
Abstract
Onset of involuntary movement patterns of the face, body and limbs are known as dyskinesia. They mostly appear in association with long-term levodopa (l-dopa) therapy in patients with Parkinson’s disease. Consequences include patient distress, caregiver embarrassment and reduced quality of life. A severe intensity of this motor complication may result in troublesome disability; however, patients typically prefer motor behaviour with slight, non-troublesome dyskinesia to ‘OFF’ states. Pharmacotherapy of dyskinesia is complex. Continuous nigrostriatal postsynaptic dopaminergic receptor stimulation may delay onset of l-dopa-associated dyskinesia, while non-physiological, ‘pulsatile’ receptor stimulation facilitates appearance of dyskinesia. In the past, there have been many clinical trial failures with compounds that were effective in animal models of dyskinesia. Only the N-methyl-d-aspartate antagonist amantadine has shown moderate antidyskinetic effects in small well-designed clinical studies. Amantadine is an old antiviral compound, which moderately improves impaired motor behaviour. Recently, there has been a resurgence of its use due to the US Food and Drug Administration approval of an extended-release (ER) amantadine formulation for treatment of l-dopa-induced dyskinesia. This pharmacokinetic innovation improved dyskinesia and ‘OFF’ states in pivotal trials, with a once-daily oral application in the evening. Amantadine ER provides higher and more continuous amantadine plasma bioavailability than conventional immediate-release formulations, which require administration up to three times daily.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Extended-release (ER) amantadine formulations ameliorate levodopa-induced dyskinesia. |
ER formulations provide higher amantadine plasma concentrations than immediate-release formulations, and are administered once a day at bedtime. |
ER amantadine application is simpler than deep brain stimulation or an infusion regimen. |
1 Introduction
Parkinson’s disease (PD) is the second most frequent chronic neurodegenerative disease worldwide, with an incidence range from 8 to 18 per 100,000 individuals [1]. One estimate is that the number of PD cases will double by 2030 [2]. PD diagnosis is rare under the age of 50 years, but the incidence of PD considerably rises beyond the age of 60 years.
The term ‘PD’ describes a disease entity. It consists of various heterogeneous subtypes that closely resemble one another [3, 4]. Motor and non-motor symptoms and progression considerably differ in each affected individual. Diagnosis of PD is mostly made with the initially transient, then permanent, manifestation of ‘cardinal’ motor symptoms: rigidity, akinesia and resting tremor. Symptoms occur after the death of approximately 50–60% of nigrostriatal dopaminergic neurons. Unspecific non-motor features, such as depression or apathy, often precede the onset of impaired motor behaviour [3, 4].
1.1 Treatment with Levodopa (l-Dopa) Induces Motor Complications
Levodopa (l-dopa) administration, combined with a dopa decarboxylase inhibitor (DDI), is the most efficacious and best tolerated dopamine-substituting drug. Sooner or later it is necessary to put PD patients on l-dopa to ameliorate closely related motor and non-motor symptoms [5].
The efficacy of l-dopa intake depends on peripheral gastrointestinal absorption, the closely related plasma appearance, its brain delivery via blood–brain barrier transporting systems and its neuronal conversion to dopamine by dopa decarboxylase in presynaptic dopaminergic neurons. All these components of oral l-dopa application are considerably influenced by the short plasma half-life of l-dopa, which lasts approximately 30–45 min [6].
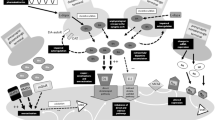
This peripheral pharmacokinetic behaviour is centrally translated into a non-physiological, ‘pulsatile’ stimulation of postsynaptic dopamine receptors. This pattern generates gene and receptor dysfunction, alters more downstream not yet well-defined neuronal activity and contributes to imbalances of neurotransmitters, such as N-methyl-d-aspartate (NMDA) or serotonin (5-HT) [7,8,9,10].
To a certain extent, presynaptic striatal dopaminergic neurons may still compensate for these fluctuations of l-dopa and related dopamine levels via a presynaptic-located autoreceptor, which regulates presynaptic dopamine synthesis and dopamine release to the synaptic cleft. However, PD progression with emerging loss of presynaptic dopaminergic neurons reduces this existing compensatory capacity.
Presynaptic synthesis, storage and regulation of dopamine release for adequate striatal postsynaptic dopamine receptor activation increasingly depends on exogenously supplemented dopaminergic agents. l-Dopa itself is also more and more converted to dopamine in 5-HT neurons, which are not controlled by dopamine-sensitive autoreceptors. Thus, use of this 5-HT pathway contributes to more pronounced peaks and troughs of dopamine concentrations in the nigrostriatal and mesolimbic system. As a result, fluctuations of motor and non-motor behaviours appear [11,12,13,14,15]. These fluctuations are one of the most relevant adverse effects of long-term l-dopa/DDI treatment.
