
Abstract
Inhibitors of catechol-O-methyltransferase (COMT) are commonly used as an adjunct to levodopa in patients with Parkinson’s disease (PD) for the amelioration of wearing-off symptoms. This narrative review aims to discuss the role of COMT inhibitors on peripheral levodopa metabolism and continuous brain delivery of levodopa, and to describe their metabolic properties. Oral application of levodopa formulations with a dopa decarboxylase inhibitor (DDI) results in fluctuating levodopa plasma concentrations, predominantly due to the short half-life of levodopa and its slowing of gastric emptying. Following transport across the blood–brain barrier and its metabolic conversion to dopamine, these peripheral ‘ups and downs’ of levodopa are reflected in fluctuating dopamine levels in the synaptic cleft between presynaptic and postsynaptic dopaminergic neurons of the nigrostriatal system. As a result, pulsatile postsynaptic dopaminergic stimulation takes place and results in the occurrence of motor complications, such as wearing-off and dyskinesia. More continuous plasma behaviour was observed after the combination of levodopa/DDI formulations with COMT inhibitors. These compounds also weaken a levodopa/DDI-related homocysteine increase, as biomarker for an impaired methylation capacity, which is involved in an elevated oxidative stress exposure. These findings favour the concept of chronic levodopa/DDI application with concomitant inhibition of COMT and monoamine oxidase, since deamination of dopamine via this enzyme also generates free radicals. This triple combination is suggested as standard levodopa application in patients with PD who need levodopa, if they will tolerate it.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Catechol-O-methyltransferase (COMT) inhibitors are well established for the treatment of wearing-off phenomena in patients with Parkinson’s disease (PD). |
Inhibition of COMT supports a more continuous brain delivery of levodopa. Inhibition of monoamine oxidase enables more continuous central dopamine levels. Both therapeutic approaches have synergistic effects for the principle of continuous dopamine substitution, which improves motor complications in patients with PD, as shown in clinical trials. |
Therefore, chronic levodopa/dopa decarboxylase application with concomitant inhibition of COMT and monoamine oxidase is suggested as standard levodopa application in patients with PD who need levodopa, if they will tolerate it. |
1 Introduction
Parkinson’s disease (PD) is the most common chronic neurodegenerative disease that affects movement behaviour. Approximately 2 % of individuals over the age of 65 years and up to 5 % of those aged over 85 years suffer from the disorder [1]. PD is mainly pathologically characterised by dopaminergic neuronal loss in the substantia nigra and consecutively by striatal dopamine loss with the accumulation of the protein α-synuclein [2–4].
However, chronic neuronal death also affects other neurotransmitter systems both in the periphery and in the brain. In the periphery, orthostatic hypotension is a result of sympathetic neurocirculatory failure, characterized by cardiac sympathetic failure. Both are in line with a reduced generation of biogenic amines in the adrenal medulla of patients with PD [5]. In the brain, the neurodegenerative process also takes place in the predominant norepinephrinergic locus ceruleus, the serotonergic Raphe nuclei and the cholinergic nucleus basalis Meynert. All of them induce dysfunction of cortical and limbic projections and disturbances of vegetative nervous system function in the region of the dorsal nucleus of the vagus nerve or the sympathetic ganglia [2–4].
Additionally, serious cytoskeletal damage is found in glutamatergic, gamma aminobutyric acid-ergic, cholinergic, noradrenergic, serotonergic and peptidergic neurons [6]. As a result, an individually pronounced and different expression of symptoms occurs in each patient with PD.
Tremor at rest, bradykinesia, rigidity and postural instability are the main features of motor impairment. These symptoms are accompanied by the onset of a wide variety of non-motor symptoms. Non-motor symptoms have gained increasing attention in previous years as further characteristic clinical features of the disease [7–9]. The term PD actually reflects a superordinate concept for a variety of different kinds of diseases. They resemble each other and do not always share the concept of the neuropathological manifestation of Lewy bodies as an essential initial step to the onset of the disease process [4, 10, 11].
1.1 The Cause of Parkinson’s Disease (PD) is Still Not Known
Various hypotheses exist as to the cause of PD, including genetic defects or gene mutations, impaired detoxification capacity, exposure to acute and chronic endogenous and exogenous toxins such as pesticides, deficiencies of mitochondrial function, infection by prion-like proteins, protein misfolding, inflammation, and decrease of neurotransmitter capacity, including monoamine storage vesicles and glutamate metabolism [12–14]. In the cascade of events leading to neuronal death, all of these hypotheses share one common step, which is an increased synthesis of free radicals. All of these mechanisms contribute to the onset of the heterogeneous forms of PD as a result of the predominant death of dopamine-synthesizing presynaptic nigrostriatal neurons [6, 12–14].
2 Objectives
This narrative review aims to discuss the role of catechol-O-methyltransferase (COMT) inhibition on peripheral levodopa metabolism, continuous brain delivery of levodopa and the metabolic advantages of COMT inhibition.
3 The Role of Dopamine in PD
3.1 Dopamine Supplementation as Therapeutic Principle
Substitution of the dopamine loss is the most essential treatment approach in PD for the alleviation of motor symptoms and of certain non-motor symptoms, for instance apathy and/or cognitive slowing. Both result from a decline in dopaminergic stimulation of the mesolimbic system and frontal brain structures. Non-motor and motor features of PD respond to dopaminergic therapy in an individually pronounced fashion. The compensation of this dopamine deficiency with the various available therapeutic modes of dopamine substitution preponderantly ameliorates, for instance, the motor symptoms akinesia and rigidity and, to a lesser extent, tremor. However, disturbances of postural reflexes do not respond to dopamine substitution [15]. This adjustment of the impaired nigrostriatal dopamine neurotransmission also prevents an adaptation of the patient with PD to the disease process itself. This is, for instance, the case for symptoms like walking with small steps only or the manifestation of bound posture. This altered movement behaviour partially results from an unconscious learning process to balance the PD-related deficits of emotion and motion execution [16]. Therefore, evidence is emerging that treatment in PD should start as early and as effectively as possible. This concept is supported by results from various long-term trials on the effects of early optimum direct or indirect adjustment of the nigrostriatal dopamine loss [17]. Direct dopamine application as the best theoretical physiological way is not possible, since dopamine itself may not pass the blood–brain barrier (BBB), in contrast to its direct metabolic precursor, levodopa. In the brain, levodopa is converted by the enzyme dopa decarboxylase to dopamine in dopaminergic and serotonergic neurons [15].
3.2 Levodopa: The Double-Edged Sword in the Treatment of PD
The most efficacious and best-tolerated drug for the treatment of PD is levodopa. The introduction of levodopa was a therapeutic breakthrough, although it is worth mentioning that levodopa as a drug for PD patients would probably not be approved in the contemporary clinical research world with its demands for safety and tolerability by the approving authorities due to too frequent onset of electrocardiographic changes, nausea and gastrointestinal disturbances [15, 18].
The effect of levodopa in PD was first reported by Birkmayer and Hornykiewicz [19], who had the courage to ask adventurous patients to take levodopa as an infusion in an adequate dosage. At that time, they tested a new treatment paradigm in a clinical research world with distinctly fewer administrative hurdles and bureaucratic overload [18]. Nowadays, the optimal use of levodopa is still under debate, predominantly due to the clinical observation of the onset of fluctuations of movement in the course of PD. These so-called motor complications are predominantly associated with levodopa due to its short plasma half-life [15].
3.3 Levodopa-Related Fluctuations of Movement
Motor complications may be roughly subdivided into OFF-phenomena, which describe the reappearance of a reduced motor performance after an ON-interval of good response to adequate dopaminergic neurotransmission, and into dyskinesia, which are involuntary movements that mostly result from an over-stimulation of the dopaminergic system.
Dyskinesia can occur during both ON and OFF periods. Classification of dyskinesia is generally performed in relation to the timing of levodopa administration. ON dyskinesia appear either (1) during the period when patients experience maximal relief from their motor symptoms, in which case they are classified as peak-dose dyskinesia [20–22], or (2) in a biphasic fashion, soon after intake of levodopa, when the patient starts to turn ON; they reappear again when the levodopa effect is wearing off and the patient begins to turns OFF. The threshold concentration for dyskinesia onset and the concentration necessary to get patients out of the OFF state increasingly converge with the progression of PD [23–25]. Therefore, the maximum levodopa plasma level after drug intake often causes peak-dose dyskinesia as the most common form of these involuntary movements [20]. As the disease progresses, patients may develop dyskinesia throughout the whole ON time, spreading over the whole body in an individually pronounced fashion [26]. Generally, PD patients better tolerate and accept mild dyskinesia more than OFF periods [27].
The risk of developing dyskinesia has been associated with a number of clinical factors. The severity of PD and the dosage of levodopa therapy, and a younger age of the patient are currently believed to be among the variables that best predict the development of dyskinesia [20, 28]. Dose, dosing strategy, and the timing of meals are further essential determining factors for the development of dyskinesia and motor complications [23, 26].
3.3.1 Motor Complications as an Essential Feature for the Progression of PD
Generally, the onset of motor fluctuations is regarded as one essential clinical milestone in the progression of PD. Peaks and troughs of plasma levodopa levels are, to a certain extent, transferred into ups and downs of dopamine concentrations [24]. This results in a pulsatile stimulation of postsynaptic dopamine receptors, which in turn supports onset of movement fluctuations [29]. Initially they are predictable and thus in relation to prior drug intake. Later they become unpredictable and show no relation to previous drug intake. Generally motor fluctuations can be brief or long term, lasting for minutes, or even hours. They cause patient disability, embarrassment and frustration and caregiver burden [20].
3.3.2 Treatment of Motor Complications: Still an Unmet Need
Therapy or even prevention of motor complications, particularly wearing-off phenomena, is still a major problem. These motor side effects of long-term levodopa therapy initiated a long debate with a focus on various hypothetical models of basal ganglia interaction and dysfunction [23, 24]. The long-term side effects of levodopa application in relation to the blocking of its metabolizing enzymes were only considered to a certain extent.
3.4 Levodopa: Modes of Oral Application
Levodopa was initially administered as an infusion, followed by an oral form without inhibition of the essential levodopa-degrading enzymes dopa decarboxylase and COMT. Treatment with oral levodopa was subsequently improved with the combination of oral levodopa formulations and dopa decarboxylase inhibitors (DDIs). The two commonly used dopa decarboxylase inhibitors are carbidopa and benserazide, both of which only act in the periphery. The next step was the introduction of COMT inhibitors [15, 19, 30].
3.5 Blocking of Levodopa Metabolism
The basic pharmacological principle underlying many of the approved drugs for PD involves enzymatic inhibition of levodopa degradation, leading to a reduction in the peripheral conversion of levodopa to dopamine. Therefore, the plasma bioavailability of each orally administered levodopa compound rises due to the extension of the plasma half-life of levodopa. Accordingly, the clinical benefit of levodopa on motor behaviour improves in patients with PD.
