
Abstract
Regorafenib (Stivarga®), a small molecule inhibitor of multiple kinases, is the first drug to be approved for the treatment of hepatocellular carcinoma (HCC) in patients who have progressed during or after sorafenib therapy. Its approval was based on the results of the randomized, double-blind, placebo-controlled, multinational, phase III RESORCE trial in patients with HCC who had progressed during sorafenib therapy. In RESORCE, regorafenib significantly prolonged overall survival (OS; primary endpoint), progression-free survival (PFS) and time to progression (TTP) compared with placebo, with the OS benefit appearing to be largely due to disease stabilization. Regorafenib had an acceptable tolerability profile. The most common treatment-related adverse events in the regorafenib group included hand-foot skin reaction, fatigue, diarrhoea and hypertension. No fatal hepatic failure was reported with regorafenib in patients with HCC in RESORCE. In conclusion, current evidence suggests that regorafenib is an important new targeted therapy option for the treatment of HCC patients who have progressed on sorafenib therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
First systemic agent approved for the treatment of patients with HCC who have previously received sorafenib therapy |
Significantly prolongs median OS, PFS and TTP compared with placebo in the pivotal phase III trial |
Acceptable tolerability profile |
1 Introduction
Globally, hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related deaths, with hepatitis C virus (HCV) infection, hepatitis B virus (HBV) infection and alcoholic cirrhosis among the most common aetiologies [1,2,3]. Most patients are diagnosed at an intermediate [e.g. Barcelona Clinic Liver Cancer (BCLC) stage B] or advanced (BCLC stage C) stage, both of which have limited therapeutic options [2, 3]. Sorafenib is the current standard of care systemic agent for the first-line treatment of advanced HCC [1, 4]. It improves the overall survival (OS) of patients with advanced HCC, with ≈ 50% of these patients achieving disease control; the remaining ≈ 50% patients experience disease progression and retreatment options for such patients are currently lacking [1].
Oral regorafenib (Stivarga®), a small molecule inhibitor of multiple kinases, is the first systemic agent to be approved in numerous countries including the USA [5] and those of the EU [6], as well as Japan [7] and China [8], for the second-line treatment of HCC in adult patients who have previously received sorafenib therapy [5, 6, 8] or who progressed after cancer treatment [7] (Sect. 6). This review focuses on the therapeutic efficacy and tolerability of regorafenib in patients with HCC, as well as summarizes its pharmacological properties. Regorafenib is also approved for the treatment of advanced gastrointestinal stromal tumours (GISTs) and colorectal cancer (CRC) [5,6,7]; however, discussion of these indications is outside the scope of this review.
2 Pharmacodynamic Properties of Regorafenib
The pharmacodynamic profile of regorafenib has been previously reviewed elsewhere [9, 10]; hence, only a brief overview is provided here. In vitro, regorafenib inhibited multiple protein kinases, including those that are involved in tumour angiogenesis (VEGFR-1, -2 and -3, TIE2, PDGFR-β, FGFR1) and oncogenesis (KIT, RET, c-RAF/RAF-1, BRAFV600E) [11]. It also altered proteins involved in MAPK signalling pathway (P-ERK1/2, P-JNK, P-c-Jun), apoptosis (Bax, Bcl-2, Bcl-X, survivin, cleaved caspase-3, -7, -8 and -9) and autophagy (Beclin-1, LC3-II) [12]. Of note, regorafenib appeared to be more pharmacologically potent than sorafenib, possibly due to its inhibition of a broader spectrum of kinases (e.g. VEGFR-1, TIE2, RET) [13]. In vitro and in vivo, the two major active metabolites of regorafenib [M-2 (N-oxide) and M-5 (N-oxide and N-desmethyl)] exhibited similar pharmacological activities to that of regorafenib (Sect. 3) [5, 6].
Regorafenib exhibited antiproliferative, antiangiogenic, antitumour and antimetastatic activity in rat or mouse models of various cancers [5, 6, 11]. In mouse HCC xenograft models, regorafenib exhibited significant (p < 0.05) tumour inhibition compared with vehicle [14, 15]. Furthermore, both regorafenib and sorafenib displayed effective antitumour activity in a patient-derived HCC xenograft mouse model, with the response to regorafenib being typically greater than that to sorafenib [15].
In terms of cardiac effects, changes from baseline in corrected QT interval or left-ventricular ejection fraction at tmax [time to maximum plasma concentration (Cmax)] were modest in patients with advanced solid tumours receiving regorafenib 160 mg once daily for the first 21 days of each 28-day cycle in a phase I trial [16]. These changes were considered unlikely to be of any clinical relevance [16].