1.2 Benefit of l-Dopa on Motor Behaviour Changes During Parkinson’s Disease (PD) Progression
l-Dopa dosing and response mirrors progression of PD to a certain extent (for review, see Müller and Möhr [14]). Early PD patients have longer-lasting beneficial l-dopa effects and only require l-dopa intake two or three times daily. At this stage, presynaptic striatal dopaminergic neurons may compensate for fluctuations of l-dopa and dopamine concentrations. However, as PD progresses, with increasing loss of presynaptic dopaminergic neurons, the compensatory capacity of fluctuating l-dopa levels goes down due to neuronal death of presynaptic dopamine-synthesising neurons.
The duration of clinical benefit, the so-called ‘motor response’ derived from each oral l-dopa dose, shortens due to the declining therapeutic window to obtain relief from motor symptoms by l-dopa. At this stage, patients initially experience return of motor impairment before the next l-dopa intake. This so-called ‘wearing-off’ phenomenon is predictable because it is related to the previous drug intake [13,14,15].
Further advance of PD requires higher l-dopa dosing; however, the resulting motor complications become unpredictable and more intense and the relationship to drug intake gets progressively lost.
1.3 Hypotheses on Dyskinesia Generation in PD Patients
In addition to the aforementioned considerations, one also assumes functional disturbances in the basal ganglia. Loss of nigral dopaminergic neurons induces abnormalities in the connectivity between the motor cortex and striatum. Another theory suggests that loss of nigrostriatal dopaminergic neurons induces postsynaptic plastic changes with supersensitivity of postsynaptic dopaminergic neurons [16]. There are also suggestions that several other non-dopaminergic systems, including glutamatergic, γ-aminobutyric acid-ergic, serotonergic, histaminergic, adenosine and cannabinoid receptors, play an important role in the development of l-dopa-induced dyskinesia (for review, see Fabbrini et al. [17] and Cerri et al. [18]).
However, the pathogenesis of l-dopa-induced dyskinesia is not well-understood. It is known that, in contrast to PD patients, humans without PD do not develop dyskinesia during long-term treatment with l-dopa. PD patients with young age at disease onset, and thus longer l-dopa lifetime exposure or high l-dopa dosages, are particularly at risk for developing dyskinesia. This was shown, for example, in the l-dopa 600 mg arm in the l-DOPA study or in the STalevo Reduction in Dyskinesia Evaluation in Parkinson's Disease (STRIDE-PD) study, in contrast to the LEvodopa in EArly Parkinson's disease (LEAP) study with its exposure to lower l-dopa doses [19,20,21].
1.4 Clinical Phenomenology of Dyskinesia
Generally, motor complications are a combination of dyskinesia and ‘OFF’ states. Both depend on the pharmacokinetic behaviour of short-lived dopamine-substituting compounds, such as l-dopa. As a result, pulsatile stimulation of nigrostriatal dopaminergic receptors takes place centrally. ‘ON’-state dyskinesia mostly appears during a period with maximal relief from motor symptoms (‘peak-dose’ dyskinesia). Other kinds of dyskinesia are diphasic; they emerge soon after l-dopa intake, when the patient turns ‘ON’, or when the l-dopa response wears ‘OFF’ again.
Various dyskinesia forms exist. The most frequent movement sequences are chorea, athetosis, dystonia, stereotypy, ballismus, or a combination of these [22,23,24,25]. The intensity of dyskinesia ranges from mild to completely disabling, and it occurs in addition to motor fluctuations during both ‘ON’ and ‘OFF’ periods. Patients themselves tend to ignore mild dyskinesia symptoms, since they induce little disability; most PD patients prefer to have small intervals with dyskinesia and remain in the ‘ON’ state rather than have less dyskinesia with more time spent ‘OFF’. Conversely, severe dyskinesia may cause considerable disability in advanced PD patients, potentially inducing pain, severe speech and swallowing problems, and contributing to weight loss. Such symptoms also may become exhausting or even life-threatening due to shortness of breath because of diaphragmatic dyskinesia [24, 26]. The intensity of dyskinesia is closely associated with stress and emotion; for example, dyskinesia worsens during exposure to all kind of stressors.
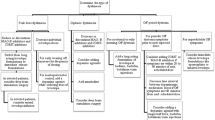
2 Current Treatment Approaches for Dyskinesia in PD Patients
2.1 Delay of Dyskinesia
It is well-known that continuous nigrostriatal postsynaptic dopamine receptor stimulation (continuous dopaminergic stimulation [CDS]) improves motor complications. Dopamine agonists have a long half-life and thereby provide CDS. This may reduce the risk of dyskinesia onset, as shown with ropinirole in a trial lasting 5 years [27].