The addition of a DDI to levodopa allows a four- to fivefold reduction of the oral levodopa dose. As a result, the frequency of levodopa-related peripheral side effects, such as nausea and vomiting, declines. DDI shifts the peripheral levodopa turnover to the COMT enzyme.
COMT is a major catabolic regulator of synaptic catecholamine neurotransmitters. COMT catalyzes the transfer of a methyl group to catecholamines and degrades dopamine, norepinephrine and epinephrine [31]. COMT is densely expressed throughout both the prefrontal cortex and the limbic system [32]. High COMT activity is also found in the liver, kidney and gut wall. The enzyme activity is controlled by the COMT gene.
The two forms of COMT (soluble COMT [S-COMT] and membrane-bound COMT [MB-COMT]) are coded by a single gene. This gene is located on the chromosome band 22q11.2 [31, 33]. S-COMT contains 221 amino acids. MB-COMT has an additional amino terminal extension of 43 (rat) or 50 (human) amino acids. The hydrophobic 17 and 24 amino acid residues in rats and humans, respectively, form an alpha-helical transmembrane domain, which is the membrane anchor. MB-COMT is not a precursor of S-COMT [33]. Constraint of COMT enzyme activity further diminishes peripheral levodopa metabolism.
This adjunct prolonging of the levodopa plasma half-life elevates its plasma appearance and, accordingly, its brain delivery. Experimental and clinical study results underline the efficacy of peripheral dual inhibition of the main levodopa-metabolizing enzymes, which reduce peripheral dopamine generation and accumulation of the levodopa metabolite 3-O-methyldopa (3-OMD) [34, 35].
3.6 3-OMD
The plasma half-life of the O-methylated levodopa derivative 3-OMD is between 15 and 24 h and depends on renal excretion [34]. 3-OMD competes with levodopa at the large neutral amino acid transport carriers of the gastrointestinal tract and of the BBB. Elevation of peripheral 3-OMD concentrations may interact with the absorption, plasma bioavailability and brain delivery of levodopa. COMT inhibition reduces 3-OMD synthesis [36–38]. Hypotheses suggest that this 3-OMD reduction contributes to a better absorption and BBB transfer of levodopa [34, 35]. This issue is still under debate. It was also suggested that at clinical concentrations 3-OMD makes a small contribution to the large total neutral amino acid pool competing with levodopa for brain entry [36, 39].
3.7 Oral Levodopa Administration and Continuous Dopaminergic Stimulation
The first therapy interval with levodopa is generally described as the honeymoon period, since it is associated with good tolerability and motor response to oral levodopa intake. The onset of predominantly levodopa-related motor complications may occur after an interval of several months or years [23]. The plasma levodopa half-life of approximately 60–90 min following oral intake determines its pharmacokinetic behaviour to a considerable extent, which is characterised by ups and downs of peripheral levodopa plasma levels [40–43].
This variability in levodopa levels is further promoted by gastrointestinal transport and absorption mechanisms. Following the swallowing process, levodopa-containing tablets must pass the stomach and reach the jejunal structures, where levodopa absorption predominantly takes place [44]. Pharmacokinetic investigations comparing oral levodopa/carbidopa application in a standardized fashion, with and without intake of the COMT inhibitor entacapone on two different days, described nearly identical concentration–time curves [40, 41, 43, 45]. However, there were differences between subjects. One possible reason is the influence of the gastric emptying rate. Slowed or delayed gastric emptying decreases plasma levodopa occurrence in general and delays peak levodopa concentrations. Thus, the gastric emptying velocity is one further essential determinant for the onset of levodopa effect on motor symptoms in patients with PD [41–43, 46].
All these peripheral mechanisms of gastrointestinal transport, absorption and pharmacokinetic behavior of levodopa predispose for occurrence of motor complications [44]. Accordingly, continuous, duodenal levodopa/carbidopa infusion pump systems reduce these peripheral levodopa plasma fluctuations. They circumvent the impact of gastric emptying. Frequency and intensity of motor complications in patients with very advanced PD considerably improve [47]. An additional further essential component of levodopa absorption may be the impact of food. High protein content, for instance in meat, eggs or fish, interacts with the gastrointestinal transporter system and limits levodopa uptake. Proteins or fat may also slow the gastric emptying velocity. These nutritional factors further facilitate an inter- and intra-individual variability of the peripheral levodopa metabolism behaviour in patients with PD [44]. Therefore motor complications may vary from day to day in terms of frequency and severity.
3.8 Central Prerequisites in the Brain for Manifestations of Motor Complications
After its transport across the BBB, levodopa is transformed and stored in vesicles of presynaptic dopamine-generating neurons or, as an alternative, in serotonin-generating neuronal cells. Progression of PD increases presynaptic neuronal degeneration and thus reduces the capacity of presynaptic dopamine storage. Moreover, control of synaptic dopamine concentrations via the presynaptic dopaminergic autoreceptors, and thus regulation of presynaptic endogenous dopamine synthesis, gets increasingly lost [25, 48]. Both mechanisms of neuronal degeneration ease the release of abnormally high dopamine concentrations into the synaptic cleft. They enable non-physiological fluctuations of striatal dopamine levels, which complement with the ups and downs of levodopa plasma levels. These processes finally support a pulsatile, irregular stimulation of postsynaptic dopamine receptors and neurons.
Further downstream, intracellular changes take place [49]. They limit adequate physiological neuronal function, which is normally based on the principle of a continuous neurotransmission of dopamine [50]. Therefore, motor complications occur in the long term. The duodenal levodopa/carbidopa gel infusion by a pump system allows a more direct, external fine tuning of levodopa and indirectly more continuous dopamine supply in nigrostriatal structures. However, this method is expensive, complex and may cause dangerous, severe gastrointestinal infections and neuropathy [51–54]. Therefore, an optimisation of the oral levodopa drug supply is warranted by routes circumventing gastrointestinal absorption via transdermal or subcutaneous application routes or the additional use of modulators of peripheral and central levodopa and dopamine metabolism [55, 56]. These drugs are inhibitors of COMT and monoamine oxidase B (MAO-B).
4 The Development of COMT Inhibitors
4.1 The Failed Retarded Release Concept
Various approaches were undertaken to prolong the efficacy of each levodopa dose and to smooth out the fluctuations of levodopa plasma levels. The aim was to enable a more continuous levodopa brain delivery. First, oral levodopa/DDI formulations were developed with an extended release profile. These tablets showed a declined clinical efficacy in comparison with the conventional levodopa/DDI tablets, when the same oral l-dopa dosage was given. No delay of onset of motor complications according to clinical study outcomes was observed. However, these clinical investigations were not designed to assess or determine OFF phenomena and dyskinesia in detail [57, 58]. There is some evidence that retarded-release levodopa formulations show some additional benefit when applied with COMT inhibitors [59]. However, this concept was not further developed in randomized clinical trials.
4.2 Pharmacokinetic Behaviour, Dosing Intervals, Levodopa and COMT Inhibition: A Complex Issue
The principle of dual enzyme inhibition of dopa decarboxylase and COMT during oral levodopa/DDI therapy was further investigated in view of an experimental animal study outcome. It described less frequent and less intense dyskinesia during treatment with levodopa/DDI combined with the COMT inhibitor entacapone, given four times daily, when the COMT inhibitor was started right from the beginning of treatment [29, 60].
One clinical attempt to translate this concept into clinical practice was undertaken with the STRIDE-PD (STalevo Reduction In Dyskinesia Evaluation) study [61]. This investigation aimed to initiate levodopa therapy with levodopa/carbidopa or levodopa/carbidopa/entacapone, given four times daily at 3.5 h intervals. The primary endpoint was defined as the interval to onset of dyskinesia. Levodopa was applied with a fixed dosing regimen. No adaption of the dosing interval or dosage was allowed with an initial appearance of motor complications. However, this is common in clinical practice [62]. The study design did not consider pharmacokinetic findings on levodopa metabolism in patients with PD. They demonstrated that repeat additional dosing of entacapone, for instance every 3 h, or of tolcapone, increased the maximum and minimum plasma levodopa concentrations [40, 41, 43, 63, 64]. This result was not found after a single application of COMT inhibitors to a levodopa formulation [43, 63–65]. Elevation of bioavailability and an increase in the peak concentrations of levodopa after repeated levodopa intake support the risk for onset of peak-dose dyskinesia.
4.3 Reasons for the Performance of STRIDE-PD
Earlier clinical studies circumvented the problem noted in the STRIDE-PD trial [66, 67]. They were performed in levodopa-naïve patients. According to the study protocols, subjects received levodopa only three times daily (tid), with a distinctly longer dosing interval between drug administration than was used in STRIDE-PD.
4.3.1 FIRST-STEP-Study
The FIRST-STEP (Favorability of Immediate-Release Levodopa/Carbidopa vs STalevo Short-Term comparison in Early Parkinson’s disease) study compared the efficacy of two different modes of levodopa application in patients with early PD who needed to initiate levodopa therapy. One study arm received conventional levodopa/carbidopa tablets. The other group of patients received levodopa/carbidopa together with the COMT inhibitor entacapone in one tablet. This multicentre, double-blind, randomized, parallel-group study administered a fixed oral levodopa dose of 300 mg/day, administered as 100 mg levodopa formulations tid at approximately 5 h intervals to 424 patients with PD. In the 39-week study, patients in the levodopa/carbidopa/entacapone arm performed significantly better than those in the levodopa/carbidopa-treated cohort, both after week 4 and throughout the remaining course of the study according to the computed sum scores of the Unified Parkinson’s Disease Rating Scale (UPDRS) part II (activities of daily living) and UPDRS part III (motor examination) as main primary outcome at the remaining study visits.
Thus, the FIRST-STEP trial only demonstrated that levodopa/carbidopa was inferior to levodopa/carbidopa/entacapone treatment, probably due to the more continuous peripheral levodopa plasma occurrence as a result of the COMT inhibition [63, 66]. The known additional levodopa/DDI efficacy-enhancing effects of entacapone, given as an extra tablet, to an existing levodopa/DDI regimen in treated patients was confirmed.
4.3.2 The ELLDOPA Study
Levodopa/carbidopa was given in different dosages (50 mg, 100 mg and 200 mg levodopa tid) in comparison with placebo tid in the ELLDOPA (Earlier versus Later LevoDOPA) study. Therefore, a plasma accumulation of levodopa was also unlikely and, if at all, occurred only after administration of the higher levodopa dosages with more pronounced ups and downs of levodopa plasma concentrations. Accordingly, dyskinesia rarely appeared and, if at all, mostly in higher dosages, for instance in the 200 mg tid arm after 39 weeks [67].