3 Pharmacokinetic Properties of Regorafenib
Following a single dose of regorafenib 160 mg (as tablets) in patients with advanced solid tumours, regorafenib reached Cmax within a median time of 4 h [5, 6]. The systemic exposure of regorafenib at steady state increased in a less than dose-proportional manner at doses > 60 mg [17]. The active metabolites, M-2 and M-5, exhibited similar concentrations as regorafenib at steady state [17]. In healthy volunteers, administration of regorafenib 160 mg with a low-fat meal (8.2 g of fat [5]) increased exposure to regorafenib, M-2 and M-5 by 36, 40 and 23%, respectively, compared with a fasted state [5, 6]. In contrast, a high-fat meal (54.6 g of fat [5]) increased exposure to regorafenib by 48% but decreased exposure to M-2 and M-5 by 20 and 51% compared with a fasted state [5, 6]. Regorafenib should therefore be administered after a low-fat meal (Sect. 6).
In vitro, plasma protein binding is high (≥ 99.5%) for regorafenib, M-2 and M-5 [5, 6]. In the liver, regorafenib primarily undergoes oxidative metabolism by CYP3A4, as well as glucuronidation by UGT1A9, to its two major active metabolites (M-2 and M-5) [5, 6] and six minor circulating metabolites [6]. Metabolites may undergo further reduction or hydrolysis by microbial flora in the gastrointestinal (GI) tract, thereby allowing the unconjugated active substance and metabolites to enter enterohepatic circulation [6].
Regorafenib is primarily eliminated via the hepatic route [5]. Following oral administration of a 120 mg dose of radiolabelled regorafenib solution, 71.2% of the radioactivity was recovered in faeces (47.2% unchanged and 23.7% as metabolites) and 19.3% was recovered in urine (mostly as glucuronides) within 12 days after administration [18]. The mean elimination half-life of regorafenib, M-2 and M-5 following a single 160 mg dose of oral regorafenib was 28, 25 and 51 h, respectively [5, 6].
The pharmacokinetics of regorafenib are not significantly affected by gender, age, race and bodyweight [5, 6]. Regorafenib dosage adjustment is not required in patients with mild or moderate hepatic impairment in the USA [5] or in patients with mild hepatic impairment in the EU (there are no recommendations for patients with moderate hepatic impairment due to limited data) [6]. Patients with hepatic impairment should be monitored closely for hepatic adverse reactions [5] and overall safety [6]. Regorafenib is not recommended in patients with severe hepatic impairment, as regorafenib has not been studied in this population [5, 6]. Dosage adjustments are not required in patients with mild, moderate or severe renal impairment. However, there is no recommended dosage of regorafenib for patients with end-stage renal disease on haemodialysis as pharmacokinetic studies of regorafenib have not been conducted in this population [5, 6].
3.1 Potential Drug Interactions
In vitro studies indicate that regorafenib is likely to inhibit CYP2C8, CYP2C9, CYP2B6, CYP3A4 and CYP2C19; M-2 is likely to inhibit CYP2C8, CYP2C9, CYP3A4 and CYP2D6; and M-5 is likely to inhibit CYP2C8 [5]. However, on the basis of clinical pharmacokinetic data, regorafenib has no clinically meaningful drug interactions with CYP2C8 [5, 6], CYP2C9 [6], CYP3A4 [5, 6] or CYP2C19 [5, 6] substrates, although as a consequence of warfarin (CYP2C9 substrate) exposure increasing by 25% upon coadministration with regorafenib, it is advised (in the USA) that the international normalized ratio is monitored more frequently in regorafenib recipients being treated with warfarin [5].
Regorafenib is a substrate of CYP3A4; thus, coadministration of regorafenib with strong inducers of CYP3A4 (e.g. rifampin, phenytoin, carbamazepine, phenobarbital, and St. John’s wort) should be avoided as regorafenib exposure may be reduced, leading to decreased efficacy [5, 6]. Coadministration of regorafenib with a strong inhibitor of CYP3A4 (e.g. clarithromycin, grapefruit juice, ketoconazole, itraconazole, nefazodone, telithromycin, posaconazole and voriconazole) should also be avoided as regorafenib exposure may be increased (with decreasing M-2 and M-5 exposures), leading to toxicity [5, 6].
In vitro studies indicate that at therapeutically relevant concentrations, regorafenib, M-2 and M-5 competitively inhibit UGT1A9 [5, 6] and UGT1A1 [5]; M-5 only inhibits UGT1A1 after reaching a steady state concentration in vivo [6]. Consequently, systemic exposure of UGT1A9 and UGT1A1 substrates may increase when coadministered with regorafenib [6].