Generally, ergoline and non-ergoline dopamine agonists directly stimulate postsynaptic dopamine receptors. They support CDS due to their long half-life (i.e. cabergoline has a half-life of 24 h) or provide long-lasting stimulation of postsynaptic dopamine receptors (i.e. lisuride with its short half-life of 2 h).
All these pharmacological approaches help to spare oral l-dopa dosing and thus reduce the consequences of intermittent l-dopa brain delivery with pulsatile stimulation of dopaminergic receptors. Careful and cautious l-dopa titration may also contribute to delayed dyskinesia onset.
2.2 Surgical Approaches
Lesion or deep brain stimulation (DBS) of the medial globus pallidus, in particular, improves dyskinesia. Both methods reduce the excitatory drive on thalamic and motor cortical nuclei [28,29,30]. However, the electric stimulation pattern of DBS also enhances the endogenous synthesis and continuous release of biogenic amines. Concomitantly, homovanillic acid concentrations increase in urine [31]. Thus, these neurochemical DBS effects hypothetically resemble CDS performed with drugs (i.e. with l-dopa or apomorphine). However, this surgical approach may have severe long-term disadvantages in the clinic. For example, DBS increases the risk of personality changes and cognitive disturbances [32].
Other surgical treatment options include continuous subcutaneous infusions of apomorphine or intestinal l-dopa gel (LCIG). Both therapies reduce ‘OFF’ time and dyskinesia [11, 33, 34]; however, they are expensive and require caregiver support for the demanding pump systems, and they are only applicable in well-selected PD patients [11, 33,34,35,36]. Accordingly, oral application of antidyskinetic agents remains the best and most easy-to-perform option for PD patients.
2.3 Approved Antidyskinetic Drugs
Reduction of the glutamatergic input on the nigrostriatal dopamine system is a proven option to reduce dyskinesia according to experimental and clinical investigations [37, 38].
One approach is pharmacological antagonism of NMDA receptors with amantadine. Amantadine is an antiviral drug approved for the treatment of dyskinesia in patients with PD receiving l-dopa [39]. Its moderate effects on motor impairment were coincidentally discovered in a female PD patient taking 200 mg daily for antiviral prophylaxis [40, 41]. Subsequently performed investigations with different amantadine formulations confirmed this initial observation (e.g. Muhlack et al. [42], Müller et al. [43, 44]).
Experimental findings describe three mechanisms of action. Amantadine modulates the dopamine system [45]. Presynaptic and postsynaptic actions at dopaminergic terminals and release of intraneuronal dopamine from extravesicular stores were shown, in addition to NMDA antagonism and mild anticholinergic effects [45,46,47].
Amantadine (1-adamantanamine) is a tricyclic amine that is mainly excreted via urine. Two immediate-release (IR) formulations have mostly been employed in the past. Amantadine hydrochloride is given orally; maximum plasma concentrations appear between 1 and 4 h postdose and the half-life is about 15 h [48]. An alternative is the salt amantadine sulphate with its oral and intravenous application route (currently available in Germany and Austria only). The conventional oral amantadine formulations are commonly titrated up to 200 mg/day [48].
A new extended-release (ER) formulation of amantadine (Gocovri™; Adamas Pharma, LLC, Emeryville, CA, USA) has also recently been approved by the US Food and Drug Administration (FDA) for the treatment of dyskinesia in PD patients on l-dopa therapy with or without concomitant dopamine-substituting medications. The recommended dosage is 274 mg once daily at bedtime, which corresponds to amantadine hydrochloride 340 mg [49].
The innovation of this new ER preparation is that oral administration of the capsule achieves high plasma drug concentrations (approximately 1500 ng/mL) throughout the day, which cannot be obtained with typical administration of amantadine IR [49].
Drawbacks of all available amantadine formulations include a certain incidence of livedo reticularis, mostly in the lower limbs, and pedal oedema. Distinctly less common, but more troublesome, are the cognitive adverse effects such as confusion, visual hallucinations and insomnia, which promptly disappear with drug discontinuation. Generally, these cognitive adverse effects are mostly reported in individuals with an underlying pre-existing cognitive dysfunction. The predominant renal amantadine excretion requires cautious use of amantadine in patients with impaired kidney function. Occasional occurrences of dry mouth and blurred vision are probably related to the mild anticholinergic properties of amantadine.
A pharmacokinetic/pharmacodynamic analysis in animal models showed that the amantadine 50% effective plasma concentration (EC50) required to significantly reduce dyskinesia is 1400 ng/mL (or 9 µM) across multiple species, from mice to non-human primates [50]. This EC50 is consistent with the known half-maximal inhibitory concentration (IC50) of amantadine for inhibition of the NMDA receptor in striatal neurons (12 µM) [50].