4.3.3 Interpretation of Both Trials
Both FIRST-STEP and ELLDOPA were designed as short-term follow-up studies. They did not assess the rate of motor complications during chronic therapy of patients with PD as a primary objective. Nevertheless, both trials showed some interesting findings regarding the onset and frequency of wearing off in patients with PD. In the FIRST-STEP trial, the number of monitored wearing-off phenomena was higher in the levodopa/carbidopa arm than in the levodopa/carbidopa/entacapone-treated patients. The frequency of noted wearing-off phenomena was rather low in relation to the size of the study population and the short observation interval. Therefore, this difference was not significant in the patients with early PD. Nevertheless, the results of these earlier studies allowed the conclusion at that time that entacapone supplementation may help to prevent the onset of wearing off due to a more continuous brain delivery of levodopa. In the ELLDOPA trial, the number of patients experiencing wearing off increased with higher levodopa/carbidopa dosing, probably due to more pronounced fluctuations of levodopa plasma levels in comparison with less pronounced levodopa fluctuations in plasma during the application of lower levodopa dosages.
4.3.4 The Failure of STRIDE-PD
STRIDE-PD confirmed that COMT inhibition improves wearing off. The interplay between pharmacokinetic plasma behaviour, shorter dosing intervals, COMT inhibition and the demands of the design with the missing possibility to adapt the levodopa dosage after initial onset of probable—mostly peak dose—dyskinesia were essential reasons for the premature onset of dyskinesia in the levodopa/carbidopa/entacapone arm in the STRIDE-PD trial [63, 68]. A more pronounced levodopa plasma accumulation took place, particularly where the higher oral levodopa dose was administered every 3.5 h [61]. This was indirectly confirmed in a further analysis of the failed STRIDE-PD trial, which described the oral levodopa dosage and thus more pronounced fluctuations of levodopa plasma levels as the main prerequisite for the onset of wearing off and dyskinesia in PD [28, 69].
4.4 The Importance of Pharmacokinetics for the Pharmacodynamics of Levodopa
It is noteworthy that certain levodopa-equivalent calculations should be scrutinized in randomized clinical trials. These so-called evidence-based medicine reviews suggest that patients receive levodopa 100 mg with one levodopa/carbidopa 100 mg formulation, levodopa 133 mg with one levodopa/25 mg carbidopa/200 mg entacapone tablet and levodopa 150 mg with one levodopa/carbidopa 100 mg combined tablet administered with tolcapone 100 mg. Therefore, one may assume a better efficacy of levodopa on motor behaviour based on higher levodopa plasma occurrence only [70]. However, this concept of equivalent dosage calculations is misleading, since the pharmacokinetic behaviour of levodopa during repeated intake and the dosing intervals are not considered in terms of pharmacodynamic effects of levodopa [40, 62]. The importance of these functional aspects of levodopa behaviour in plasma and delivery to the brain were shown in a pharmacokinetic trial. Within a standardised design, patients received the following via oral administration: (1) levodopa 100 mg plus carbidopa 25 mg in the morning and 4.5 h later again; (2) 1 week later, they received the same oral administration of levodopa plus entacapone 200 mg each; and (3) 1 week later the same oral levodopa dosage with tolcapone 100 mg each. Interestingly, the plasma bioavailability did not significantly differ between all three conditions. More continuous levodopa plasma behaviour was observed during additional entacapone/tolcapone intake. Thus, a less pronounced fall of levodopa, higher minimum levodopa concentrations and a lower fluctuations index appeared during additional COMT inhibition. All caused a beneficial effect on motor response during COMT inhibition [63]. Therefore, individually adapted dosing intervals during levodopa fractionation are essential in clinical practice to minimize levodopa plasma fluctuations and to optimise the motor response to levodopa [41, 63, 71].
5 COMT Inhibition and Gastrointestinal Absorption of Levodopa
A further difference exists between the levodopa/DDI administration with and without COMT inhibition. Generally, levodopa uptake depends on gastric emptying time, gastrointestinal absorption and transport via the gastrointestinal amino acid transporter system, as mentioned previously. Patients with PD often receive combination therapy involving multiple daily dosing of a particular compound and additional supplementation with other drugs, which share modes of action. Efficacy of all administered compounds depends on patient compliance, the nature of the delivery system, physicochemical properties of the drug and physiological considerations. Therefore, interactions between these compounds are likely. They may affect the rate at which the drug is absorbed throughout the gastrointestinal tract, then its bioavailability and metabolism [42, 44, 70].
Consistent COMT inhibition promotes the synthesis of more basic levodopa metabolites, i.e. the tyrosine aminotransferase-dependent substrates dihydroxyphenylpyruvate acetate and trihydroxyphenylacetate. Therefore, COMT inhibition may model the environmental pH and thus intestinal conditions for the duodenal absorption rate of levodopa. COMT appears in higher concentrations in the cells of the gastrointestinal tract. The physicochemical properties of a drug also affect its absorption through the gastrointestinal tract. Compounds, including levodopa, are weak bases or weak acids or are the salts of them and, as such, demonstrate pH-dependent solubility. The pH partition hypothesis asserts that the passage rate of a drug through a membrane depends on the environmental pH and the acid-base dissociation constant (pKa) of the drug. Drugs with low pKa are not ionized in the stomach and subsequently are rapidly absorbed. On passage to the small intestine, with its comparatively increased pH, the rate of ionisation changes and absorption subsequently slows.
The converse is true for drugs with a higher pKa value. This influences the bioavailability of hydrophilic drug formulations. They have a narrow window of absorption, limited predominantly to the stomach or the upper intestine.
Absorption is also limited by low pKa values and/or the site of active transport absorption mechanism, for instance in the case of levodopa [72]. Additionally, the absorption behaviour of oral levodopa/DDI tablets also depends on gastrointestinal transit rates, since uptake of levodopa occurs mainly in the proximal third of the small intestine (duodenum/jejunum) but not in the stomach. Intestinal levodopa absorption is rapid, but the plasma bioavailability of levodopa is only 30 % as a result of prior degradation to dopamine by DDI and to a lesser extent to 3-OMD by COMT, i.e. in the gut cells. The longer levodopa remains in the stomach and the small intestine, the more extensively it is metabolized and becomes less available for absorption [34]. A formulation sharing the peripheral absorption site profile of levodopa, is sodium octanoate. It is used as [13C] marked substrate in breath tests, which are non-invasive, feasible, alternative methods without ionizing radiation to assess the gastric emptying velocity of solids and liquids. After intake, [13C]-sodium octanoate is rapidly absorbed from the proximal intestine and carried to the liver via the portal venous system. There it is oxidized and eliminated as CO2 in the breath, reflecting gastric emptying as the rate-limiting step of the process. Significant relations between levodopa plasma concentrations and the outcomes of the [13C]-octanoic acid breath test were shown. There was no impact of COMT inhibition on gastric emptying time. However, the COMT inhibitor increased the recovery rate of the salt [13C]-sodium-octanoate [72]. Therefore, one may assume that levodopa is better absorbed during COMT inhibition due to a more basic environment, which improves the absorption of the acid levodopa. This may further enhance the absorption and the bioavailability of levodopa due to COMT inhibition. The COMT inhibitors tolcapone (C14H11NO5) and entacapone (C14H15N3O5) are both weak acids and have low aqueous solubility at acidic pH, which increases considerably in basic pH conditions. In turn, this supports absorption of both COMT inhibitors themselves in a basic environment. Tolcapone is significantly more lipophilic than entacapone at physiological pH values, therefore it passes the BBB as precondition for its central actions [63, 73, 74]. Further additional metabolic aspects of COMT inhibition exist.
6 Metabolic Aspects of COMT Inhibition
COMT is a widespread enzyme in the human body. One of its main tasks is the adding of methyl groups to a wide variety of compounds [34].
6.1 Chronic Methylation of Toxins and Drugs
Generally, metabolism also generates harmful substances during reduction, oxidation, conjugation and excretion. If substances such as drugs are present in higher concentrations than normal or are not expected to be present or produced, they will be metabolised [75]. Important enzymes for the hepatic microsomal detoxification degradation are in the cytochrome P450 system. Transferases, i.e. UDP-glucuronosyltransferase, glutathione S-transferase, N- and O-methyltransferase are further examples [75, 76]. These enzymes are responsible for the turnover of certain drugs and endogenous and exogenous toxins. Toxins often lose their dangerous effect if a methyl group is added [34, 77]. In case of overstraining this kind of detoxification by chronic exposure or excessive occurrence of too high concentrations of the methylated substrate, this chronic reaction leads to an up-regulation of homocysteine synthesis as a biomarker for an elevated methyl group consumption [34, 76].
6.2 Examples for Drug-Induced Limitations of Methylation Capacity
The degradation by methylation of many anticonvulsive and centrally acting drugs, such as valproic acid or levodopa, also consumes methyl groups. Accordingly, chronic therapy with these compounds induces a homocysteine increase [78, 79]. Subsequently, a decline of methyl group-donating vitamins, such as folic acid or vitamins B6 or B12, occurs, as these vitamins promote the conversion of homocysteine to the methyl group donor methionine again. In the long run, a drug-induced deficiency of methyl group-donating vitamins occurs [75]. As a consequence of these metabolic changes during long-term administration of levodopa, an acceleration of ageing-associated brain atrophy, small vessel disease, cognition deterioration and peripheral nerve dysfunction may hypothetically appear [46, 80]. These symptoms are found in the course of PD, particularly when patients are receiving a high-dose levodopa/DDI regimen [16, 81].
6.3 The Role of Levodopa Turnover
In the presence of a DDI, the degradation of levodopa is predominantly shifted to O-methylation of levodopa to 3-OMD by COMT. COMT transfers a methyl group from the donor methionine. The resulting derivative S-adenosylmethionine is transformed into the short-living S-adenosyl-homocysteine and then to homocysteine [16, 81].
6.4 Homocysteine as a Marker for the Capacity of Methylation Processes
The up-regulation of homocysteine production reflects an inappropriate or reduced capacity for metabolism of other methylation processes. N-methyltransferase and O-methyltransferase, for instance COMT, have a broad detoxification potential that is regulated by a limited availability of methyl groups. If chronic drug degradation via COMT, like in the case of levodopa, consumes methyl groups, endogenous or exogenous toxins will no longer be detoxified by methylation processes in an adequate manner. Accordingly, the vulnerability for exposition against endogenous xenobiotics or exogenous substances, such as rural toxins and pesticides, increases [16, 81]. Against this background, it is interesting that chronic pesticide exposure is under investigation as a PD onset and progression-supporting phenomenon. However, researchers do not yet consider a possible impact of chronic levodopa/DDI exposure in PD patients on their findings [82].
6.5 The Methylation Potential and the Reversible Homocysteine Degradation to Methionine
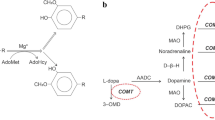
A homocysteine increase also changes the ratio between the methyl group donor methionine, its metabolic intermediates S-adenosylmethionine and S-adenosyl-L-homocysteine, and finally homocysteine. This ratio is defined as methylation potential. The methylation potential also describes the flow of methyl groups between cells. Chronic homocysteine elevation is associated with higher levels of S-adenosyl-L-homocysteine and a low methylation potential. Thus, the re-methylation capacity declines. An increase of S-adenosyl-L-homocysteine also supports a further feedback inhibition of the S-adenosylmethionine-dependent methyltransferases, including the DNA methyltransferases. Thus, a low methylation potential is related to a decreased DNA methylation capacity, which may also weaken methylation-dependent gene regulation [75]. For instance, hyperhomocysteinemia exerts highly selective inhibitory effects on cyclin A transcription through a hypomethylation-related mechanism, which blocks cell cycle progression and regeneration [75]. In addition to remethylation of homocysteine, a further pathway for homocysteine decrease is an irreversible turnover of homocysteine to cysteine (Fig. 1) [16, 76, 83, 84].