As regorafenib is a substrate of UGT1A9 [5, 6], concomitant use of regorafenib with a strong inhibitor of UGT1A9 (e.g. mefenamic acid, diflunisal and niflumic acid) should be avoided in the EU as studies assessing how exposure to regorafenib and its metabolites may be impacted at steady state have not been conducted [6].
Regorafenib may increase systemic exposure of breast-cancer-resistance protein (BCRP) substrates (e.g. methotrexate, fluvastatin, atorvastatin) [5, 6]. Close monitoring for signs and symptoms of increased exposure [6] or exposure-related toxicity [5] of BCRP substrates is recommended.
When regorafenib is coadministered with neomycin, regorafenib exposure is unchanged but M-2 and M-5 exposures are decreased by ≈ 80%, which may decrease the efficacy of regorafenib [5, 6]. Inhibitors and inducers of P-glycoprotein (P-gp) and BCRP may affect M-2 and M-5 exposures as they are substrates of P-gp and BCRP [6]. Bile salt-sequestering agents (e.g. cholestyramine) may interfere with the absorption or reabsorption of regorafenib by forming insoluble complexes with the drug, and thus may decrease the efficacy of regorafenib. The clinical significance of interactions with neomycin, inhibitors and inducers of P-gp and BCRP, and bile salt-sequestering agents is unknown [6].
4 Therapeutic Efficacy of Regorafenib
This section focuses on the efficacy of oral regorafenib in the treatment of HCC in adults with disease progression on sorafenib, as evaluated in a multinational, randomized, double-blind, placebo-controlled, phase III study (RESORCE) [19]. Based on the findings of earlier phase I [17] and II [20] trials, RESORCE used regorafenib at a dosage of 160 mg for the first 21 days of each 28-day cycle [19].
The RESORCE trial included patients with HCC (BCLC stage B or C), with ≥ 1 measurable lesion, who could not benefit from resection, local ablation or chemoembolization [19]. Eligible patients had evidence of radiological disease progression during previous sorafenib therapy, had tolerated ≥ 400 mg/day of sorafenib for ≥ 20 days during the last cycle and had received the last dose of sorafenib ≤ 10 weeks before randomization. Patients were required to have Child-Pugh (CP) class A liver function, Eastern Cooperative Oncology Group performance status (ECOG-PS) of 0 or 1, adequate bone marrow, liver and renal function, and a predicted life expectancy of ≥ 3 months. Exclusion criteria included previous systemic molecular targeted therapy other than sorafenib, cardiovascular problems, CNS metastasis or significant bleeding ≤ 30 days before randomization [19].
Eligible patients were randomized to receive 160 mg of regorafenib or placebo once daily for the first 21 days of each 28-day cycle plus best supportive care [19]. Treatment interruption or two step-wise dose reductions (120 then 80 mg) were allowed if 160 mg was not tolerated; however, treatment was discontinued if patients required further dose reduction. The reduced dose could be up-titrated to a maximum of 160 mg once the lower dose was tolerated. Treatment continued until disease progression [as defined by modified Response Evaluation Criteria in Solid Tumours (mRECIST)], clinical progression (i.e. ECOG-PS of ≥ 3 or symptomatic deterioration such as increased liver function tests), unacceptable toxicity or death had occurred. The treating physician could discontinue treatment or continue it beyond disease progression if in the patient’s best interest. Randomization was stratified by geographical region (Asia vs. rest of world), presence of macrovascular invasion (yes vs. no) and extrahepatic disease (yes vs.no), ECOG-PS (0 vs. 1) and α-fetoprotein (AFP) level (< 400 vs. ≥ 400 ng/mL) [19].
At baseline, overall, most patients were CP class A (98%) and had advanced disease (BCLC stage C) (87%), extrahepatic disease (72%), ≥ 2 target lesions (83%), an ECOG-PS of 0 (66%) and HCC caused by HBV, alcohol use and/or HCV (each 21–38%); 38% of patients were from Asia (enrolment was restricted to 40% of the total population), 43% had AFP level of ≥ 400 ng/mL and 29% had macrovascular invasion [19]. The regorafenib and placebo groups were balanced for these and other baseline characteristics, including the pattern of progression on sorafenib therapy (which can influence outcomes to second-line treatment); while on sorafenib, 40 and 41% of patients in the respective groups had developed new extrahepatic lesions, 44 and 45% had developed new intrahepatic lesions and 81 and 80% had experienced growth of existing lesions [19].