In small clinical studies, an IR formulation of amantadine hydrochloride improved dyskinesia. However, the long-term effect of this amantadine IR preparation is still under debate [38, 51]. The reported average benefit of amantadine IR application on dyskinesia was 4.9 months versus 1.3 months for placebo. PD patients who switched to placebo following a mean of 3.4 years on treatment with amantadine IR experienced worsening of dyskinesia within a median of 7 days [38, 51, 52].
Most PD patients show no problems during daily intake of amantadine IR doses between 81 and 161 mg. However, higher dosing, which may provide greater antidyskinetic benefit, is less well-tolerated. Usually clinicians administer amantadine IR in divided doses, even though this compound has a relatively long half-life (approximately 17 h) [50], to try to reduce the onset of adverse effects.
Randomised, placebo-controlled, double-blind pivotal trials showed the antidyskinetic efficacy of amantadine ER, i.e. in the ADS-5102 programme (Table 1). An additional clinical benefit was the concomitant reduction in ‘OFF’ times. Both pivotal phase III ADS-5102 studies on the 274 mg capsule had identical inclusion criteria within a similar study design [53,54,55]. However, only the EASE LID (ADS-5102 Extended Release Capsules for the Treatment of Levodopa Induced Dyskinesia) trial investigated the antidyskinetic effect over a longer study interval. Generally, the ER formulation had a good safety profile and was well-tolerated. Visual hallucinations were found to be the most frequent, clinically relevant adverse effect. Pooled analyses confirmed this outcome, its long-term benefit, and its safety and tolerability profile [49, 54, 56,57,58,59]. Table 1 reports essential excerpts of the three clinical pivotal amantadine ER trials. The higher dosing and modified ER delivery of amantadine ameliorated dyskinesia and ‘OFF’ times in l-dopa-treated PD patients. As already mentioned, amantadine is not only a NMDA antagonist. One may assume that the observed ‘OFF’-time reduction may also result from its previously discussed modes of action [60]. In comparison to the available generic amantadine salts, the innovation of this therapy does not result from a new mode of action but from another pharmacokinetic, and thus pharmacodynamic, behaviour.
3 Relevance for the Maintenance of PD Patients
3.1 Amantadine: A Well-Known PD Drug with Additional Properties
Generally, amantadine is looked upon as an old, well-known compound with a modest efficacy for the treatment of motor behaviour in PD patients. Amantadine is also employed in other indications, for instance it may help to improve fatigue in patients with multiple sclerosis or with traumatic brain injury [61,62,63]; however, this is mostly ‘off-label’ due to the lack of so-called ‘evidence-based medicine’ trials. Accordingly, the effects of amantadine on vigilance, attention or alertness have also been described in PD patients, but randomised controlled trials (RCTs) failed to confirm these effects [44, 64,65,66,67]. Conventional amantadine salts are normally administered in the first half of the day to prevent sleep disturbances during the night.
3.2 Advantages of Extended-Release Amantadine
The amantadine ER capsule was applied in the evening in clinical trials. One must speculate why this ER amantadine did not considerably alter sleep as a clinically relevant adverse effect in the pivotal study programme [50, 56,57,58,59]. One may assume that the more continuous amantadine delivery may be responsible, despite the higher plasma concentrations of amantadine [50]. Another advantage of the amantadine ER formulation is the once-daily administration regimen, which simplifies the sometimes rather complex drug intake scenario, particularly in advanced PD patients with their well-known adherence problems [68]. This is not a well-recognised issue in the study world, with its close patient monitoring in combination with well-selected study participants. Nevertheless, long-term acceptance and use of ER formulations of amantadine in the real world of PD patient care will show their real value.
The successful amantadine ER study programme also underlines that higher dosing with accordingly higher plasma concentrations of an applied compound may contribute to better efficacy, particularly in the treatment of chronic disorders. Regulatory authorities support dosing restrictions for safety and tolerability reasons. However these dosing limitations may prevent additional benefits in the real world, as shown in this case with antidyskinetic effects with higher amantadine plasma bioavailability.
A standardised head-to-head comparison of a higher and more frequent dosing regimen with amantadine IR versus amantadine ER administration once a day may now also be considered. However, the value of once-daily administration of this amantadine ER formulation in the real world should not be underestimated. Here, generally, drug treatment of advanced PD patients becomes more and more complex and requires an individually titrated drug combination regimen with close and continuous patient surveillance [5]. Pivotal studies have demonstrated that this novel amantadine formulation is effective against dyskinesia in PD patients and a switch from conventional amantadine to ER amantadine was efficacious and well-tolerated [54].