Simplified schema of homocysteine turnover. 3-OMD 3-O-methyldopa, CH 3 methyl group
6.6 Homocysteine Transformation to Cysteine and Synthesis of Antioxidants
This transsulfuration reaction metabolizes homocysteine to cystathionine by the enzyme cystathionine β-synthase [84]. Normal cystathionine β-synthase activity is essential for the generation of the cystathionine metabolite cysteine. Levels of this amino acid increase following the irreversible vitamin B6-dependent degradation of homocysteine. Accordingly, a cysteine elevation occurs in levodopa-treated patients [85]. Cysteine, l-glycine and glutamine acid are the essential parts of the antioxidant glutathione, which is also known as the tripeptide γ-glutamyl-cysteine-glycine. Its levels reflect the thiol redox state, which is a fundamental mediator of numerous cell processes. Glutathione is available in reduced monomeric and in oxidized dimeric forms, named GSSG, when thiol groups were reduced [86, 87]. Glutathione is abundant in the cytosol, nuclei and mitochondria, which are essential determinants of neuronal excitability and viability.
6.7 Glutathione Metabolism and Free Radical Generation
If glutathione scavenges free radicals, it will not be transformed to its metabolite cysteine-glycine. As consequence of this, the fall of cysteine-glycine following the application of a compound may be looked upon as an indirect biomarker for the induction of oxidative stress by this agent [88]. This was found following levodopa application with and without COMT inhibition. It is known that free radicals consume antioxidants like glutathione, which is subsequently converted to the derivative GSSG, the dimer of glutathione [88, 89]. An up-regulation of glutathione production may additionally encounter increased free radical appearance. Accordingly, a cysteine decay following a drug intake also indirectly reflects antioxidant consumption due to free radical scavenging. Unphysiological and too high free radical concentrations are involved in irregular harmful cellular metabolism, altered communication between cells and progression of neuronal degeneration [88, 89]. Generally, the activity of certain enzymes involved in glutathione metabolism may be associated with oxidative stress reduction, but other pathways also cause free radicals.
7 Monoamine Oxidase and Oxidative Stress
One of the pathways that causes free radical production is the mitochondrial monoamine oxidase, which is important for glial and mitochondrial dopamine degradation. Two types exist. Preponderantly, the subtype MAO-B is responsible for the oxidative deamination of dopamine. This reaction is supplemented by a reduction of molecular oxygen to hydrogen peroxide, a reactive oxygen species. If either the synthesis of reactive oxygen species is increased or the levels of antioxidants are reduced, oxidative stress will increase [13, 14, 86, 87].
7.1 Findings in Patients with PD
Chronic and high dosing of levodopa elevated homocysteine concentrations in levodopa-treated PD patients, but not in levodopa-naïve patients and healthy controls. These findings further confirm that levodopa, rather than the disease per se, induces hyperhomocysteinemia [90]. In addition to the levodopa dose, the treatment duration and disease severity and duration may also contribute to the elevation of homocysteine levels. This homocysteine rise was particularly found during high-dosage levodopa treatment with duodenal—or chronic oral—levodopa intake in PD patients [91]. This concomitant homocysteine increase during levodopa treatment is not only under suspicion as being associated with onset of neuropsychiatric symptoms but also of accelerating the ensuing peripheral axonal and central neurodegeneration, predominantly in the nigrostriatal, dopaminergic system [52, 91–97]. It may also contribute to elevated mortality rates from arteriosclerotic diseases and small vessel disease identified post-mortem in PD brains [80, 98].
7.2 COMT Inhibitors Lower or Even Prevent Homocysteine Rise
One further approach for homocysteine reduction is COMT inhibition on a regular basis, when levodopa/DDI treatment is performed and patients tolerate the COMT inhibitor. Since the combination of levodopa/DDI and COMT inhibitors reduces O-methylation of levodopa, it also decreases homocysteine levels [99]. A small prospective pivotal trial showed that addition of tolcapone to a stable anti-parkinsonian drug regime reduced homocysteine and its precursor S-adenosyl-L-homocysteine [100]. American prospective investigations with the COMT inhibitor entacapone failed, probably due to folate supplementation in the American diet, leading to a milder increase in homocysteine than expected [101, 102]. European observational non-prospective studies showed lower homocysteine in entacapone-treated patients. Thus, folate supplementation in the North American diet may also explain the heterogeneity of results [99, 100, 102–109].
8 Consequences of the Functional and Metabolic Advantages of COMT Inhibition for Levodopa Therapy in PD Patients
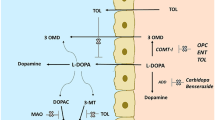
All these considerations and findings would hypothetically favour the concept of chronic levodopa/DDI application with immediate concomitant COMT and MAO inhibition once levodopa has to be introduced in the therapy of PD as the most efficacious therapeutic compound [17, 18, 110] (Fig. 2). This combination is suggested as standard for levodopa application in PD therapy.

Suggestion for an optimised dopamine substitution with levodopa. BBB blood–brain barrier, COMT-I catechol-O-methyltransferase inhibitor, COMT-I* not available central-acting catechol-O-methyltransferase inhibitor without use restrictions, DDI dopa decarboxylase inhibitor, MAO-I monoamine oxidase inhibitor, vitamins methyl group donating vitamins, plus indicates advantage
8.1 Levodopa/Dopa Decarboxylase Inhibitor (DDI) plus Inhibition of COMT and MAO: Reasons
There are two essential reasons for the implementation of levodopa therapy with DDI, COMT inhibitors and MAO-B inhibitors, which actually inhibit also MAO-A during repeated dosing [110]. A precondition is that this combination is safe and tolerable for the patient, which mostly is a result of individual exposure of the patient to this combination in clinical practice.
8.2 Continuous Dopamine Substitution
The first reason is that this concept is supported by a more continuous levodopa brain delivery on the basis of less fluctuating levodopa plasma levels with COMT inhibition and a more stable dopamine concentration in the synaptic cleft provided by MAO-B inhibition, with subsequent sparing of levodopa in the long term [111]. Whether the probably fluctuating COMT inhibition over the day, which requires intake of the available COMT inhibitors, is a certain drawback in terms of continuous levodopa pharmacokinetic behaviour is not yet known. However, both pharmacologic principles complement each other in terms of more continuous dopaminergic stimulation [112, 113]. A central-acting, safe COMT inhibitor without application restrictions would be of further advantage but is not currently available.
8.2.1 Less Oxidative Stress
The second reason is that the combination of both MAO-B inhibition and COMT inhibition also decreases oxidative stress induced via the metabolism of dopamine via MAO-B and the decrease of free radical generation triggered by homocysteine elevation at least in the periphery (Fig. 1) [112, 113]. A supplemental intake of methyl group-donating vitamins is recommended.
9 Available COMT Inhibitors
In the late 1980s, several compounds with a nitrocatechol structure were developed as potent, selective and reversible COMT inhibitors. They were considered to be second-generation COMT inhibitors, namely tolcapone and entacapone. Following positive results from large clinical trials in patients with fluctuating PD symptoms, both were introduced into clinical practice for PD in the late 1990s for the treatment of wearing off. Their pharmacological properties have been reviewed in detail [35]. From the pharmacological point of view, tolcapone appears to be more efficacious than entacapone, with higher inhibition of COMT activity, central action and a longer duration of action after oral administration to rats and humans [73, 74, 114]. The most common observed side effect of the available COMT inhibitors is harmless discoloration of the urine. An additional clinically relevant adverse event of COMT inhibition is diarrhoea, sometimes occurring even up to 2–4 months following treatment initiation [115].
9.1 Entacapone
The only peripherally acting COMT inhibitor, entacapone, was initially given as an extra tablet with each levodopa/DDI dose [115]. It improved the efficacy of levodopa on motor impairment with the focus on reduction of OFF-time in patients with fluctuating PD [116–124].
9.1.1 Safety and Tolerability
Phase III studies and post-marketing surveillance showed the safety, tolerability and efficacy of levodopa in combination with the DDIs, carbidopa or benserazide, and entacapone even with co-administration of selegiline, dopamine agonists and antidepressants such as imipramine [116–124].
9.1.2 Regulatory Affairs
One entacapone 200 mg tablet is taken with each levodopa/DDI dose. The maximum recommended dose in Europe is 200 mg ten times daily, i.e. 2,000 mg of entacapone. This increased number of tablets may reduce compliance [115]. This disadvantage of entacapone therapy was improved with the introduction of the triple fixed-dose combination of levodopa/carbidopa/entacapone (Stalevo®) [45].
9.1.3 Stalevo®
Patients with advanced PD must frequently take levodopa, sometimes up to every 2 h. This levodopa ‘fractionation’ reduces temporary loss of efficacy, which is associated with reappearance of motor symptoms. The introduction of Stalevo® was an essential step forward from the older levodopa/DDI plus an extra entacapone tablet regimen, as the frequency of tablet intake was reduced. The pill size was also distinctly smaller, which further eased swallowing and improved patients’ acceptance of the drug. Dysphagia is a well recognized symptom of PD. Patients experiencing this symptom are not expected to comply with oral administration or to obtain optimal bioavailability of levodopa [45]. One drawback was that only few levodopa strengths were initially available, which, from the treating physician’s perspective, limited the ability to individually adapt titration form the original levodopa/DDI. This situation improved with the introduction of additional Stalevo® formulations, which enabled individual adaptation of levodopa in a range between 50 and 200 mg in 25 mg levodopa equivalents [45, 125–129]. Switching from levodopa plus entacapone coadministration to Stalevo® in patients with advanced PD provides additional benefit in clinical practice, and compliance problems may be reduced. Hypothetically, the improved timing of enzyme blockade with synchronous ingestion of both enzyme inhibitors is better for the pharmacokinetic levodopa behaviour; however, this has not yet been proven in a pharmacokinetic trial.