At the cut-off time for the final analysis, the mean treatment durations were 5.9 and 3.3 months in the regorafenib and placebo groups, and the mean dosages were 144.1 and 157.4 mg/day [19]. An exposure-response analysis of the RESORCE trial revealed that the efficacy of regorafenib was not significantly correlated with its exposure [21].
After a median follow-up of 7.0 months (cut-off date 29 February 2016), regorafenib significantly prolonged median OS compared with placebo (primary endpoint; Table 1) [19]. This finding was supported by an updated analysis (cut-off date 23 January 2017) in which median OS was 10.7 and 7.9 months in the regorafenib and placebo groups [hazard ratio (HR) 0.61; 95% CI 0.50–0.75; p < 0.0001] (data from an abstract [22]).
Regorafenib significantly improved median progression-free survival (PFS), median time to progression (TTP), objective response rate (ORR) and disease control rate compared with placebo when measured by mRECIST (Table 1), as well as when measured by RECIST version 1.1 [19]. In regorafenib and placebo recipients, the median duration of response were 3.5 versus 2.7 months and the median duration of stable disease were 5.5 versus 3.1 months; 49 versus 23% of patients demonstrated evidence of target lesion shrinkage [19]. A numerically greater proportion of regorafenib than placebo recipients had stable disease (54 vs. 32%) and it appeared that disease stabilization was largely accountable for the prolonged OS and PFS in the regorafenib group [23].
In preplanned subgroup analyses, HRs for regorafenib versus placebo favoured regorafenib for OS, PFS and TTP, regardless of factors such as patient age (< 65 or ≥ 65 years), sex, stratification factors, CP score (A5 or A6) and HCC aetiology (HBV, HCV or alcohol use) [19]. However, significance (based on 95% CIs) was not reached for OS in some groups (female, ECOG score 1, CP score A6, no extrahepatic disease, HCV infection and alcohol use), which had limited patient numbers (n = 69–199) [19].
In exploratory analyses (available as abstracts), regorafenib significantly improved post-progression survival relative to placebo irrespective of the progression pattern during sorafenib therapy [24] and provided consistent OS benefit compared with placebo regardless of prior sorafenib dose (800 vs. < 800 mg/day) [25], baseline c-Met protein and AFP levels (low or high) [26] or baseline platelet count (> 150 or ≤ 150 × 109 /L) [27]. Development of new extrahepatic lesions on previous sorafenib therapy, or high baseline levels of c-Met and AFP may be associated with a poor prognosis [24, 26]. A higher baseline platelet count was associated with a poor prognosis during first-line sorafenib therapy but not during second-line regorafenib therapy [27].
Regorafenib did not appear to adversely affect health-related quality of life (HR-QOL), as measured by the Functional Assessment of Cancer Therapy (FACT)-General, FACT-Hepatobiliary (FACT-Hep), EuroQOL (EQ)-5D and EQ-VAS questionnaires [19]. Although some scores were significantly (p ≤ 0.0006) lower in regorafenib than placebo recipients (namely FACT-Hep total and trial outcome index subscale scores), between-group differences did not reach the established minimally important threshold for these or any of the other HR-QOL measures [19].
5 Tolerability of Regorafenib
Oral regorafenib had an acceptable tolerability profile in patients with HCC who had progressed on previous sorafenib treatment [19]. The tolerability profile of regorafenib in the RESORCE study was consistent with that seen in patients with CRC [9] or GISTs [10]. Exploratory analyses (available as abstracts) of the RESORCE trial showed that treatment-emergent adverse events (TEAEs) with regorafenib therapy in Chinese patients were consistent with those seen in overall population [28] and that the incidence of grade ≥ 3 TEAEs for regorafenib versus placebo was generally similar regardless of prior sorafenib dose (800 vs. < 800 mg/day) [25].
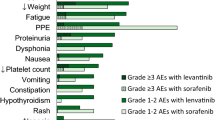
In the RESORCE trial, the incidence of treatment-related adverse events (TRAEs) of any grade was ≈ 2-fold higher in the regorafenib group than in the placebo group (93 vs. 52%) [19]. TRAEs occurring in > 10% of regorafenib recipients and with a > 5% higher incidence than in placebo recipients included hand-foot skin reaction (HFSR), diarrhoea, fatigue, anorexia, hypertension and oral mucositis (Fig. 1). The incidence of grade 3 or 4 TRAEs in patients receiving regorafenib or placebo was 50 versus 17%, with the most common being HFSR (13 vs. 1%), hypertension (13 vs. 3%), blood bilirubin increase (7 vs. 2%) and fatigue (6 vs. 2%) [19].