3.3 Further Potential Antidyskinetic Drugs
There is still an unmet need to develop further treatments against l-dopa-induced dyskinesia in PD patients. As already mentioned, dyskinesia ameliorates with low exposure or better tolerability to all kind of stressors, or even, to a lesser extent, with placebo [69]. The antidyskinetic efficacy of drugs (i.e. the atypical antipsychotic clozapine with its benzodiazepine-like metabolites, or other sleep-inducing or antidepressant compounds, such as buspirone, mirtazapine, quetiapine and topiramate) has been investigated in small trials or in case series. They report both positive and negative outcomes [70,71,72,73,74,75]. Due to the heterogeneity of dyskinesia and PD itself, currently no new, more generally valid treatment approach is likely to appear on the horizon for the treatment of PD patients in the clinic. Experimental trials in animal PD models with l-dopa-induced dyskinesia repeatedly report beneficial effects of various approaches, probably due to the standardised and uniform generation of dyskinesia. However, the subsequently performed clinical trials if performed at all, often fail to reproduce this beneficial effect. A future realistic alternative may be an easy-to-handle subcutaneous l-dopa pump application [76]. Such a device should deliver l-dopa in a similar way to an insulin pump application in diabetes mellitus, i.e. in a continuous manner adapted to the patients’ needs. Currently, subcutaneous infusion of apomorphine or enteral infusion of an l-dopa-containing gel are available; both provide continuous drug delivery to the brain [33, 77].
4 Conclusion
The causes of l-dopa-induced dyskinesia are not well-understood and probably have a multifactorial origin. Their clinical presentation is heterogeneous and, accordingly, their treatment is complex. Positive experimental outcomes with promising compounds have typically failed to translate to use in the clinic in the past. Dyskinesia therapy in PD patients includes cautious and careful titration of PD drugs or implementation of costly techniques, including DBS or continuous infusion of dopamine-substituting compounds. In contrast, daily one-time application of amantadine ER formulations is simple. It showed antidyskinetic efficacy in clinical trials, but the real value of this pharmacokinetic innovation of an old PD drug will be evaluated in the real world of maintenance of PD patients in the future.
References
de Rijk MC, Launer LJ, Berger K, Breteler MM, Dartigues JF, Baldereschi M, et al. Prevalence of Parkinson’s disease in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000;54(11 Suppl 5):S21–3.
Bach JP, Riedel O, Klotsche J, Spottke A, Dodel R, Wittchen HU. Impact of complications and comorbidities on treatment costs and health-related quality of life of patients with Parkinson’s disease. J Neurol Sci. 2012;314(1–2):41–7.
Berg D, Godau J, Seppi K, Behnke S, Liepelt-Scarfone I, Lerche S, et al. The PRIPS study: screening battery for subjects at risk for Parkinson’s disease. Eur J Neurol. 2013;20(1):102–8.
Przuntek H, Müller T, Riederer P. Diagnostic staging of Parkinson’s disease: conceptual aspects. J Neural Transm (Vienna). 2004;111(2):201–16.
Müller T, Öhm G, Eilert K, Möhr K, Rotter S, Haas T, et al. Benefit on motor and non-motor behavior in a specialized unit for Parkinson’s disease. J Neural Transm (Vienna). 2017;124(6):715–20.
Contin M, Riva R, Martinelli P, Albani F, Baruzzi A. Effect of age on the pharmacokinetics of oral levodopa in patients with Parkinson’s disease. Eur J Clin Pharmacol. 1991;41(5):463–6.
Boraud T, Bezard E, Bioulac B, Gross CE. Dopamine agonist-induced dyskinesias are correlated to both firing pattern and frequency alterations of pallidal neurones in the MPTP-treated monkey. Brain. 2001;124:546–57.
Bezard E, Brotchie JM, Gross CE. Pathophysiology of levodopa-induced dyskinesia: potential for new therapies. Nat Rev Neurosci. 2001;2(8):577–88.
Stocchi F, Olanow CW. Continuous dopaminergic stimulation in early and advanced Parkinson’s disease. Neurology. 2004;62(1 Suppl 1):S56–63.
Widnell K. Pathophysiology of motor fluctuations in Parkinson’s disease. Mov Disord. 2005;20(Suppl 11):S17–22.
Witjas T, Kaphan E, Azulay JP, Blin O, Ceccaldi M, Pouget J, et al. Nonmotor fluctuations in Parkinson’s disease: frequent and disabling. Neurology. 2002;59(3):408–13.
Melamed E, Hefti F, Wurtman RJ. Nonaminergic striatal neurons convert exogenous l-dopa to dopamine in parkinsonism. Ann Neurol. 1980;8(6):558–63.
Fahn S. The spectrum of levodopa-induced dyskinesias. Ann Neurol. 2000;47(4 Suppl 1):S2–9.
Müller T, Möhr JD. Long-term management of Parkinson’s disease using levodopa combinations. Expert Opin Pharmacother. 2018;19(9):1003–11.