9.2 Tolcapone
Tolcapone was introduced before entacapone and, similarly, may also induce dyskinesia to a considerable extent, dependent on the design of the trial. Repeated dosing of levodopa is known to result in an increase of maximum concentration and bioavailability in plasma. Tolcapone also possesses centrally acting properties, but a tolcapone trial in levodopa-naive patients failed [130]. The hypothesis was that striatal dopamine is metabolized by COMT and MAO-B and thus central COMT inhibition with tolcapone alone or in combination with MAO-B inhibition might provide a symptomatic benefit for patients not receiving levodopa. This pilot study investigated the tolerability, safety and efficacy of tolcapone alone and in combination with oral selegiline in untreated patients with early PD. Patients were randomized to receive tolcapone 200 mg tid or placebo for the 8 weeks of the study. Open-label oral selegiline (5 mg in the morning and midday) was administered to all patients during the second 4 weeks of the study. There was no difference between treatment groups according to the investigator’s assessment of tolerability at week 4. During the initial 4 weeks, 95 % of patients treated with tolcapone and 98 % of those receiving placebo experienced good tolerability. A decrease in tolerability occurred in the tolcapone group during the second 4 weeks of the study following the addition of selegiline. No symptomatic benefit was associated with tolcapone alone or in combination with oral selegiline in these otherwise untreated patients. However, this trial points out that even central COMT inhibitors are only efficacious on motor behaviour in combination with levodopa. In patients with more advanced PD, switch-over studies with the competitor entacapone, which acts only in the periphery, showed that tolcapone with its additional central effects on dopaminergic neurotransmission is more efficacious in terms of reduction of PD symptoms. However, the outcomes of the best randomized controlled trial available were not conclusive. In this study, the primary outcome was number of patients (proportion) with ≥1 h ON time response. The results were entacapone 32 (43 %) and tolcapone 40 (53 %) (p = 0.191). Although the results were statistically not significant, it was suggested that the tendency favouring tolcapone was consistent and probably not a result of chance [131]. The addition of tolcapone also reduced motor symptoms to a similar extent in comparison with the dopamine agonists, bromocriptine and pergolide, in open-label trials. Quality of life scores were significant better under tolcapone due to fewer dopamine agonist-related side effects. Moreover, titration was not necessary with tolcapone but was with the dopamine agonists, which additionally biased this outcome. These trials have limited value, since they were under-powered to detect clinically relevant differences between tolcapone and the dopamine agonist [132–135].
9.2.1 Safety Issues
One hypothesizes that mutations in the UDP-glucuronosyltransferase 1A9 gene, which leads to defective glucuronidation activity, predispose for COMT inhibitor-induced hepatotoxicity [136]. After three fatal cases of hepatotoxicity, tolcapone was temporarily withdrawn nearly all over the world from November 1998 until April 2004. It was approved again; however, its prescription now demands a strict control of liver enzyme activity on a regular basis, both in Europe and to a lesser extent in the USA. For instance, the administration of tolcapone is now restricted to prescription and supervision by physicians experienced in the management of advanced PD. Additionally, liver function tests must be carried out on a regular basis. If the dose of tolcapone is raised to 200 mg tid, liver enzymes should be checked prior to initiating the higher dose, and the monitoring scheme should be reset from the beginning. A further criterion is that patients with PD must fail to respond or be intolerant of other COMT inhibitors before starting tolcapone. Contraindications include severe dyskinesia, previous history of non-traumatic rhabdomyolysis, hyperthermia or the neuroleptic malignant syndrome symptom complex. Side effects of tolcapone are similar to those of other dopaminergic compounds. Onset and or aggravation of dyskinesia, nausea, vomiting, anorexia, insomnia, orthostatic symptoms and hallucinations were the most common adverse events in clinical trials. The most frequent non-dopaminergic adverse event was diarrhoea, occurring sometimes even 2–4 months after following treatment initiation. This may be due to the hypothetical inhibition of serotonin 5-HT metabolism in the gastrointestinal tract in some patients [137]. Headache, increased sweating and associated xerostomia, probably due to aggravation of dyskinesia in clinical trials, abdominal pain and—similar to entacapone—harmless urine discoloration due to the yellow colour of tolcapone were further side effects, which were more frequently reported than in the placebo arm in clinical trials.
9.3 Tolcapone or Entacapone in Clinical Practice
The discussion on the liver toxicity of tolcapone and the need for a previous failed response or intolerance of entacapone intake still bias prescribing preference towards entacapone. Tolcapone only requires an additional intake of three tablets to an existing levodopa/DDI regime. Tolcapone is superior to entacapone in combination with the Duodopa® pump system [138].
9.4 Unmet Needs in the Context of Therapy with COMT Inhibitors
There need for the development of further COMT inhibitors with novel and better pharmacodynamic profiles is still unmet. These compounds should lead to more sustained levodopa levels in patients with PD and should have a lower frequency of drug administration. One example is opicapone.
9.5 Opicapone
Opicapone (also known as BIA 9-1067, manufactured by BIAL-Portela & Ca) is a long-acting, purely peripheral third-generation nitrocatechol COMT inhibitor. The compound possesses a high binding affinity to the enzyme with a slow dissociation constant. Opicapone produces a stronger and more prolonged inhibitory effect upon erythrocyte S-COMT than that reported for tolcapone and entacapone [139–146]. At 6 h after levodopa/benserazide administration, the 3-OMD concentration was still 25 % of the concentration in the controls. A dose of opicapone (100 mg/kg) used in monkeys corresponded to the disposition observed in humans for a dose of 100 mg. Opicapone in the cynomolgus monkey was shown to double the systemic exposure of levodopa, with a shift in time to maximum concentration (t max) to a later time, but without significantly affecting maximum concentration of levodopa (see also Table 1).
9.5.1 Opicapone in Healthy Subjects
Opicapone provides a sustained COMT inhibition in erythrocytes [145, 147–149]. A single-centre, randomized, double-blind, gender-balanced, placebo-controlled study in healthy subjects administered with once-daily opicapone 25, 50 or 75 mg/day or placebo for 11 days and levodopa/carbidopa 100/25 mg, entacapone 200 mg or placebo tid showed that mean levodopa area under the concentration–time curve (AUC) plasma values were higher when levodopa/carbidopa was administered with any opicapone dose group than when administered concomitantly with entacapone. Maximum S-COMT inhibition was higher with all opicapone doses than with entacapone [149]. Single rising oral doses of opicapone 10–1,200 mg were studied under a double-blind, randomised, placebo-controlled design. Eight sequential groups of eight subjects were enrolled. Within each group, six subjects were randomised to receive opicapone and two subjects to receive placebo. Opicapone/placebo was administered after a 10 h overnight fast. The extent and rate of systemic exposure (AUC and maximum plasma concentration) to opicapone increased in an approximately dose-proportional manner. Despite the relatively short half-life of opicapone (1–4 h), inhibition of S-COMT activity in erythrocytes was long-lasting, ranging from 6 % (10 mg) to 55 % (1,200 mg) at 72 h post-dose. Maximum S-COMT inhibition occurred between 1 and 6 h post-dose and was 34.5, 71.7, 93.8, 96.3, 100, 100, 100 and 100 with the doses of 10, 25, 50, 100, 200, 400, 800 and 1,200 mg, respectively. Urine levels of opicapone and its metabolites usually remained below the limit of quantification, showing that the kidney is not the primary route of excretion. Opicapone was well tolerated at all doses tested. Similar effects were found during repeated dosing [149–151].
9.5.2 Opicapone in Patients with PD
The efficacy of opicapone has been demonstrated in patients with PD who were taking levodopa/carbidopa or levodopa/benserazide, as well as in patients with fluctuating PD symptoms.
Once-daily 5, 15 and 30 mg doses were applied to PD patients with motor fluctuations treated with standard-release 100/25 mg levodopa/carbidopa or levodopa/benserazide in a multicentre, double-blind, randomised, placebo-controlled study in four parallel groups. Subjects were sequentially and randomly assigned to be administered, once daily, during the 21- to 28-day maintenance phase with placebo or opicapone 5, 15 and 30 mg. They performed two levodopa challenge tests, one on the morning of the day after admission and another following the maintenance phase.
They also completed a diary to record their ON/OFF periods. In comparison with placebo, levodopa plasma exposure increased 24.73, 53.93 and 65.61 % following opicapone 5, 15 or 30 mg, respectively. Maximum S-COMT inhibition ranged from 52 % (5 mg) to 80 % (30 mg opicapone). The exploratory analysis showed improvement of various motor outcomes, including a dose-dependent change in absolute OFF time corresponding to a percentage decrease of 0.77, 4.16, 29.55 and 32.71 % with placebo and opicapone 5, 15 and 30 mg, respectively [152].
Another trial investigated the efficacy and safety of opicapone 25 and 50 mg administered once daily in comparison with placebo, in PD patients receiving levodopa treatment and with wearing-off motor fluctuations. This pivotal phase III, multinational, multicentre, double-blind, placebo-controlled and parallel-group study randomized patients to placebo (N [number of participants in each arm] = 135) to opicapone 25 mg (N = 125) or opicapone 50 mg (N = 147). The double-blind phase lasted 14–15 weeks. The primary efficacy endpoint was the change from baseline in absolute OFF-time, based on patient diaries. Mean reduction in absolute OFF-time in both opicapone 25 and 50 mg groups was greater than in the placebo group (1.7, 2.0 and 1.1 h, respectively). There was a high placebo response; nevertheless, opicapone 50 mg but not 25 mg was significantly better than placebo (p = 0.0084). Opicapone once daily was safe and well tolerated [153] (see also Table 1).
9.5.3 Future Advantages of Opicapone in Clinical Practice
Despite that latter negative outcome in the so-called BIPARK II study in the opicapone 25 mg arm, this compound has one essential advantage over the available COMT inhibitors. It only requires the additional intake of one tablet in a group of patients with considerable compliance problems [153–155]. Due to its pharmacologic profile, one may speculate that opicapone may provide a more sustained and thus less fluctuating COMT inhibition, which will probably further enhance the metabolic advantages of COMT inhibition during chronic levodopa/DDI therapy in PD patients.
10 Conclusions
Concomitant COMT inhibition during chronic levodopa/DDI therapy in patients with PD improves the efficacy of levodopa, reduces fluctuating levodopa plasma levels and ameliorates motor complications, particularly wearing-off phenomena. Entacapone and tolcapone are established drugs for COMT inhibition, whereas opicapone, with its once-daily application, is still in clinical trials.
COMT inhibition counteracts levodopa-associated homocysteine increase, which is a biomarker for a limited methylation capacity and supports oxidative stress generation.
References
Rajput AH, Birdi S. Epidemiology of Parkinson’s disease. Parkinsonism Relat Disord. 1997;3(4):175–86.
Brooks DJ. Examining Braak’s hypothesis by imaging Parkinson’s disease. Mov Disord. 2010;25(Suppl 1):S83–8.
Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55(3):181–4.
Milber JM, Noorigian JV, Morley JF, Petrovitch H, White L, Ross GW, et al. Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology. 2012;79(24):2307–14.
Stoddard SL. The adrenal medulla and Parkinson’s disease. Rev Neurosci. 1994;5(4):293–307.
Przuntek H, Müller T, Riederer P. Diagnostic staging of Parkinson’s disease: conceptual aspects. J Neural Transm. 2004;111(2):201–16.
Lim SY, Fox SH, Lang AE. Overview of the extranigral aspects of Parkinson disease. Arch Neurol. 2009;66(2):167–72.
Lim SY, Lang AE. The nonmotor symptoms of Parkinson’s disease—an overview. Mov Disord. 2010;25(Suppl 1):S123–30.
Siderowf A, Lang AE. Premotor Parkinson’s disease: concepts and definitions. Mov Disord. 2012;27(5):608–16.