Treatment-related adverse events of any grade occurring in > 10% of regorafenib recipients and with a > 5% higher incidence than in placebo recipients in the RESORCE trial [19]. HFSR hand-foot skin reaction, PL placebo, REG regorafenib, ↑ increase
Several TRAEs were laboratory abnormalities, with the most common with regorafenib being increased blood bilirubin (Fig. 1), increased aspartate aminotransferase (AST; 13 vs. 8% with placebo), increased alanine aminotransferase (ALT; 8 vs. 4%), hypophosphataemia (6 vs. 1%) and thrombocytopenia (5 vs. 1%) [19]. Among those of grade 3 or 4 severity, the most frequent with an incidence numerically greater with regorafenib than with placebo were increased blood bilirubin (7 vs. 2%) and hypophosphataemia (5 vs. 2%) [19].
In RESORCE, 54% of regorafenib and 10% of placebo recipients had dose interruptions or reductions because of TRAEs [19]. TRAEs led to treatment discontinuation in 10 and 4% of regorafenib and placebo recipients; the most common adverse events leading to discontinuation in the respective groups were HFSR (2 vs. 0%) and increases in AST (2 vs. 2%) or ALT (1 vs. 0%). Serious TRAEs occurred in 10% of regorafenib and 3% of placebo recipients. Treatment-related deaths were reported in seven regorafenib recipients (one each due to myocardial infarction, gastric perforation, encephalopathy, upper GI haemorrhage, intracranial haemorrhage, general disorders and unknown reasons) and two placebo recipients (both due to hepatic failure) [19].
5.1 Adverse Events of Special Interest
In randomized trials across approved indications (CRC, GIST or HCC), hepatotoxicity, infections, haemorrhage, GI perforation or fistula, dermatological toxicity, hypertension, cardiac ischemia and infarction, and reversible posterior leukoencephalopathy syndrome have been reported in regorafenib recipients. Warnings and precautions pertaining to these events are included in the manufacturer’s prescribing information [5,6,7]. For instance, the US prescribing information for regorafenib carries a boxed warning regarding hepatotoxicity, based on fatal hepatic failures seen in patients with CRC or GIST receiving the drug [5]. A similar warning/precaution also appears in the EU [6] and Japanese [7] prescribing information. No fatal hepatic failure was reported with regorafenib in patients with HCC in RESORCE, where hepatobiliary disorders occurred in 11% of regorafenib and 18% of placebo recipients [19]. However, hepatic function should be monitored before and during treatment with regorafenib, and hepatotoxicity should be managed by treatment interruption, dosage modification or discontinuation of regorafenib, depending upon the severity and persistence of hepatotoxicity [5, 6].
In RESORCE, skin and subcutaneous adverse events occurred in 65.5 and 30.6% of regorafenib and placebo recipients, with the most common being HFSR [23]. Strategies for managing dermatological toxicities include dose modification, treatment discontinuation, and supportive therapies for symptomatic relief [5]. Infections were reported in 31.3% of regorafenib and 18.1% of placebo recipients, with death from infections reported in 1.3 and 0%, respectively [23]. Regorafenib should be withheld from patients with grade 3 or 4 infections, or worsening infection of any grade, but can be resumed at the same dosage after resolution of infection [5].
In RESORCE, treatment-related hypertension, although common with regorafenib (Fig. 1), was mostly of grade 1 or 2 severity [19]. Blood pressure should be adequately controlled before initiating regorafenib therapy and should be monitored during the course of therapy [5]. In terms of other adverse events (any grade) of special interest with regorafenib, the overall incidence of cardiac ischemia (2.4 vs. 0.5% with placebo), arrhythmia (3.7 vs. 2.1%) and GI perforation (1.3 vs. 1.6%) was low [23]. Haemorrhagic events occurred in fewer than 20% of regorafenib or placebo recipients (17.6 vs. 16.1%) and were not frequently grade ≥ 3 (5.1 vs. 8.3% of recipients) [23]. The regorafenib dose should be interrupted, reduced or permanently discontinued depending on the severity and/or persistence of these adverse events [5, 6].