Stocchi F, Vacca L, Ruggieri S, Olanow CW. Intermittent vs continuous levodopa administration in patients with advanced Parkinson disease: a clinical and pharmacokinetic study. Arch Neurol. 2005;62(6):905–10.
Bravi D, Mouradian MM, Roberts JW, Davis TL, Sohn YH, Chase TN. Wearing-off fluctuations in Parkinson’s disease: contribution of postsynaptic mechanisms. Ann Neurol. 1994;36(1):27–31.
Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goetz CG. Levodopa-induced dyskinesias. Mov Disord. 2007;22(10):1379–89.
Cerri S, Siani F, Blandini F. Investigational drugs in phase I and phase II for levodopa-induced dyskinesias. Expert Opin Investig Drugs. 2017;26(7):777–91.
Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351(24):2498–508.
Stocchi F, Rascol O, Kieburtz K, Poewe W, Jankovic J, Tolosa E, et al. Initiating levodopa/carbidopa therapy with and without entacapone in early Parkinson disease: the STRIDE-PD study. Ann Neurol. 2010;68(1):18–27.
Verschuur CVM, Suwijn SR, Boel JA, Post B, Bloem BR, van Hilten JJ, LEAP Study Group, et al. Randomized delayed-start trial of levodopa in Parkinson’s disease. N Engl J Med. 2019;380(4):315–24.
Luquin MR, Scipioni O, Vaamonde J, Gershanik O, Obeso JA. Levodopa-induced dyskinesias in Parkinson’s disease: clinical and pharmacological classification. Mov Disord. 1992;7(2):117–24.
Marconi R, Lefebvre-Caparros D, Bonnet AM, Vidailhet M, Dubois B, Agid Y. Levodopa-induced dyskinesias in Parkinson’s disease phenomenology and pathophysiology. Mov Disord. 1994;9(1):2–12.
Adler CH. Relevance of motor complications in Parkinson’s disease. Neurology. 2002;58(4 Suppl 1):S51–6.
Jankovic J. Motor fluctuations and dyskinesias in Parkinson’s disease: clinical manifestations. Mov Disord. 2005;20(Suppl 11):S11–6.
Rice JE, Antic R, Thompson PD. Disordered respiration as a levodopa-induced dyskinesia in Parkinson’s disease. Mov Disord. 2002;17(3):524–7.
Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE, et al. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med. 2000;342(20):1484–91.
Krack P, Pollak P, Limousin P, Hoffmann D, Xie J, Benazzouz A, et al. Subthalamic nucleus or internal pallidal stimulation in young onset Parkinson’s disease. Brain. 1998;121(Pt 3):451–7.
Vitek JL. Deep brain stimulation for Parkinson’s disease. A critical re-evaluation of STN versus GPi DBS. Stereotact Funct Neurosurg. 2002;78(3–4):119–31.
Vitek JL, Bakay RA, Freeman A, Evatt M, Green J, McDonald W, et al. Randomized trial of pallidotomy versus medical therapy for Parkinson’s disease. Ann Neurol. 2003;53(5):558–69.
Figee M, de Koning P, Klaassen S, Vulink N, Mantione M, van den Munckhof P, et al. Deep brain stimulation induces striatal dopamine release in obsessive-compulsive disorder. Biol Psychiatry. 2014;75(8):647–52.
Harati A, Muller T. Neuropsychological effects of deep brain stimulation for Parkinson’s disease. Surg Neurol Int. 2013;4(Suppl 6):S443–7.
Katzenschlager R, Poewe W, Rascol O, Trenkwalder C, Deuschl G, Chaudhuri KR, et al. Apomorphine subcutaneous infusion in patients with Parkinson’s disease with persistent motor fluctuations (TOLEDO): a multicentre, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2018;17(9):749–59.
Klostermann F, Jugel C, Bomelburg M, Marzinzik F, Ebersbach G, Müller T. Severe gastrointestinal complications in patients with levodopa/carbidopa intestinal gel infusion. Mov Disord. 2012;27(13):1704–5.
Nyholm D, Jansson R, Willows T, Remahl IN. Long-term 24-hour duodenal infusion of levodopa: outcome and dose requirements. Neurology. 2005;65(9):1506–7.
Stocchi F, Vacca L, De Pandis MF, Barbato L, Valente M, Ruggieri S. Subcutaneous continuous apomorphine infusion in fluctuating patients with Parkinson’s disease: long-term results. Neurol Sci. 2001;22(1):93–4.
Verhagen Metman L, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol. 1999;56(11):1383–6.
Verhagen ML, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson’s disease. Neurology. 1998;50(5):1323–6.
Davies WL, Grunert RR, Haff RF, McGahen JW, Neumayer EM, Paulshock M, et al. Antiviral activity of 1-adamantanamine (amantadine). Science. 1964;144(3620):862–3.