Dickson DW. Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med. 2012;2(8):a009258.
Weiner WJ. There is no Parkinson disease. Arch Neurol. 2008;65(6):705–8.
Blandini F. Neural and immune mechanisms in the pathogenesis of Parkinson’s disease. J Neuroimmune Pharmacol. 2013;8(1):189–201.
Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging. 2001;18(9):685–716.
Naoi M, Maruyama W, Yi H, Inaba K, Akao Y, Shamoto-Nagai M. Mitochondria in neurodegenerative disorders: regulation of the redox state and death signaling leading to neuronal death and survival. J Neural Transm. 2009;116(11):1371–81.
Riederer P, Gerlach M, Müller T, Reichmann H. Relating mode of action to clinical practice: dopaminergic agents in Parkinson’s disease. Parkinsonism Relat Disord. 2007;13(8):466–79.
Müller T. Detoxification and antioxidative therapy for levodopa-induced neurodegeneration in Parkinson’s disease. Expert Rev Neurother. 2013;13(6):707–18.
Müller T. Drug therapy in patients with Parkinson’s disease. Transl Neurodegener. 2012;1(1):1–10.
Müller T. Pharmacokinetic/pharmacodynamic evaluation of rasagiline mesylate for Parkinson’s disease. Expert Opin Drug Metab Toxicol. 2014;10(10):1423–32.
Birkmayer W, Hornykiewicz O. The effect of l-3,4-dihydroxyphenylalanine (=DOPA) on akinesia in parkinsonism. 1961. Wien Klin Wochenschr. 2001;113(22):851–4.
Müller T, Russ H. Levodopa, motor fluctuations and dyskinesia in Parkinson’s disease. Expert Opin Pharmacother. 2006;7(13):1715–30.
Rodnitzky RL, Narayanan NS. Amantadine’s role in the treatment of levodopa-induced dyskinesia. Neurology. 2014;82(4):288–9.
Stocchi F, Tagliati M, Olanow CW. Treatment of levodopa-induced motor complications. Mov Disord. 2008;23(Suppl 3):S599–612.
Nutt JG, Chung KA, Holford NH. Dyskinesia and the antiparkinsonian response always temporally coincide: a retrospective study. Neurology. 2010;74(15):1191–7.
Pearce RK, Heikkila M, Linden IB, Jenner P. l-dopa induces dyskinesia in normal monkeys: behavioural and pharmacokinetic observations. Psychopharmacology (Berl). 2001;156(4):402–9.
Cenci MA, Konradi C. Maladaptive striatal plasticity in l-DOPA-induced dyskinesia. Prog Brain Res. 2010;183:209–33.
Thomas A, Iacono D, Luciano AL, Armellino K, Di Iorio A, Onofrj M. Duration of amantadine benefit on dyskinesia of severe Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2004;75(1):141–3.
Politis M, Wu K, Molloy S, Bain G, Chaudhuri KR, Piccini P. Parkinson’s disease symptoms: the patient’s perspective. Mov Disord. 2010;25(11):1646–51.
Olanow CW, Kieburtz K, Rascol O, Poewe W, Schapira AH, Emre M, et al. Factors predictive of the development of Levodopa-induced dyskinesia and wearing-off in Parkinson’s disease. Mov Disord. 2013;28(8):1064–71.
Smith LA, Jackson MJ, Hansard MJ, Maratos E, Jenner P. Effect of pulsatile administration of levodopa on dyskinesia induction in drug-naive MPTP-treated common marmosets: effect of dose, frequency of administration, and brain exposure. Mov Disord. 2003;18(5):487–95.
Foley P, Mizuno Y, Nagatsu T, Sano A, Youdin MBH, McGeer P, et al. The l-DOPA story—an early Japanese contribution. Parkinsonism Relat Disord. 2000;6(1):1.
Pivac N, Pregelj P, Nikolac M, Zupanc T, Nedic G, Muck SD, et al. The association between catechol-O-methyl-transferase Val108/158Met polymorphism and suicide. Genes Brain Behav. 2011;10(5):565–9.
Schosser A, Calati R, Serretti A, Massat I, Kocabas NA, Papageorgiou K, et al. The impact of COMT gene polymorphisms on suicidality in treatment resistant major depressive disorder—a European multicenter study. Eur Neuropsychopharmacol. 2012;22(4):259–66.
Wardle MC, Hart AB, Palmer AA, de Wit H. Does COMT genotype influence the effects of d-amphetamine on executive functioning? Genes Brain Behav. 2013;12(1):13–20.
Kaakkola S. Clinical pharmacology, therapeutic use and potential of COMT inhibitors in Parkinson’s disease. Drugs. 2000;59(6):1233–50.
Mannisto PT, Tuomainen P, Tuominen RK. Different in vivo properties of three new inhibitors of catechol O-methyltransferase in the rat. Br J Pharmacol. 1992;105(3):569–74.
Müller T, Kolf K, Ander L, Woitalla D, Muhlack S. Catechol-O-methyltransferase inhibition improves levodopa-associated strength increase in patients with Parkinson disease. Clin Neuropharmacol. 2008;31(3):134–40.
Tornwall M, Kaakkola S, Tuomainen P, Kask A, Mannisto PT. Comparison of two new inhibitors of catechol O-methylation on striatal dopamine metabolism: a microdialysis study in rats. Br J Pharmacol. 1994;112(1):13–8.
Zurcher G, Colzi A, Da PM. Ro 40-7592: inhibition of COMT in rat brain and extracerebral tissues. J Neural Transm Suppl. 1990;32:375–80.
Nutt JG, Carter JH, Lea ES, Woodward WR. Motor fluctuations during continuous levodopa infusions in patients with Parkinson’s disease. Mov Disord. 1997;12(3):285–92.
Kuoppamaki M, Korpela K, Marttila R, Kaasinen V, Hartikainen P, Lyytinen J, et al. Comparison of pharmacokinetic profile of levodopa throughout the day between levodopa/carbidopa/entacapone and levodopa/carbidopa when administered four or five times daily. Eur J Clin Pharmacol. 2009;65(5):443–55.
Müller T, Erdmann C, Muhlack S, Bremen D, Przuntek H, Woitalla D. Inhibition of catechol-O-methyltransferase contributes to more stable levodopa plasma levels. Mov Disord. 2006;21(3):332–6.
Müller T, Erdmann C, Bremen D, Schmidt WE, Muhlack S, Woitalla D, et al. Impact of gastric emptying on levodopa pharmacokinetics in Parkinson disease patients. Clin Neuropharmacol. 2006;29(2):61–7.
Müller T, Erdmann C, Muhlack S, Bremen D, Przuntek H, Goetze O, et al. Pharmacokinetic behaviour of levodopa and 3-O-methyldopa after repeat administration of levodopa/carbidopa with and without entacapone in patients with Parkinson’s disease. J Neural Transm. 2006;113(10):1441–8.
Müller T. The impact of COMT-inhibition on gastrointestinal levodopa absorption in patients with Parkinson’s disease. Clin Med Insights Ther. 2010;2:155–68.
Müller T. Levodopa/carbidopa and entacapone in the treatment of Parkinson’s disease: efficacy, safety and patient preference. Patient Prefer Adherence. 2009;3:51–9.
Müller T. Motor complications, levodopa metabolism and progression of Parkinson’s disease. Expert Opin Drug Metab Toxicol. 2011;7(7):847–55.
Nyholm D, Nilsson Remahl AI, Dizdar N, Constantinescu R, Holmberg B, Jansson R, et al. Duodenal levodopa infusion monotherapy vs oral polypharmacy in advanced Parkinson disease. Neurology. 2005;64(2):216–23.
Ekesbo A, Rydin E, Torstenson R, Sydow O, Laengstrom B, Tedroff J. Dopamine autoreceptor function is lost in advanced Parkinson’s disease. Neurology. 1999;52(1):120–5.
Cenci MA. Dopamine dysregulation of movement control in l-DOPA-induced dyskinesia. Trends Neurosci. 2007;30(5):236–43.
Calabresi P, Di FM, Ghiglieri V, Picconi B. Molecular mechanisms underlying levodopa-induced dyskinesia. Mov Disord. 2008;23(Suppl 3):S570–9.
Jugel C, Ehlen F, Taskin B, Marzinzik F, Müller T, Klostermann F. Neuropathy in Parkinson’s disease patients with intestinal levodopa infusion versus oral drugs. PLoS One. 2013;8(6):e66639.
Klostermann F, Jugel C, Müller T, Marzinzik F. Malnutritional neuropathy under intestinal levodopa infusion. J Neural Transm. 2012;119(3):369–72.
Meiler B, Andrich J, Müller T. Rapid switch from oral antiparkinsonian combination drug therapy to duodenal levodopa infusion. Mov Disord. 2008;23(1):145–6.
Klostermann F, Jugel C, Bomelburg M, Marzinzik F, Ebersbach G, Müller T. Severe gastrointestinal complications in patients with levodopa/carbidopa intestinal gel infusion. Mov Disord. 2012;27(13):1704–5.
Kleedorfer B, Lees AJ, Stern GM. Subcutaneous and sublingual levodopa methyl ester in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1991;54(4):373.
Lee YH, Kim KH, Yoon IK, Lee KE, Chun IK, Rhie JY, et al. Pharmacokinetic evaluation of formulated levodopa methyl ester nasal delivery systems. Eur J Drug Metab Pharmacokinet. 2014;39(4):237–42.
Dupont E, Burgunder JM, Findley LJ, Olsson JE, Dorflinger E. Tolcapone added to levodopa in stable parkinsonian patients: a double-blind placebo-controlled study. Tolcapone in Parkinson’s Disease Study Group II (TIPS II). Mov Disord. 1997;12(6):928–34.
Block G, Liss C, Reines S, Irr J, Nibbelink D. Comparison of immediate-release and controlled release carbidopa/levodopa in Parkinson’s disease. A multicenter 5-year study. The CR First Study Group. Eur Neurol. 1997;37(1):23–7.
Piccini P, Brooks DJ, Korpela K, Pavese N, Karlsson M, Gordin A. The catechol-O-methyltransferase (COMT) inhibitor entacapone enhances the pharmacokinetic and clinical response to Sinemet CR in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2000;68(5):589–94.
Smith LA, Jackson MJ, Al-Barghouthy G, Rose S, Kuoppamaki M, Olanow W, et al. Multiple small doses of levodopa plus entacapone produce continuous dopaminergic stimulation and reduce dyskinesia induction in MPTP-treated drug-naive primates. Mov Disord. 2005;20(3):306–14.
Stocchi F, Rascol O, Kieburtz K, Poewe W, Jankovic J, Tolosa E, et al. Initiating levodopa/carbidopa therapy with and without entacapone in early Parkinson disease: the STRIDE-PD study. Ann Neurol. 2010;68(1):18–27.
Müller T. Pharmacokinetic considerations for the use of levodopa in the treatment of Parkinson disease: focus on levodopa/carbidopa/entacapone for treatment of levodopa-associated motor complications. Clin Neuropharmacol. 2013;36(3):84–91.
