
Abstract
Rucaparib (Rubraca™) is an oral, small molecule, poly (ADP-ribose) polymerase inhibitor being developed by Clovis Oncology, Inc. (Boulder, CO, USA) for the treatment of solid tumours. It has been approved in the USA as monotherapy for the treatment of patients with deleterious BRCA mutation (germline and/or somatic) associated advanced ovarian cancer who have been treated with two or more chemotherapies. A marketing authorization application for rucaparib for the same indication has been submitted to the European Medicines Agency. Rucaparib is also under phase II or III investigation in ovarian, breast and prostate cancer. This article summarizes the milestones in the development of rucaparib leading to this first approval for ovarian cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Rucaparib (Rubraca™) is an oral, small molecule inhibitor of poly (ADP-ribose) polymerase (PARP) enzymes, including PARP-1, -2 and -3 [1, 2]. It is being developed by Clovis Oncology, Inc. (Boulder, CO, USA) for the treatment of solid tumours. PARP enzymes play a key role in DNA repair and inhibition of these enzymes forms toxic PARP-DNA complexes in cells, resulting in DNA damage, ultimately leading to cell death [1, 2]. PARP inhibition is synthetically lethal in cells with homologous recombination deficiency (HRD), such as those with mutation in the BRCA1 and/or BRCA2 genes or high genomic loss of heterozygosity (LOH) [2, 3].
Oral rucaparib has been approved in the USA as monotherapy for the treatment of patients with deleterious BRCA mutation (germline and/or somatic) associated advanced ovarian cancer who have been treated with two or more chemotherapies [1]. Patients are selected for rucaparib treatment based on a US FDA-approved companion diagnostic (Sect. 2.5). Rucaparib received accelerated approval based on objective response rate (ORR) and duration of response seen in phase II trials (Sect. 2.3.1). Continued approval of rucaparib in this indication may be contingent upon verification and description of clinical benefit in confirmatory trials [1]. A marketing authorization application for rucaparib in ovarian cancer has been submitted to the European Medicines Agency.
Rucaparib is also under phase II or III investigation in ovarian, breast and prostate cancer. Use of an intravenous formulation of rucaparib for the treatment of malignant melanoma was explored early in the development, prior to the acquisition of rucaparib by Clovis Oncology [4], and has since been discontinued.
1.1 Company Agreements
Rucaparib was originated by Cancer Research Technology owned by Cancer Research UK and was licensed to Agouron Pharmaceuticals (later Pfizer).

Key milestones in the development of oral rucaparib monotherapy for the treatment of ovarian cancer. Est estimated completion date, MAA marketing authorization application, NDA new drug application
In June 2011, Clovis Oncology entered into a license agreement with Pfizer under which Clovis Oncology gained rights for global development and commercialisation of rucaparib [5]. Pfizer has received an upfront fee and is eligible for milestone and royalty payments from Clovis Oncology [5]. Pfizer is not involved in the development of rucaparib any longer, but the agreement will remain effective until expiration of the royalty and sublicense revenue obligations of Clovis Oncology to Pfizer.
In April 2012, Clovis Oncology gained from AstraZeneca exclusive rights to patents and patent applications for PARP inhibitors (including rucaparib) in certain indications. AstraZeneca has received an upfront fee and is eligible for milestone and royalty payments from Clovis Oncology [6]. Clovis Oncology signed a long-term manufacturing agreement for rucaparib with Lonza in October 2016 [7].
2 Scientific Summary
2.1 Pharmacodynamics
Rucaparib is a potent PARP inhibitor at nanomolar concentrations [8], with potentially greater PARP inhibitory activity than olaparib or niraparib [9]. Following a single oral or intraperitoneal dose of rucaparib in tumour-bearing mice, the drug accumulated and was retained in tumours, and inhibited PARP enzymes for 7 days [10]. Intravenous [11] or oral [12] rucaparib inhibited PARP-1 enzyme activity in peripheral blood lymphocytes from patients with metastatic melanoma [11], or advanced ovarian or breast cancer [12]. A single oral dose of 92 mg inhibited the enzyme activity by a mean ≥90% from pretreatment levels at 24 h post-dose; however, a continuous dosing schedule was necessary to maintain the inhibition [12].