Schwab RS, England AC Jr, Poskanzer DC, Young RR. Amantadine in the treatment of Parkinson’s disease. JAMA. 1969;208(7):1168–70.
Schwab RS, England AC Jr. Amantadine HCL (Symmetrel) and its relation to levo-dopa in the treatment of Parkinson’s disease. Trans Am Neurol Assoc. 1969;94:85–90.
Muhlack S, Musch P, Konietzka S, Woitalla D, Przuntek H, Muller T. Impact of oral fast release amantadine on movement performance in patients with Parkinson’s disease. Pharmaceutics. 2010;2(3):313–20.
Müller T, Kuhn W, Quack G, Przuntek H. Intravenous application of amantadine and antiparkinsonian efficacy in Parkinsonian patients. J Neural Transm Suppl. 1995;46:407–13.
Müller T, Kuhn W, Schulte T, Przuntek H. Intravenous amantadine sulphate application improves the performance of complex but not simple motor tasks in patients with Parkinson’s disease. Neurosci Lett. 2003;339(1):25–8.
Bailey EV, Stone TW. The mechanism of action of amantadine in Parkinsonism: a review. Arch Int Pharmacodyn Ther. 1975;216(2):246–62.
Chase TN, Bibbiani F, Oh JD. Striatal glutamatergic mechanisms and extrapyramidal movement disorders. Neurotoxicol Res. 2003;5(1–2):139–46.
Nastuk WL, Su P, Doubilet P. Anticholinergic and membrane activities of amantadine in neuromuscular transmission. Nature. 1976;264(5581):76–9.
Aoki FY, Sitar DS. Clinical pharmacokinetics of amantadine hydrochloride. Clin Pharmacokinet. 1988;14(1):35–51.
Elmer LW, Juncos JL, Singer C, Truong DD, Criswell SR, Parashos S, et al. Pooled analyses of phase III studies of ADS-5102 (amantadine) extended-release capsules for dyskinesia in Parkinson’s disease. CNS Drugs. 2018;32(4):387–98.
Hauser RA, Pahwa R, Wargin WA, Souza-Prien CJ, McClure N, Johnson R, et al. Pharmacokinetics of ADS-5102 (amantadine) extended release capsules administered once daily at bedtime for the treatment of dyskinesia. Clin Pharmacokinet. 2019;58(1):77–88.
Thomas A, Iacono D, Luciano AL, Armellino K, Di Iorio A, Onofrj M. Duration of amantadine benefit on dyskinesia of severe Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2004;75:141–3.
Ory-Magne F, Corvol JC, Azulay JP, Bonnet AM, Brefel-Courbon C, Damier P, et al. Withdrawing amantadine in dyskinetic patients with Parkinson disease: the AMANDYSK trial. Neurology. 2014;82(4):300–7.
Hauser RA, Pahwa R, Wargin WA, Souza-Prien CJ, McClure N, Johnson R, et al. Pharmacokinetics of ADS-5102 (amantadine) extended release capsules administered once daily at bedtime for the treatment of dyskinesia. Clin Pharmacokinet. 2019;58(1):77–88.
Isaacson SH, Fahn S, Pahwa R, Tanner CM, Espay AJ, Trenkwalder C, et al. Parkinson’s patients with dyskinesia switched from immediate release amantadine to open-label ADS-5102. Mov Disord Clin Pract. 2018;5(2):183–90.
Oertel W, Eggert K, Pahwa R, Tanner CM, Hauser RA, Trenkwalder C, et al. Randomized, placebo-controlled trial of ADS-5102 (Amantadine) Extended-Release Capsules for Levodopa-Induced Dyskinesia in Parkinson’s Disease (EASE LID 3). Mov Disord. 2017;32(12):1701–9.
Hauser RA, Pahwa R, Tanner CM, Oertel W, Isaacson SH, Johnson R, et al. ADS-5102 (Amantadine) Extended-Release Capsules for Levodopa-Induced Dyskinesia in Parkinson’s Disease (EASE LID 2 Study): interim results of an open-label safety study. J Parkinsons Dis. 2017;7(3):511–22.
Pahwa R, Tanner CM, Hauser RA, Sethi K, Isaacson S, Truong D, et al. Amantadine extended release for levodopa-induced dyskinesia in Parkinson’s disease (EASED study). Mov Disord. 2015;30(6):788–95.
Pahwa R, Tanner CM, Hauser RA, Isaacson SH, Nausieda PA, Truong DD, et al. ADS-5102 (Amantadine) Extended-Release Capsules for Levodopa-Induced Dyskinesia in Parkinson Disease (EASE LID study): a randomized clinical trial. JAMA Neurol. 2017;74(8):941–9.