Muhlack S, Herrmann L, Salmen S, Müller T. Fewer fluctuations, higher maximum concentration and better motor response of levodopa with catechol-O-methyltransferase inhibition. J Neural Transm. 2014;121(11):1357–66.
Müller T, Woitalla D, Schulz D, Peters S, Kuhn W, Przuntek H. Tolcapone increases maximum concentration of levodopa. J Neural Transm. 2000;107(1):113–9.
Jorga KM. Pharmacokinetics, pharmacodynamics, and tolerability of tolcapone: a review of early studies in volunteers. Neurology. 1998;50(5 Suppl 5):S31–8.
Hauser RA, Panisset M, Abbruzzese G, Mancione L, Dronamraju N, Kakarieka A. Double-blind trial of levodopa/carbidopa/entacapone versus levodopa/carbidopa in early Parkinson’s disease. Mov Disord. 2009;24(4):541–50.
Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351(24):2498–508.
Nyholm D, Askmark H, Aquilonius SM. Stalevo reduction in dyskinesia evaluation in Parkinson’s disease results were expected from a pharmacokinetic viewpoint. Ann Neurol. 2011;69(2):424.
Olanow CW, Kieburtz K, Stocchi F. Initiating levodopa therapy for Parkinson’s disease. Mov Disord. 2014;29(3):430.
Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE. Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov Disord. 2010;25(15):2649–53.
LeWitt PA, Jennings D, Lyons KE, Pahwa R, Rabinowicz AL, Wang J, et al. Pharmacokinetic–pharmacodynamic crossover comparison of two levodopa extension strategies. Mov Disord. 2009;24(9):1319–24.
Müller T, Woitalla D, Goetze O, Erdmann C. Entacapone improves absorption of a coadministered salt in patients with Parkinson’s disease. Mov Disord. 2008;23(10):1458–61.
Ceravolo R, Piccini P, Bailey DL, Jorga KM, Bryson H, Brooks DJ. 18F-dopa PET evidence that tolcapone acts as a central COMT inhibitor in Parkinson’s disease. Synapse. 2002;43(3):201–7.
Russ H, Müller T, Woitalla D, Rahbar A, Hahn J, Kuhn W. Detection of tolcapone in the cerebrospinal fluid of parkinsonian subjects. Naunyn Schmiedebergs Arch Pharmacol. 1999;360(6):719–20.
De Bonis ML, Tessitore A, Pellecchia MT, Longo K, Salvatore A, Russo A, et al. Impaired transmethylation potential in Parkinson’s disease patients treated with l-Dopa. Neurosci Lett. 2010;468(3):287–91.
Cacciapuoti F. Hyper-homocysteinemia: a novel risk factor or a powerful marker for cardiovascular diseases? Pathogenetic and therapeutical uncertainties. J Thromb Thrombolysis. 2011;32(1):82–8.
Zhang L, Jin Y, Chen M, Huang M, Harvey RG, Blair IA, et al. Detoxication of structurally diverse polycyclic aromatic hydrocarbon (PAH) o-quinones by human recombinant catechol-O-methyltransferase (COMT) via O-methylation of PAH catechols. J Biol Chem. 2011;286(29):25644–54.
Chuang YC, Chuang HY, Lin TK, Chang CC, Lu CH, Chang WN, et al. Effects of long-term antiepileptic drug monotherapy on vascular risk factors and atherosclerosis. Epilepsia. 2012;53(1):120–8.
Müller T. Role of homocysteine in the treatment of Parkinson’s disease. Expert Rev Neurother. 2008;8(6):957–67.
Schwartz RS, Halliday GM, Cordato DJ, Kril JJ. Small-vessel disease in patients with Parkinson’s disease: a clinicopathological study. Mov Disord. 2012;27(12):1506–12.
Müller T, van Laar T, Cornblath DR, Odin P, Klostermann F, Grandas FJ, et al. Peripheral neuropathy in Parkinson’s disease: levodopa exposure and implications for duodenal delivery. Parkinsonism Relat Disord. 2013;19(5):501–7.
Tanner CM, Ross GW, Jewell SA, Hauser RA, Jankovic J, Factor SA, et al. Occupation and risk of parkinsonism: a multicenter case–control study. Arch Neurol. 2009;66(9):1106–13.
Zhang YD, Ke XY, Shen W, Liu Y. Relationship of homocysteine and gene polymorphisms of its related metabolic enzymes with Alzheimer’s disease. Chin Med Sci J. 2005;20(4):247–51.
Zhu BT. Catechol-O-Methyltransferase (COMT)-mediated methylation metabolism of endogenous bioactive catechols and modulation by endobiotics and xenobiotics: importance in pathophysiology and pathogenesis. Curr Drug Metab. 2002;3(3):321–49.
Müller T, Kuhn W. Cysteine elevation in levodopa-treated patients with Parkinson’s disease. Mov Disord. 2009;24(6):929–32.
Ho PI, Ashline D, Dhitavat S, Ortiz D, Collins SC, Shea TB, et al. Folate deprivation induces neurodegeneration: roles of oxidative stress and increased homocysteine. Neurobiol Dis. 2003;14(1):32–42.
Zeevalk GD, Razmpour R, Bernard LP. Glutathione and Parkinson’s disease: is this the elephant in the room? Biomed Pharmacother. 2008;62(4):236–49.
Müller T, Muhlack S. Cysteinyl-glycine reduction as marker for Levodopa induced oxidative stress in Parkinson’s disease patients. Mov Disord. 2011;26(3):543–6.
Müller T, Muhlack S. Levodopa-related cysteinyl-glycine and cysteine reduction with and without catechol-O-methyltransferase inhibition in Parkinson’s disease patients. J Neural Transm. 2014;121(6):643–8.
Müller T, Werne B, Fowler B, Kuhn W. Nigral endothelial dysfunction, homocysteine, and Parkinson’s disease. Lancet. 1999;354(9173):126–7.
Müller T, Jugel C, Ehret R, Ebersbach G, Bengel G, Muhlack S, et al. Elevation of total homocysteine levels in patients with Parkinson’s disease treated with duodenal levodopa/carbidopa gel. J Neural Transm. 2011;118(9):1329–33.
Lee ES, Chen H, Soliman KF, Charlton CG. Effects of homocysteine on the dopaminergic system and behavior in rodents. Neurotoxicology. 2005;26(3):361–71.
Nakaso K, Yasui K, Kowa H, Kusumi M, Ueda K, Yoshimoto Y, et al. Hypertrophy of IMC of carotid artery in Parkinson’s disease is associated with l-DOPA, homocysteine, and MTHFR genotype. J Neurol Sci. 2003;207(1–2):19–23.
O’Suilleabhain PE, Sung V, Hernandez C, Lacritz L, Dewey RB Jr, Bottiglieri T, et al. Elevated plasma homocysteine level in patients with Parkinson disease: motor, affective, and cognitive associations. Arch Neurol. 2004;61(6):865–8.
Postuma RB, Lang AE. Homocysteine and levodopa: should Parkinson disease patients receive preventative therapy? Neurology. 2004;63(5):886–91.
Rogers JD, Sanchez-Saffon A, Frol AB, Diaz-Arrastia R. Elevated plasma homocysteine levels in patients treated with levodopa: association with vascular disease. Arch Neurol. 2003;60(1):59–64.
Toth C, Brown MS, Furtado S, Suchowersky O, Zochodne D. Neuropathy as a potential complication of levodopa use in Parkinson’s disease. Mov Disord. 2008;23(13):1850–9.
Ben Shlomo Y, Marmot MG. Survival and cause of death in a cohort of patients with parkinsonism: possible clues to aetiology? J Neurol Neurosurg Psychiatry. 1995;58(3):293–9.
Müller T, Muhlack S. Peripheral COMT inhibition prevents levodopa associated homocysteine increase. J Neural Transm. 2009;116(10):1253–6.
Müller T, Kuhn W. Tolcapone decreases plasma levels of S-adenosyl-l-homocysteine and homocysteine in treated Parkinson’s disease patients. Eur J Clin Pharmacol. 2006;62(6):447–50.
Postuma RB, Espay AJ, Zadikoff C, Suchowersky O, Martin WR, Lafontaine AL, et al. Vitamins and entacapone in levodopa-induced hyperhomocysteinemia: a randomized controlled study. Neurology. 2006;66(12):1941–3.
Zesiewicz TA, Wecker L, Sullivan KL, Merlin LR, Hauser RA. The controversy concerning plasma homocysteine in Parkinson disease patients treated with levodopa alone or with entacapone: effects of vitamin status. Clin Neuropharmacol. 2006;29(3):106–11.
Lamberti P, Zoccolella S, Iliceto G, Armenise E, Fraddosio A, DeMari M, et al. Effects of levodopa and COMT inhibitors on plasma homocysteine in Parkinson’s disease patients. Mov Disord. 2005;20(1):69–72.
Müller T, Woitalla D, Muhlack S. Inhibition of catechol-O-methyltransferase modifies acute homocysteine rise during repeated levodopa application in patients with Parkinson’s disease. Naunyn Schmiedebergs Arch Pharmacol. 2011;383(6):627–33.
Nevrly M, Kanovsky P, Vranova H, Langova K, Hlustik P. Effect of entacapone on plasma homocysteine levels in Parkinson’s disease patients. Neurol Sci. 2010;31(5):565–9.
Nissinen E, Nissinen H, Larjonmaa H, Vaananen A, Helkamaa T, Reenila I, et al. The COMT inhibitor, entacapone, reduces levodopa-induced elevations in plasma homocysteine in healthy adult rats. J Neural Transm. 2005;112(9):1213–21.
Valkovic P, Benetin J, Blazicek P, Valkovicova L, Gmitterova K, Kukumberg P. Reduced plasma homocysteine levels in levodopa/entacapone treated Parkinson patients. Parkinsonism Relat Disord. 2005;11(4):253–6.
Zoccolella S, Iliceto G, de Mari M, Livrea P, Lamberti P. Management of l-Dopa related hyperhomocysteinemia: catechol-O-methyltransferase (COMT) inhibitors or B vitamins? Results from a review. Clin Chem Lab Med. 2007;45(12):1607–13.
Zoccolella S, Lamberti P, Armenise E, de Mari M, Lamberti SV, Mastronardi R, et al. Plasma homocysteine levels in Parkinson’s disease: role of antiparkinsonian medications. Parkinsonism Relat Disord. 2005;11(2):131–3.
Bartl J, Müller T, Grunblatt E, Gerlach M, Riederer P. Chronic monoamine oxidase-B inhibitor treatment blocks monoamine oxidase-A enzyme activity. J Neural Transm. 2014;121(4):379–83.
Przuntek H, Conrad B, Dichgans J, Kraus PH, Krauseneck P, Pergande G, et al. SELEDO: a 5-year long-term trial on the effect of selegiline in early Parkinsonian patients treated with levodopa. Eur J Neurol. 1999;6(2):141–50.