Rucaparib showed PARP-dependent and -independent cytotoxic mechanisms in cancer cells [8, 13]. In vitro, rucaparib was more cytotoxic than olaparib [14]. The cytotoxicity of rucaparib is increased in cancer cells with mutations in the BRCA1/2 genes and other DNA repair genes [13, 15, 16]. Rucaparib reduced tumour growth in mouse xenograft models of human cancer with [10, 15, 16] or without [1] BRCA mutations. An HRD signature (Sect. 2.3.1) and a two-gene (CYB5R2, GSTT1) signature [17] have been shown to predict response to rucaparib.
Rucaparib showed synergistic or additive interactions with commonly used chemotherapeutic agents, in vitro and in vivo [13, 18,19,20,21]. It sensitized cancer cells to platinum therapy [16, 22], chemoradiotherapy [23], irradiation [24, 25] and radiopharmaceuticals [25, 26]. Rucaparib is known to induce vasodilation, which may increase accumulation of cytotoxic drugs in cancer cells by increasing tumour perfusion [27,28,29].

Chemical structure of rucaparib
Mechanisms of resistance to rucaparib therapy include recovery of HRD [30] and defects in non-homologous end joining [31]. Rucaparib is efficiently transported by multidrug efflux transporters, such as BRCP and P-glycoprotein, and therefore, they can restrict oral bioavailability and brain penetration of rucaparib [32, 33]. Rucaparib may partially offset its efficacy by activating the phosphatidylinositol 3-kinase (PI3K)/AKT signalling pathway; overexpression of the INPP4B gene, which negatively regulates this pathway, enhanced the antitumor activity of rucaparib in cancer cells [34].
2.2 Pharmacokinetics
All rucaparib pharmacokinetic data discussed herein are from patients with cancer. Rucaparib showed linear, dose-proportional and time-independent pharmacokinetics over a dose range of 240–840 mg twice daily [1]. Following the recommended 600 mg twice daily dosage, the median Tmax [time to maximal plasma concentration (Cmax)] was 1.9 h. When taken with a high-fat meal, Cmax and the area under the plasma concentration-time curve from time 0 to 24 h (AUC0–24) were increased by 20 and 38%, respectively, and Tmax was delayed by 2.5 h relative to dosing under fasted condition; however, the effect is not considered clinically significant and rucaparib can be taken with or without food. Rucaparib immediate-release tablet has a mean absolute bioavailability of 36% [1]. Following administration of 600 mg twice daily in a 28-day cycle, rucaparib steady state was observed on day 15 of cycle 1 [3]. At steady state, accumulation of rucaparib was 3.5 to 6.2 fold at tested dose levels [1].
Rucaparib had a volume of distribution of 113–262 L following a single intravenous dose of 12–40 mg [1]. At therapeutic concentrations, in vitro plasma protein binding of rucaparib was 70% in human plasma, and the drug preferentially distributed into red blood cells (blood to plasma concentration ratio 1.83). In vitro, rucaparib was metabolized primarily by cytochrome P450 (CYP) enzyme CYP2D6 and to a lesser extent by CYP1A2 and CYP3A4. Rucaparib steady-state concentrations did not differ markedly across CYP2D6 or CYP1A2 genotype subgroups [1].
Oral rucaparib had a mean terminal half-life of 17–19 h after a single 600 mg dose and an apparent clearance of 15.3–79.2 L/h after multiple doses of 600 mg twice daily [1]. After a single intravenous dose of rucaparib, 11% of the dose was recovered in the urine over 24 h, indicating that this is not the major excretion route of the drug [11].
Mild or moderate renal impairment increased rucaparib exposure by 15 and 32%, respectively, versus normal renal function; mild hepatic impairment did not markedly affect the pharmacokinetics of rucaparib [1]. Starting dosage adjustment is not recommended in these patients. The pharmacokinetics of rucaparib in patients with severe renal impairment (creatinine clearance <30 mL/min) and in those with moderate to severe hepatic impairment (total bilirubin >1.5 times the upper limit of normal) are unknown, and therefore, starting dosage recommendation is not available for these patients [1].
Interactions of rucaparib with enzymes and transporters were evaluated in vitro. Rucaparib is a substrate and inhibitor of BCRP and P-glycoprotein [1]. It inhibits (CYP1A2, CYP2C19, CYP2C9, CYP3A, and CYP2C8, CYP2D6, UGT1A1 to a lesser extent), induces (CYP1A2) or down-regulates (CYP2B6, CYP3A4) several CYP enzymes. Rucaparib inhibits several transporter proteins potently (MATE1 and MATE2-K), moderately (OCT1) or weakly (MRP4, OATP1B1, OATP1B3, OAT1 and OAT3). The clinical relevance of these interactions is not clear. Concomitant administration of rucaparib with proton pump inhibitors has no clinically meaningful change in rucaparib steady-state exposure [1].