Pahwa R, Isaacson S, Jimenez-Shaheed J, Malaty IA, Deik A, Johnson R, et al. Impact of dyskinesia on activities of daily living in Parkinson’s disease: results from pooled phase 3 ADS-5102 clinical trials. Parkinsonism Relat Disord. 2019;60(3):118–25.
Wolf E, Seppi K, Katzenschlager R, Hochschorner G, Ransmayr G, Schwingenschuh P, et al. Long-term antidyskinetic efficacy of amantadine in Parkinson’s disease. Mov Disord. 2010;25(10):1357–63.
Miller P, Soundy A. The pharmacological and non-pharmacological interventions for the management of fatigue related multiple sclerosis. J Neurol Sci. 2017;381:41–54.
Tur C. Fatigue management in multiple sclerosis. Curr Treat Options Neurol. 2016;18(6):26.
Gramish JA, Kopp BJ, Patanwala AE. Effect of amantadine on agitation in critically ill patients with traumatic brain injury. Clin Neuropharmacol. 2017;40(5):212–6.
Müller HF, Dastoor DP, Klingner A, Cole M, Boillat J. Amantadine in senile dementia: electroencephalographic and clinical effects. J Am Geriatr Soc. 1979;27(1):9–16.
Garssen MP, Schmitz PI, Merkies IS, Jacobs BC, van der Meche FG, van Doorn PA. Amantadine for treatment of fatigue in Guillain–Barre syndrome: a randomised, double blind, placebo controlled, crossover trial. J Neurol Neurosurg Psychiatry. 2006;77(1):61–5.
Kraus MF, Smith GS, Butters M, Donnell AJ, Dixon E, Yilong C, et al. Effects of the dopaminergic agent and NMDA receptor antagonist amantadine on cognitive function, cerebral glucose metabolism and D2 receptor availability in chronic traumatic brain injury: a study using positron emission tomography (PET). Brain Inj. 2005;19(7):471–9.
Sailer M, Heinze HJ, Schoenfeld MA, Hauser U, Smid HG. Amantadine influences cognitive processing in patients with multiple sclerosis. Pharmacopsychiatry. 2000;33(1):28–37.
Malek N, Grosset DG. Medication adherence in patients with Parkinson’s disease. CNS Drugs. 2015;29(1):47–53.
Goetz CG, Laska E, Hicking C, Damier P, Muller T, Nutt J, et al. Placebo influences on dyskinesia in Parkinson’s disease. Mov Disord. 2008;23(5):700–7.
Bonifati V, Fabrizio E, Cipriani R, Vanacore N, Meco G. Buspirone in levodopa-induced dyskinesias. Clin Neuropharmacol. 1994;17(1):73–82.
Durif F, Debilly B, Galitzky M, Morand D, Viallet F, Borg M, et al. Clozapine improves dyskinesias in Parkinson disease: a double-blind, placebo-controlled study. Neurology. 2004;62(3):381–8.
Katzenschlager R, Manson AJ, Evans A, Watt H, Lees AJ. Low dose quetiapine for drug induced dyskinesias in Parkinson’s disease: a double blind cross over study. J Neurol Neurosurg Psychiatry. 2004;75(2):295–7.
Kobylecki C, Burn DJ, Kass-Iliyya L, Kellett MW, Crossman AR, Silverdale MA. Randomized clinical trial of topiramate for levodopa-induced dyskinesia in Parkinson’s disease. Parkinsonism Relat Disord. 2014;20(4):452–5.
Meco G, Fabrizio E, Di RS, Alessandri A, Pratesi L. Mirtazapine in l-dopa-induced dyskinesias. Clin Neuropharmacol. 2003;26(4):179–81.
Schaeffer E, Pilotto A, Berg D. Pharmacological strategies for the management of levodopa-induced dyskinesia in patients with Parkinson’s disease. CNS Drugs. 2014;28(12):1155–84.
Ramot Y, Nyska A, Maronpot RR, Shaltiel-Karyo R, Tsarfati Y, Manno RA, et al. Ninety-day local tolerability and toxicity study of ND0612, a novel formulation of levodopa/carbidopa, administered by subcutaneous continuous infusion in minipigs. Toxicol Pathol. 2017;45(6):764–73.
Westin J, Nyholm D, Palhagen S, Willows T, Groth T, Dougherty M, et al. A pharmacokinetic-pharmacodynamic model for duodenal levodopa infusion. Clin Neuropharmacol. 2011;34(2):61–5.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received in relation to this article.
Conflict of interest
TM. and J.-D.M. have no relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the article. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Rights and permissions
About this article

Cite this article
Müller, T., Möhr, JD. Recent Clinical Advances in Pharmacotherapy for Levodopa-Induced Dyskinesia. Drugs 79, 1367–1374 (2019). https://doi.org/10.1007/s40265-019-01170-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-019-01170-5