6 Dosage and Administration of Regorafenib
Regorafenib is indicated in several countries, including the USA [5] and those of the EU [6], for the treatment of HCC in patients who have been previously treated with sorafenib. In Japan, regorafenib is indicated for the treatment of unresectable HCC that exacerbated after cancer chemotherapy [7]. Regorafenib is also approved in China for the second-line treatment of HCC [8]. The approved dosage of oral regorafenib is four tablets (4 × 40 mg) taken once daily for the first 21 days of each 28-day cycle [5,6,7]. Regorafenib should be administered with water after a low-fat meal (< 30% fat). Regorafenib treatment should be continued until disease progression or unacceptable toxicity occurs. Dose interruption/modification or discontinuation of regorafenib may be required for the management of adverse events [5,6,7]. Local prescribing information should be consulted for details regarding drug interactions, warning and precautions, dosage adjustments for adverse events and use of regorafenib in special patient populations.
7 Place of Regorafenib in the Management of Hepatocellular Carcinoma
HCC is one of the most frequent types of liver cancer and is difficult to diagnose at early stages due to its atypical radiological appearance [2]. Curative therapies, such as liver transplant, surgical resection and local ablation, are applicable only to patients with early stages of HCC (BCLC stage 0 or A) [2,3,4]. Treatment options for patients with intermediate or advanced HCC have until recently been limited to locoregional therapies (e.g. chemoembolization) or systemic treatment with sorafenib, with no targeted therapy options for patients who progressed on sorafenib [2].
With an improved understanding of the molecular pathogenesis of HCC, various targeted therapies have been assessed for use in HCC, either as first-line treatments or for use as a second-line treatment after sorafenib [2, 3, 29]. Regorafenib became the second drug (after sorafenib) to receive regulatory approval in HCC and the first to be approved in the second-line setting (Sect. 6). Its approval was based on the pivotal RESORCE trial (Sect. 4), in which regorafenib significantly improved OS (as well as other measures, such as PFS and ORR) versus placebo in HCC patients who had progressed on sorafenib. This benefit appeared to be due largely to patients achieving disease stabilization and was evident across all predefined patient subgroups. RESORCE had narrow eligibility criteria (i.e. patients with good functional status and liver function) and, therefore, results may not be generalizable to the real world. Thus, the efficacy of regorafenib in the real-world setting would be of interest.
The tolerability profile of regorafenib in patients with HCC in RESORCE was acceptable and consistent with that seen in patients with CRC or GISTs (Sect. 5); however, excluding patients who could not tolerate sorafenib from the trial may have lessened the likelihood of tolerability issues. The most common TRAEs with regorafenib were consistent with those seen with sorafenib (i.e. HFSR, diarrhoea, hypertension and fatigue) and were generally manageable with dose interruptions/modifications (Sect. 5). Although underlying liver dysfunction is more common in patients with HCC, the incidence of hepatic adverse events in HCC patients (Sect. 5.1) was similar to that seen in patients with CRC or GIST [19, 23]. The adverse events associated with regorafenib did not appear to adversely affect HR-QOL in patients with HCC (Sect. 4), although long-term safety studies of regorafenib in this population will be useful.
Regorafenib is structurally similar to sorafenib but may have more potent pharmacological activity [13]. The results of RESORCE indicate that the sequential use of two multikinase inhibitors (regorafenib after progression on sorafenib) with partially overlapping, but distinct, kinase inhibitor profiles may provide OS and other clinical benefits and have acceptable tolerability [19, 23]. The US NCCN guidelines recommend regorafenib for patients with unresectable HCC who are not transplant candidates and who have progressed on or after sorafenib therapy provided they have preserved liver function (CP class A) [30]. The EASL guidelines also recommend regorafenib as a second-line treatment for those who have tolerated and progressed on sorafenib and who have preserved liver function (CP class A) [31]. Furthermore, the Asian Pacific Association for the Study of the Liver guidelines recognize the efficacy of regorafenib in HCC patients who have progressed on sorafenib therapy [32].
Regorafenib was estimated not to be cost effective in the second-line treatment of HCC in a Markov model-based pharmacoeconomic analysis conducted from a US healthcare system perspective using data from RESORCE; regorafenib was associated with additional quality-adjusted life-years gained over best supportive care, but at an additional cost [33]. The National Institute for Health and Care Excellence appraisal also did not consider regorafenib to be a cost-effective treatment option in patients with advanced unresectable HCC who have had sorafenib in England [34]. However, the pharmacoeconomic value of regorafenib for this indication may improve by identifying subpopulations that will gain maximum benefit from regorafenib treatment [33]. To date, there are no validated predictors of treatment response for patients with HCC [19]. In the exploratory analyses of the RESORCE trial, circulating miRNA moieties appeared to have a predictive or prognostic effect for OS [35] and plasma protein biomarkers such as LOX-1 and ANG-1 were identified as potential predictors for OS and TTP [36] with regorafenib treatment. Further biomarker research to identify predictors of response to regorafenib would therefore be of interest.