Features and properties of rucaparib
Alternative names | AG 014699; AG-14699; CO-338; PF-01367338; PF-1367338; RUBRACA; Rucaparib camsylate |
Class | 3-Ring heterocyclic compounds, antineoplastics, azepines, indoles, small molecules |
Mechanism of action | Poly(ADP-ribose) polymerase inhibitors |
Route of administration | Oral |
Pharmacodynamics | Shows antitumour activity in cells with mutations in the BRCA1/2 and other DNA repair genes |
Pharmacokinetics | Displays linear, dose-proportional, time-independent pharmacokinetics; Tmax 1.9 h |
Most frequent adverse events | Nausea, fatigue (including asthenia), vomiting, anaemia, abdominal pain, dysgeusia, constipation, decreased appetite, diarrhoea, thrombocytopenia, dyspnoea, increase in creatinine, ALT, AST and cholesterol, and decrease in haemoglobin, lymphocytes, platelets and neutrophils |
WHO ATC code | L01X-X55 (Rucaparib) |
EphMRA ATC code | L1 (Antineoplastics) |
Chemical name | 8-fluoro-2-{4-[(methylamino)methyl]phenyl}-1,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one ((1S,4R)-7,7-dimethyl-2-oxobicyclo[2.2.1]hept-1-yl)methanesulfonic acid salt |
2.3 Therapeutic Trials
2.3.1 Ovarian Cancer
Rucaparib efficacy data are available from fully published articles [3, 12], US prescribing information [1], a conference presentation [35], posters [36, 37] and an abstract [38].
In a pooled analysis of ARIEL2 (NCT01891344) and Study 10 (NCT01482715), rucaparib treatment produced an ORR [assessed by the investigator using Response Evaluation Criteria In Solid Tumours (RECIST) 1.1] of 54% (95% CI 44–64) in 106 evaluable patients with high-grade ovarian cancer who had progressed on ≥2 prior platinum-based chemotherapies [1, 35]. Complete and partial responses were seen in 9 and 45% of patients, respectively [1, 35]. The median duration of response was 9.2 months (95% CI 6.6–11.6) [1, 35]. The median progression-free survival (PFS) was 10.0 months (95% CI 7.3–12.5) and 41% of patients were progression-free at 12 months [35]. ARIEL2 was a two-part, single-group, open-label, multinational, phase II trial [3] and Study 10 was a phase I/II trial [36]. Women with recurrent ovarian cancer with a deleterious BRCA1/2 mutation (germline or somatic [3] or germline [36]) were among those included in these trials. All patients received rucaparib 600 mg twice daily as monotherapy until disease progression or unacceptable toxicity [1]. This was the recommended phase II/III dose identified in the phase I portion of the Study 10 in patients with ovarian or other solid tumours (n = 56) [38].
In pooled subgroup analyses of ARIEL2 and Study 10, ORRs did not differ (95% CI overlapped) between germline and somatic mutations (53.4 and 46.2%), BRCA1 and BRCA2 mutations (53.7 and 53.8%), 2 and ≥2 prior chemotherapy (68.3 and 53.8%) or 2 and ≥2 prior platinum-based regimen (65.0 and 53.8%) subgroups [35]. In platinum-refractory, -resistant and -sensitive subgroups, the ORR (95% CI) was 0% (0.0–41.0), 25.0% (8.7–49.1) and 65.8% (54.3–76.1), respectively. In patients with a progression-free interval (after the latest platinum therapy) of <6, ≥6 to 12, and >12 months, the ORR (95% CI) was 18.5% (6.3–38.1), 62.5% (48.6–75.1) and 73.9% (51.6–89.8), respectively [35].
ARIEL2 (part 1) results suggest that an HRD signature can be a predictive biomarker of response to rucaparib in women with ovarian cancer [3]. Patients were classified into three predefined subgroups: deleterious BRCA1/2 germline or somatic mutation (BRCA-mutant; n = 40); BRCA wild-type with high or low LOH [high and low LOH, respectively; n = 82 and 70]. The median duration of rucaparib treatment was 5.7 months. The median PFS (95% CI) was 12.8 months (9.0–14.7) in the BRCA-mutant, 5.7 months (5.3–7.6) in the high LOH and 5.2 months (3.6–5.5) in the low LOH subgroups. The median PFS was significantly longer in the BRCA mutant [hazard ratio (HR) 0.27; 95% CI 0.16–0.44; p < 0.0001)] and high LOH (0.62; 0.42–0.90; p = 0.011) subgroups, compared with the low LOH subgroup. The proportion of patients with PFS (95% CI) at 12 months in these subgroups was 50% (33–65), 28% (18–39), and 10% (4–19), respectively [3].
