ClinicalTrials.gov. A Study to Evaluate Safety and Tolerability of Aducanumab in Participants With Alzheimer's Disease Who Had Previously Participated in the Aducanumab Studies 221AD103, 221AD301, 221AD302 and 221AD205 2020. https://clinicaltrials.gov/ct2/show/NCT04241068?term=aducanumab&draw=2&rank=2. Accessed 15 Jul 2021.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and conflict of interest
During the peer review process Biogen, the manufacturer of the agent under review, was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Sohita Dhillon is a contracted employee of Adis International Ltd/Springer Nature and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
The original article has been updated: Due to ESM update.
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dhillon, S. Aducanumab: First Approval. Drugs 81, 1437–1443 (2021). https://doi.org/10.1007/s40265-021-01569-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01569-z