In conclusion, regorafenib is the first systemic agent available for the treatment of HCC patients who have progressed with sorafenib therapy. Providing survival benefit with acceptable tolerability in these patients, regorafenib meets a previously unmet need and is thus an important treatment option.
Data Selection Regorafenib HCC: 319 records identified
Duplicates removed | 65 |
Excluded during initial screening (e.g. press releases; news reports; not relevant drug/indication; preclinical study; reviews; case reports; not randomized trial) | 189 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase I/II trials) | 29 |
Cited efficacy/tolerability articles | 10 |
Cited articles not efficacy/tolerability | 26 |
Search Strategy: EMBASE, MEDLINE and PubMed from 1946 to present. Clinical trial registries/databases and websites were also searched for relevant data. Key words were regorafenib, Stivarga, hepatocellular carcinoma. Records were limited to those in English language. Searches last updated 21 May 2018 | |
References
Kondo M, Numata K, Hara K, et al. Treatment of advanced hepatocellular carcinoma after failure of sorafenib treatment: subsequent or additional treatment interventions contribute to prolonged survival postprogression. Gastroenterol Res Pract. 2017. https://doi.org/10.1155/2017/5728946.
Ding XX, Zhu QG, Zhang SM, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715–30.
Desai JR, Ochoa S, Prins PA, et al. Systemic therapy for advanced hepatocellular carcinoma: an update. J Gastrointest Oncol. 2017;8(2):243–55.
Allaire M, Nault JC. Advances in management of hepatocellular carcinoma. Curr Opin Oncol. 2017;29(4):288–95.
Bayer HealthCare Pharmaceuticals Inc. Stivarga® (regorafenib): US prescribing information 2017. http://www.fda.gov/. Accessed 26 Dec 2017.
European Medicines Agency. Stivarga: summary of product characteristics. 2017. http://www.ema.europa.eu/. Accessed 10 Dec 2017.
Bayer HealthCare Pharmaceuticals Inc. Stivarga® (regorafenib): Japanese prescribing information 2017. 2017.
Bayer HealthCare Pharmaceuticals Inc. Bayer receives approval in China for Stivarga® (regorafenib) for the second-line systemic treatment of liver cancer. 2017.
Shirley M, Keating GM. Regorafenib: a review of its use in patients with advanced gastrointestinal stromal tumours. Drugs. 2015;75(9):1009–17.
Carter NJ. Regorafenib: a review of its use in previously treated patients with progressive metastatic colorectal cancer. Drugs Aging. 2014;31(1):67–78.
Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129(1):245–55.
Carr BI, Cavallini A, Lippolis C, et al. Fluoro-Sorafenib (regorafenib) effects on hepatoma cells: growth inhibition, quiescence, and recovery. J Cell Physiol. 2013;228(2):292–7.
Rey JB, Launay-Vacher V, Tournigand C. Regorafenib as a single-agent in the treatment of patients with gastrointestinal tumors: an overview for pharmacists. Target Oncol. 2015;10(2):199–213.
Tai WT, Chu PY, Shiau CW, et al. STAT3 mediates regorafenib-induced apoptosis in hepatocellular carcinoma. Clin Cancer Res. 2014;20(22):5768–76.
Kissel M, Berndt S, Fiebig L, et al. Antitumor effects of regorafenib and sorafenib in preclinical models of hepatocellular carcinoma. Oncotarget. 2017;8(63):107096–108.
Jones RL, Bendell JC, Smith DC, et al. A phase I open-label trial evaluating the cardiovascular safety of regorafenib in patients with advanced cancer. Cancer Chemother Pharmacol. 2015;76(4):777–84.
Mross K, Frost A, Steinbild S, et al. A phase I dose-escalation study of regorafenib (BAY 73-4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(9):2658–67.
Gerisch M, Hafner FT, Lang D, et al. Mass balance, metabolic disposition, and pharmacokinetics of a single oral dose of regorafenib in healthy human subjects. Cancer Chemother Pharmacol. 2018;81(1):195–206.
Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66.
Bruix J, Tak W-Y, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412–9.
Solms A, Reinecke I, Fiala-Buskies S, et al. Exposure-response relationship of regorafenib efficacy in patients with hepatocellular carcinoma. Eur J Pharm Sci. 2017;109S:S149–53.
Merle P, Granito A, Huang Y-H, et al. Updated overall survival (OS) analysis from the international, phase 3, randomized, placebo-controlled RESORCE trial of regorafenib for patients with hepatocellular carcinoma (HCC) who progressed on sorafenib treatment [abstract no. O-031]. In: 11th international liver cancer association annual conference. 2017.