In ARIEL2 (part 1), consistent with PFS, confirmed ORRs (by RECIST 1.1) were significantly higher in the BRCA-mutant (80%; p < 0.0001) and high LOH (29%; p = 0.0033) subgroups than in the low LOH subgroup (10%) [3]. A similar trend in ORRs were seen when RECIST 1.1 plus cancer antigen (CA)-125 criteria were applied. The median duration of response was also significantly (p < 0.05) longer in the BRCA-mutant (9.2 months) and high LOH (10.8 months) subgroups than in the low LOH subgroup (5.6 months). ORRs were generally similar irrespective of whether the BRCA mutation was germline or somatic, or BRCA1 or BRCA2. Among patients with BRCA wild-type tumours, genomic LOH predicted response to rucaparib with a significantly greater sensitivity (78%) than a mutation in non-BRCA homologous recombination genes (11%; p < 0.0001) or methylation of BRCA1 or RAD51C (48%; p < 0.021) [3]. The HRD signature identified in part 1 will be further refined (using a higher LOH cut-off) and prospectively applied to ARIEL2 (part 2) and ARIEL3 (Sect. 2.6) [3, 37].
In the phase II portion of the Study 10, rucaparib produced an ORR of 67 and 77% by RECIST 1.1 and RECIST 1.1 plus CA-125 criteria, respectively, in patients with ovarian cancer (n = 39 evaluable) [36]. In subgroup analyses, the ORR (by RECIST 1.1) was 67 and 73% in patients with BRCA1 (n = 27) or BRCA2 (n = 11) mutation, 64 and 82% in patients with a progression-free interval (after the latest platinum therapy) of 6–12 (n = 25) and >12 (n = 11) months, and 69% in those who had received ≥3 prior chemotherapy regimens (n = 13). The disease control rate (complete or partial response plus stable disease for >24 weeks) was 87% [36].
In an investigator-initiated trial (NCT00664781), a continuous dosing schedule of oral rucaparib (92–480 mg once daily or 240–600 twice daily within a 21-day cycle) was associated with disease control (complete or partial response plus stable disease for ≥12 weeks) in 12 of 13 patients with ovarian cancer [12]. This was an open-label, phase II trial in which 78 women with advanced breast or ovarian cancer harbouring proven germline BRCA1/2 mutations were treated with intermittent or continuous schedules of intravenous or oral rucaparib [12].
2.3.2 Other Malignancies
In an investigator-initiated phase II trial (NCT01074970), rucaparib (intravenous and then oral) plus cisplatin given after preoperative neoadjuvant therapy did not improve 2-year disease-free survival in patients with triple negative breast cancer [n = 128] (abstract presentation [39]). The dosage of rucaparib administered in this study was substantially lower than the recommended phase II/III dose of oral rucaparib 600 mg twice daily (Sect. 2.3.1) [39].
In the RUCAPANC trial (NCT02042378), oral rucaparib was associated with clinical benefit in some patients with relapsed pancreatic cancer and a known deleterious germline or somatic BRCA mutation [n = 19] (poster presentation [40]). The investigator-assessed ORR (by RECIST 1.1) was 15.8% (95% CI 3.4–39.6) and the disease control rate (complete or partial response plus stable disease for ≥12 weeks) was 31.6% (95% CI 12.6–56.6). RUCAPANC was an open-label, phase II trial in which patients received rucaparib 600 mg twice daily in 28-day cycles until disease progression [40].
In a phase II study sponsored by Pfizer, an intravenous formulation of rucaparib in combination with temozolomide was associated with confirmed partial response (17.4% of patients; 95% CI 7.9–31.6) and stable disease for ≥24 weeks (17.4%; 7.9–31.6) in some patients with metastatic melanoma (n = 46) [4]. The median PFS and overall survival was 3.5 (95% CI 2.0–6.2) and 9.9 (6.2–14.7) months, respectively. The 1-year survival rate was 40%. Rucaparib was administered at 12 mg/m2 1 h before temozolomide 200 mg/m2 (starting dosages) on days 1–5 in a 28-day cycle [4].