Lyytinen J, Kaakkola S, Ahtila S, Tuomainen P, Teravainen H. Simultaneous MAO-B and COMT inhibition in l-Dopa-treated patients with Parkinson’s disease. Mov Disord. 1997;12(4):497–505.
Müller T, Kuhn W, Przuntek H. Therapy with central active catechol-O-methyltransferase (COMT)-inhibitors: is addition of monoamine oxidase (MAO)-inhibitors necessary to slow progress of neurodegenerative disorders? J Neural Transm Gen Sect. 1993;92(2–3):187–95.
Apud JA, Mattay V, Chen J, Kolachana BS, Callicott JH, Rasetti R, et al. Tolcapone improves cognition and cortical information processing in normal human subjects. Neuropsychopharmacology. 2007;32(5):1011–20.
Müller T. Entacapone. Expert Opin Drug Metab Toxicol. 2010;6(8):983–93.
Brooks DJ, Sagar H. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind, six month study. J Neurol Neurosurg Psychiatry. 2003;74(8):1071–9.
Brooks DJ, Agid Y, Eggert K, Widner H, Ostergaard K, Holopainen A. Treatment of end-of-dose wearing-off in Parkinson’s disease: stalevo (levodopa/carbidopa/entacapone) and levodopa/DDCI given in combination with Comtess/Comtan (entacapone) provide equivalent improvements in symptom control superior to that of traditional levodopa/DDCI treatment. Eur Neurol. 2005;53(4):197–202.
Kieburtz K, Hubble J. Benefits of COMT inhibitors in levodopa-treated parkinsonian patients: results of clinical trials. Neurology. 2000;55(11 Suppl 4):S42–5.
Olanow CW, Kieburtz K, Stern M, Watts R, Langston JW, Guarnieri M, et al. Double-blind, placebo-controlled study of entacapone in levodopa-treated patients with stable Parkinson disease. Arch Neurol. 2004;61(10):1563–8.
Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M. Efficacy and safety of entacapone in Parkinson’s disease patients with suboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand. 2002;105(4):245–55.
Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology. 1998;51(5):1309–14.
Ruottinen HM, Rinne UK. A double-blind pharmacokinetic and clinical dose-response study of entacapone as an adjuvant to levodopa therapy in advanced Parkinson’s disease. Clin Neuropharmacol. 1996;19(4):283–96.
Ruottinen HM, Rinne UK. Effect of one month’s treatment with peripherally acting catechol-O-methyltransferase inhibitor, entacapone, on pharmacokinetics and motor response to levodopa in advanced parkinsonian patients. Clin Neuropharmacol. 1996;19(3):222–33.
Ruottinen HM, Rinne UK. Entacapone prolongs levodopa response in a one month double blind study in parkinsonian patients with levodopa related fluctuations. J Neurol Neurosurg Psychiatry. 1996;60(1):36–40.
Hauser RA. Levodopa/carbidopa/entacapone (Stalevo). Neurology. 2004;62(1 Suppl 1):S64–71.
Koller W, Guarnieri M, Hubble J, Rabinowicz AL, Silver D. An open-label evaluation of the tolerability and safety of Stalevo (carbidopa, levodopa and entacapone) in Parkinson’s disease patients experiencing wearing-off. J Neural Transm. 2005;112(2):221–30.
Myllyla V, Haapaniemi T, Kaakkola S, Kinnunen E, Hartikainen P, Nuutinen J, et al. Patient satisfaction with switching to Stalevo: an open-label evaluation in PD patients experiencing wearing-off (Simcom Study). Acta Neurol Scand. 2006;114(3):181–6.
Seeberger LC, Hauser RA. Levodopa/carbidopa/entacapone in Parkinson’s disease. Expert Rev Neurother. 2009;9(7):929–40.
Sethi KD, Hauser RA, Isaacson SH, McClain T. Levodopa/carbidopa/entacapone 200/50/200 mg (Stalevo 200) in the treatment of Parkinson’s disease: a case series. Cases J. 2009;2:7134.
Hauser RA, Molho E, Shale H, Pedder S, Dorflinger EE. A pilot evaluation of the tolerability, safety, and efficacy of tolcapone alone and in combination with oral selegiline in untreated Parkinson’s disease patients. Tolcapone De Novo Study Group. Mov Disord. 1998;13(4):643–7.
Entacapone to Tolcapone Switch Study Investigators. Entacapone to tolcapone switch: multicenter double-blind, randomized, active-controlled trial in advanced Parkinson’s disease. Mov Disord. 2007;22(1):14–9.
Ries V, Selzer R, Eichhorn T, Oertel WH, Eggert K. Replacing a dopamine agonist by the COMT-inhibitor tolcapone as an adjunct to l-dopa in the treatment of Parkinson’s disease: a randomized, multicenter, open-label, parallel-group study. Clin Neuropharmacol. 2010;33(3):142–50.
Inzelberg R, Carasso RL, Schechtman E, Nisipeanu P. A comparison of dopamine agonists and catechol-O-methyltransferase inhibitors in Parkinson’s disease. Clin Neuropharmacol. 2000;23(5):262–6.
Koller W, Lees A, Doder M, Hely M. Randomized trial of tolcapone versus pergolide as add-on to levodopa therapy in Parkinson’s disease patients with motor fluctuations. Mov Disord. 2001;16(5):858–66.
Agid Y, Destee A, Durif F, Montastruc JL, Pollak P. Tolcapone, bromocriptine, and Parkinson’s disease. French Tolcapone Study Group. Lancet. 1997;350(9079):712–3.
Martignoni E, Cosentino M, Ferrari M, Porta G, Mattarucchi E, Marino F, et al. Two patients with COMT inhibitor-induced hepatic dysfunction and UGT1A9 genetic polymorphism. Neurology. 2005;65(11):1820–2.
Goetze O, Nikodem AB, Wiezcorek J, Banasch M, Przuntek H, Müller T, et al. Predictors of gastric emptying in Parkinson’s disease. Neurogastroenterol Motil. 2006;18(5):369–75.
Nyholm D, Johansson A, Lennernas H, Askmark H. Levodopa infusion combined with entacapone or tolcapone in Parkinson disease: a pilot trial. Eur J Neurol. 2012;19(6):820–6.
Dingemanse J, Jorga KM, Schmitt M, Gieschke R, Fotteler B, Zurcher G, et al. Integrated pharmacokinetics and pharmacodynamics of the novel catechol-O-methyltransferase inhibitor tolcapone during first administration to humans. Clin Pharmacol Ther. 1995;57(5):508–17.
Kaakkola S, Gordin A, Mannisto PT. General properties and clinical possibilities of new selective inhibitors of catechol O-methyltransferase. Gen Pharmacol. 1994;25(5):813–24.
Maltete D, Cottard AM, Mihout B, Costentin J. Erythrocytes catechol-O-methyl transferase activity is up-regulated after a 3-month treatment by entacapone in parkinsonian patients. Clin Neuropharmacol. 2011;34(1):21–3.
Tuomainen P, Reenila I, Mannisto PT. Validation of assay of catechol-O-methyltransferase activity in human erythrocytes. J Pharm Biomed Anal. 1996;14(5):515–23.
Goncalves D, Alves G, Fortuna A, Soares-da-Silva P, Falcao A. An HPLC-DAD method for the simultaneous quantification of opicapone (BIA 9-1067) and its active metabolite in human plasma. Analyst. 2013;138(8):2463–9.
Goncalves D, Alves G, Soares-da-Silva P, Falcao A. Bioanalytical chromatographic methods for the determination of catechol-O-methyltransferase inhibitors in rodents and human samples: a review. Anal Chim Acta. 2012;710:17–32.
Kiss LE, Ferreira HS, Torrao L, Bonifacio MJ, Palma PN, Soares-da-Silva P, et al. Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase. J Med Chem. 2010;53(8):3396–411.
Palma PN, Bonifacio MJ, Loureiro AI, Soares-da-Silva P. Computation of the binding affinities of catechol-O-methyltransferase inhibitors: multisubstate relative free energy calculations. J Comput Chem. 2012;33(9):970–86.
Almeida L, Rocha JF, Falcao A, Palma PN, Loureiro AI, Pinto R, et al. Pharmacokinetics, pharmacodynamics and tolerability of opicapone, a novel catechol-O-methyltransferase inhibitor, in healthy subjects: prediction of slow enzyme-inhibitor complex dissociation of a short-living and very long-acting inhibitor. Clin Pharmacokinet. 2013;52(2):139–51.
Bonifacio MJ, Sutcliffe JS, Torrao L, Wright LC, Soares-da-Silva P. Brain and peripheral pharmacokinetics of levodopa in the cynomolgus monkey following administration of opicapone, a third generation nitrocatechol COMT inhibitor. Neuropharmacology. 2014;77:334–41.
Rocha JF, Almeida L, Falcao A, Palma PN, Loureiro AI, Pinto R, et al. Opicapone: a short lived and very long acting novel catechol-O-methyltransferase inhibitor following multiple dose administration in healthy subjects. Br J Clin Pharmacol. 2013;76(5):763–75.
Nunes T, Rocha JF, Pinto R, Machado R, Wright LC, Falcao A, et al. Pharmacokinetics, pharmacodynamics and tolerability of opicapone, a novel COMT inhibitor, during first administration to healthy male subjects [abstract]. Parkinsonism Relat Disord. 2012;18S2, S81–S159.
Rocha JF, Nunes T, Vaz-da-Silva M, Machado R, Wright LC, Falcao A, et al. Pharmacokinetics, pharmacodynmics and tolerability of opicapone, a novel COMT inhibitor, during multiple dose rise regimen in healthy male subjects [abstract]. Parkinsonism Relat Disord. 2013;18S2, S81–S159.
Feirreira JJ, Rocha JF, Falcao A, Pinto R, Nunes T. Effect of opicapone multiple-dose regimens on levodopa pharmacokinetics, motor response, and erythrocyte-COMT activity in Parkinson’s patients co-administered with levodopa/dopa-decarboxylase inhibitor [abstract]. J Neurol Sci. 2013;333:e109–51.
Lees AJ, Ferreira JJ, Costa R, Rocha JF, Oliveira C, Lopes N. Efficacy and safety of opicapone, a new COMT-inhibitor, for the treatment of motor fluctuations in Parkinson’s Disease patients: BIPARK-II study. J Neurol Sci. 2013;333:e109–51.
Grosset D. Therapy adherence issues in Parkinson’s disease. J Neurol Sci. 2010;289(1–2):115–8.
Richy FF, Pietri G, Moran KA, Senior E, Makaroff LE. Compliance with pharmacotherapy and direct healthcare costs in patients with Parkinson’s disease: a retrospective claims database analysis. Appl Health Econ Health Policy. 2013;11(4):395–406.
Disclosures
Professor Dr. Thomas Müller has performed studies, served on advisory boards and held lectures for Roche, Novartis, Orion and MEDA in the past 20 years. This manuscript was not funded by any of these companies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Müller, T. Catechol-O-Methyltransferase Inhibitors in Parkinson’s Disease. Drugs 75, 157–174 (2015). https://doi.org/10.1007/s40265-014-0343-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-014-0343-0