European Medicines Agency. Stivarga: EPAR-public assessment report 2017. http://www.ema.europa.eu/. Accessed 10 Dec 2017.
Bruix J, Merle P, Granito A, et al. Analysis of overall survival (OS) by pattern of progression of hepatocellular carcinoma (HCC) during prior sorafenib treatment in the randomized phase 3 RESORCE trial comparing regorafenib with placebo [abstract no. OP073]. Hepatol Int. 2017;11(Suppl 1):S63.
Finn RS, Merle P, Granito A, et al. Outcomes with sorafenib (SOR) followed by regorafenib (REG) or placebo (PBO) for hepatocellular carcinoma (HCC): results of the international, randomized phase 3 RESORCE trial [abstract no. 344]. J Clin Oncol. 2017;35(4 Suppl).
Teufel M, Köchert K, Meinhardt G, et al. Efficacy of regorafenib (REG) in patients with hepatocellular carcinoma (HCC) in the phase III RESORCE trial according to alpha-fetoprotein (AFP) and c-Met levels as predictors of poor prognosis [abstract no. 4078]. J Clin Oncol. 2017;35(15 Suppl).
Meinhardt G, De Sanctis Y, LeBerre M-A, et al. Overall survival (OS) by platelet count at baseline in patients with hepatocellular carcinoma (HCC) treated with sorafenib (SOR) in the SHARP and AP trials and regorafenib (REG) in the RESORCE trial [abstract no. 706P]. Ann Oncol. 2017;28(Suppl 5):240–1.
Han G, Qin S, Song T, et al. Efficacy and safety of regorafenib (REG) versus placebo (PBO) in Chinese patients with hepatocellular carcinoma (HCC) progressing on sorafenib (SOR): subgroup analysis of the international, randomized phase 3 RESORCE trial [abstract no. PL015]. Hepatol Int. 2017;11(Suppl 1):S9–10.
Llovet JM, Hernandez-Gea V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res. 2014;20(8):2072–9.
National Comprehensive Cancer Network. Hepatobiliary cancers (NCCN clinical practice guidelines in oncology). 2017. http://www.nccn.org/. Accessed 20 Dec 2017.
European Association for the Study of the Liver. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018. https://doi.org/10.1016/j.jhep.2018.03.019.
Omata M, Cheng AL, Kokudo N, et al. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: a 2017 update. Hepatol Int. 2017;11(4):317–70.
Parikh ND, Singal AG, Hutton DW. Cost effectiveness of regorafenib as second-line therapy for patients with advanced hepatocellular carcinoma. Cancer. 2017;123(19):3725–31.
National Institute for health and Care Excellence. Technical appraisal guidance: regorafenib for previously treated advanced hepatocellular carcinoma 2018. http://www.hcvguidelines.org/. Accessed 01 May 2018.
Teufel M, Seidel H, Kochert K, et al. Circulating miRNA biomarkers predicting regorafenib (REG) clinical benefit in patients with hepatocellular carcinoma (HCC) in the RESORCE trial [abstract no. 705P]. Ann Oncol. 2017;28(Suppl 5):240.
Teufel M, Kochert K, Meinhardt G, et al. Protein biomarkers as predictors of outcome with regorafenib (REG) in patients (pts) with hepatocellular carcinoma (HCC) in the RESORCE trial [abstract no. 625PD]. Ann Oncol. 2017;28(Suppl 5):213.
Acknowledgements
During the peer review process, the manufacturer of regorafenib was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflict of interest
Young-A Heo and Yahiya Y. Syed are salaried employees of Adis/Springer, are responsible for the article content and declare no relevant conflicts of interest.
Additional information
The manuscript was reviewed by: G. G. Di Costanzo, Department of Transplantation, Liver Unit, Cardarelli Hospital, Naples, Italy; M. Fukudo, Department of Hospital Pharmacy and Pharmacology, Asahikawa Medical University, Hokkaido, Japan; N. Personeni, Medical Oncology and Hematology Unit, Humanitas Cancer Center, Humanitas Clinical and Research Center/Department of Biomedical Sciences, Humanitas University, Milan, Italy; L. Rimassa, Medical Oncology and Hematology Unit, Humanitas Cancer Center, Humanitas Clinical and Research Center, Milan, Italy.
Rights and permissions
About this article

Cite this article
Heo, YA., Syed, Y.Y. Regorafenib: A Review in Hepatocellular Carcinoma. Drugs 78, 951–958 (2018). https://doi.org/10.1007/s40265-018-0932-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-018-0932-4