Key clinical trials of rucaparib
Drug(s) | Indication | Phase | Status | Location | Trial identifier | Sponsors |
|---|---|---|---|---|---|---|
Rucaparib, chemotherapy | Ovarian cancer | III | Recruiting | Multinational | NCT02855944 (ARIEL4) | Clovis Oncology |
Rucaparib, placebo | Ovarian cancer | III | Ongoing | Multinational | NCT01968213 (ARIEL3) | Clovis Oncology |
Rucaparib | Ovarian cancer | II | Ongoing | Multinational | NCT01891344 (ARIEL2) | Clovis Oncology |
Rucaparib | Ovarian cancer, solid tumours | I/II | Ongoing | Multinational | NCT01482715 (Study 10) | Clovis Oncology |
Rucaparib | Ovarian or breast cancer | II | Completed | UK | NCT00664781 | Cancer Research UK |
Rucaparib + bevacizumab | Ovarian cancer | II | Planned | NA | NA | NA |
Rucaparib + atezolizumab | Ovarian cancer | Ib | Planned | NA | NA | Clovis Oncology, Genentech |
Rucaparib, abiraterone acetate, enzalutamide, docetaxel | Prostate cancer | III | Recruiting | Multinational | NCT02975934 (TRITON3) | Clovis Oncology |
Rucaparib | Prostate cancer | II | Recruiting | Multinational | NCT02952534 (TRITON2) | Clovis Oncology |
Rucaparib | Breast cancer | II | Ongoing | UK | EudraCT2014-003319-12 (RIO) | The Royal Marsden NHS Foundation Trust, The Institute of Cancer Research |
Rucaparib | Breast cancer | II | Recruiting | France | NCT02505048 (RUBY) | UNICANCER |
Rucaparib + cisplatin, cisplatin | Breast cancer | II | Ongoing | USA | NCT01074970 | Hoosier Cancer Research Network |
Rucaparib | Pancreatic cancer | II | Completed | Multinational | NCT02042378 (RUCAPANC) | Clovis Oncology |
Rucaparib | Solid tumours | I | Recruiting | Hungary | NCT02986100 (AME) | Clovis Oncology |
Rucaparib + carboplatin | Solid tumours | I | Completed | UK | NCT01009190 | Clovis Oncology |
2.4 Adverse Events
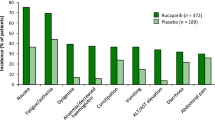
Monotherapy with oral rucaparib 600 mg twice daily had a manageable tolerability profile in women with high-grade ovarian cancer who had progressed on ≥2 prior platinum-based chemotherapies in the pooled analysis of ARIEL2 and Study 10 (n = 377) [1, 35]. The median duration of treatment was 5.5 months [1] and the median dose intensity was 0.92 [35]. Treatment-related all-grade and grade ≥3 adverse events were reported in 96 and 47% of patients, respectively [35]. Adverse events led to dose interruption or reduction in 62% of patients, most commonly because of anaemia (27%) and fatigue/asthenia (22%) [1]. Adverse events led to treatment discontinuation in 10% of patients, most commonly because of fatigue/asthenia (2%) [1]. Eight patients (2%) died because of progression of malignancy and one patient because of sepsis, which was assessed by the investigator as not related to rucaparib [35].
In the pooled analysis, the most common (incidence ≥20% for all grade) treatment-emergent adverse events (all grade; grade 3–4) were: nausea (77%; 5%), asthenia/fatigue (77%; 11%), vomiting (46%; 4%), anaemia (44%; 25%), constipation (40%; 2%), decreased appetite (39%; 3%), dysgeusia (39%; 0.3%), diarrhoea (34%; 2%), abdominal pain (32%; 3%), dyspnoea (21%; 0.5%) and thrombocytopenia (21%; 5%) [35]. Laboratory abnormalities reported in ≥35% of patients included (any worsening grade; shift to grade 3–4): an increase in creatinine (92%; 1%), ALT (74%; 13%), AST (73%; 5%) or cholesterol (40%; 2%), and a decrease in haemoglobin (67%; 23%), lymphocytes (45%; 7%), platelets (39%; 6%) or neutrophils (35%; 10%) [1]. AST and ALT levels normalised over time with continued treatment [35].
In the pooled analysis, myelodysplastic syndrome/acute myeloid leukaemia were reported in <1% of patients [35]. Therefore, patients should be monitored for haematological toxicity at baseline and monthly thereafter [1]. Based on its mechanism of action and data from animal studies, rucaparib can cause embryo-foetal toxicity when administered to pregnant women [1]. Hence, contraception is advised in women of reproductive potential during and 6 months after rucaparib treatment [1].
At steady state, rucaparib 600 mg twice daily increased Fridericia’s formula-corrected QT interval from baseline by a mean 14.9 ms in patients with solid tumours [1].
2.5 Companion Diagnostic
Foundation Medicine has developed a companion diagnostic (FoundationFocus™ CDx BRCA ) for rucaparib [41]. It is a next-generation sequencing-based in vitro device for the qualitative detection of BRCA1/2 sequence alteration in ovarian tumour tissue [41]. The test is approved by the US FDA for selecting patients for the treatment of ovarian cancer with rucaparib [1, 41].
2.6 Ongoing Clinical Trials
The pivotal programme for rucaparib in ovarian cancer is ongoing and includes two phase III trials, ARIEL3 (NCT01968213) [3] and ARIEL4 (NCT02855944) [35]. These trials are evaluating rucaparib in maintenance (ARIEL3) or treatment (ARIEL4) settings. ARIEL4 will compare rucaparib with standard chemotherapy. ARIEL2 and Study 10 are also still ongoing. An investigator-initiated phase II trial is planned to evaluate rucaparib in combination with bevacizumab (as a first-line maintenance therapy) and a phase Ib trial is planned in collaboration with Genentech to investigate the combination of rucaparib with atezolizumab in women with advanced ovarian cancer [42].
With regard to other indications, phase II (TRITON2; NCT02952534) and phase III (TRITON3; NCT02975934) trials are investigating rucaparib in metastatic castration-resistant prostate cancer. In addition to NCT01074970, two other investigator-initiated phase II trials are evaluating rucaparib in breast cancer [RIO (EudraCT2014-003319-12); RUBY (NCT02505048)]. The RIO trial is investigating the activity of rucaparib using Ki67 as a surrogate marker in patients with primary, sporadic, triple-negative or BRCA1/2-positive breast cancer [43]. A phase I trial is investigating the absorption, metabolism and excretion of rucaparib following a single oral dose in patients with advanced solid tumours (NCT02986100). An additional phase I trial is investigating drug interactions of rucaparib with caffeine, digoxin, omeprazole, vitamin K or warfarin in patients with solid tumours (NCT02740712).
3 Current Status
Rucaparib received its first global approval on the 19th of December 2016 for ovarian cancer in the USA.
References
Clovis Oncology Inc. Prescribing information for RubracaTM (rucaparib) tablets, for oral use. 2016. http://www.fda.gov. Accessed 9 Jan 2017.
Jenner ZB, Sood AK, Coleman RL. Evaluation of rucaparib and companion diagnostics in the PARP inhibitor landscape for recurrent ovarian cancer therapy. Future Oncol. 2016;12(12):1439–56.
Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(1):75–87.
Plummer R, Lorigan P, Steven N, et al. A phase II study of the potent PARP inhibitor, rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemother Pharmacol. 2013;71(5):1191–9.
Clovis Oncology Inc. Clovis Oncology, Inc. receives license for worldwide development and commercialization rights to Pfizer’s oral and IV PARP inhibitor PF-01367338 [media release]. 2011. http://www.clovisoncology.com.
Clovis Oncology. SEC filings: 10K annual report for 2013. 2013. http://www.clovisoncology.com. Accessed 21 Feb 2017.
Lonza. Lonza and Clovis Oncology sign strategic long-term manufacturing agreement to secure supply of rucaparib [media release]. 2016. http://www.lonza.com.
Murai J, Huang SY, Renaud A, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13(2):433–43.
Kern KA, Zhang S, Shalinsky DR, et al. Comparative PARP enzyme inhibition of PF-01367338, olaparib, and MK-4827 [abstract no. e13552]. J Clin Oncol. 2011;29(Suppl 15). doi:10.1200/jco.2011.29.15_suppl.e13552
Murray J, Thomas H, Berry P, et al. Tumour cell retention of rucaparib, sustained PARP inhibition and efficacy of weekly as well as daily schedules. Br J Cancer. 2014;110(8):1977–84.
Plummer R, Jones C, Middleton M, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14(23):7917–23.
Drew Y, Ledermann J, Hall G, et al. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br J Cancer. 2016;114(12):723–30.
Ihnen M, Zu Eulenburg C, Kolarova T, et al. Therapeutic potential of the poly(ADP-ribose) polymerase inhibitor rucaparib for the treatment of sporadic human ovarian cancer. Mol Cancer Ther. 2013;12(6):1002–15.
Chuang HC, Kapuriya N, Kulp SK, et al. Differential anti-proliferative activities of poly(ADP-ribose) polymerase (PARP) inhibitors in triple-negative breast cancer cells. Breast Cancer Res Treat. 2012;134(2):649–59.
Robillard L, Lin K, Lopez-Casas PP, et al. Preclinical efficacy of the PARP inhibitor rucaparib (CO-338/AG014699/PF-01367338) in pancreatic cancer models with homologous recombination deficiencies (HRD) [abstract no. 249]. Eur J Cancer. 2014;50(Suppl 6):83.
Drew Y, Mulligan EA, Vong W-T, et al. Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst. 2011;103(4):334–46.
Al Rubaish SS, Xiong Y, Marchion D, et al. Development of a rucaparib response signature that shows in vitro predictive value to the PARP inhibitors, ABT-888 and olaparib in ovarian cancer cells [abstract no. 54]. Gynecol Oncol. 2014;133(Suppl 1):22.
Audrey LC, Vanessa C, Marie K. The PARP inhibitor, Rucaparib enhance the sensitivity to Trabectedin in soft tissue sarcomas cell lines and in patient xenograft model [abstract no. 3002]. Cancer Res. 2016;76(Suppl 14). doi:10.1158/1538-7445.AM2016-3002
Falzacappa MV, Ronchini C, Faretta M, et al. The combination of the PARP inhibitor rucaparib and 5FU is an effective strategy for treating acute leukemias. Mol Cancer Ther. 2015;14(4):889–98.
Daniel RA, Rozanska AL, Mulligan EA, et al. Central nervous system penetration and enhancement of temozolomide activity in childhood medulloblastoma models by poly(ADP-ribose) polymerase inhibitor AG-014699. Br J Cancer. 2010;103(10):1588–96.
Hoch U, Charych D. Combining the long-acting topoisomerase 1-inhibitor etirinotecan pegol with the PARP inhibitor rucaparib to provide anti-tumor synergy without increased toxicity [abstract no. 33]. Eur J Cancer. 2014;50(Suppl 6):16–7.
Kotsopoulos IC, Begbie JA, Mukhopadhyay A, et al. In vitro single agent rucaparib cytotoxicity and cisplatin chemo-potentiation in cervical cancer cell lines [abstract no. IGCS-0702]. Int J Gynecol Cancer. 2016;26(Suppl 3):374.
Porcelli L, Quatrale AE, Mantuano P, et al. Optimize radiochemotherapy in pancreatic cancer: PARP inhibitors a new therapeutic opportunity. Mol Oncol. 2013;7(3):308–22.
Hunter JE, Willmore E, Irving JA, et al. NF-kappaB mediates radio-sensitization by the PARP-1 inhibitor, AG-014699. Oncogene. 2012;31(2):251–64.
Nile DL, Rae C, Hyndman IJ, et al. An evaluation in vitro of PARP-1 inhibitors, rucaparib and olaparib, as radiosensitisers for the treatment of neuroblastoma. BMC Cancer. 2016;16(1):621.
Cullinane C, Waldeck K, Eu P, et al. The PARP inhibitor, rucaparib enhances the antitumor activity of 177Lu-DOTA-octreotate radionuclide therapy in preclinical models of neuroendocrine tumor [abstract no. 1800]. Cancer Res. 2015;75(Suppl 15). doi:10.1158/1538-7445.AM2015-1800
McCrudden CM, O’Rourke MG, Cherry KE, et al. Vasoactivity of rucaparib, a PARP-1 inhibitor, is a complex process that involves myosin light chain kinase, P2 receptors, and PARP itself. PLoS One. 2015;10(2):e0118187.
Ali M, Kamjoo M, Thomas HD, et al. The clinically active PARP inhibitor AG014699 ameliorates cardiotoxicity but does not enhance the efficacy of doxorubicin, despite improving tumor perfusion and radiation response in mice. Mol Cancer Ther. 2011;10(12):2320–9.
Ali M, Telfer BA, McCrudden C, et al. Vasoactivity of AG014699, a clinically active small molecule inhibitor of poly(ADP-ribose) polymerase: a contributory factor to chemopotentiation in vivo? Clin Cancer Res. 2009;15(19):6106–12.
McCormick A, Nakjang S, Donoghue P, et al. The role of homologous recombination recovery in cisplatin and rucaparib resistance in ovarian cancer [abstract no. ESGO-0857]. Int J Gynecol Cancer. 2015;25(Suppl 9):627.
McCormick A, Donoghue P, Dixon M, et al. Ovarian cancers harbour defects in non-homologous end joining resulting in resistance to rucaparib. Clin Cancer Res. 2016. doi:10.1158/1078-0432.ccr-16-0564.
Durmus S, Sparidans RW, van Esch A, et al. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) restrict oral availability and brain accumulation of the PARP inhibitor rucaparib (AG-014699). Pharm Res. 2015;32(1):37–46.
Parrish KE, Cen L, Murray J, et al. Efficacy of PARP inhibitor rucaparib in orthotopic glioblastoma xenografts is limited by ineffective drug penetration into the central nervous system. Mol Cancer Ther. 2015;14(12):2735–43.
Sun Y, Ding H, Liu X, et al. INPP4B overexpression enhances the antitumor efficacy of PARP inhibitor AG014699 in MDA-MB-231 triple-negative breast cancer cells. Tumour Biol. 2014;35(5):4469–77.
Kristeleit RS, Shapira-Frommer R, Oaknin A, et al. Clinical activity of the poly(ADP-ribose) polymerase (PARP) inhibitor rucaparib in patients (pts) with high-grade ovarian carcinoma (HGOC) and a BRCA mutation (BRCAmut): analysis of pooled data from study 10 (parts 1, 2a, and 3) and ARIEL2 (parts 1 and 2) [abstract no. 856O]. In: European Society for Medical Oncology 2016 Congress. 2016.
Shapira-Frommer R, Oza A, Domchek SM, et al. A phase 2 open-label, multicenter study of single-agent rucaparib in the treatment of patients with relapsed ovarian cancer and a deleterious BRCA mutation [abstract no. 2746 plus poster]. Eur J Cancer. 2015;51(Suppl 3):S545.
Coleman RL, Swisher EM, Oza AM, et al. Refinement of prespecified cutoff for genomic loss of heterozygosity (LOH) in ARIEL2 part 1: a phase II study of rucaparib in patients (pts) with high grade ovarian carcinoma (HGOC) [abstract no. 5540 plus poster]. J Clin Oncol. 2016;34(Suppl).
Kristeleit RS, Burris HA, LoRusso P, et al. Phase 1/2 study of oral rucaparib: final phase 1 results [abstract no. 2573]. J Clin Oncol. 2014;32(Suppl 5).
Miller K, Tong Y, Jones DR, et al. Cisplatin with or without rucaparib after preoperative chemotherapy in patients with triple negative breast cancer: final efficacy results of Hoosier Oncology Group BRE09-146 [abstract no. 1082]. J Clin Oncol. 2015;33(Suppl 15).
Domchek SM, Hendifar AE, McWilliams RR, et al. RUCAPANC: an open-label, phase 2 trial of the PARP inhibitor rucaparib in patients (pts) with pancreatic cancer (PC) and a known deleterious germline or somatic BRCA mutation [abstract no. 4110 plus poster]. J Clin Oncol. 2016;34(Suppl).
Foundation Medicine Inc. FoundationFocus™ CDxBRCA: draft summary of technical information. 2017. http://www.fda.gov. Accessed 20 Feb 2017.
Clovis Oncology. Clovis Oncology announces Q2 2016 operating results and corporate update [media release]. 8 Aug 2016. http://www.clovisoncology.com.
Toms C, Chopra N, Houlton L, et al. Window study of the PARP inhibitor rucaparib in patients with primary triple negative or BRCA1/2 related breast cancer (RIO) [abstract no. 219TiP]. Ann Oncol. 2016;27(Suppl 6).
Disclosure
The preparation of this review was not supported by any external funding. During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the author on the basis of scientific completeness and accuracy. Y.Y. Syed is a salaried employee of Adis, Springer SBM.
Author information
Authors and Affiliations
Corresponding author
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Rights and permissions
About this article

Cite this article
Syed, Y.Y. Rucaparib: First Global Approval. Drugs 77, 585–592 (2017). https://doi.org/10.1007/s40265-017-0716-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0716-2