
Abstract
Gliomas are a heterogeneous group of brain tumors with limited therapeutic options. However, identification of BRAF V600E mutations in a subset of gliomas has provided a genomic-targeted approach for management of these diseases. In this review, we aimed to review the role of BRAF V600E in gliomagenesis, to characterize concurrent genomic alterations and their potential prognostic implications, and to review comprehensively the efficacy data of BRAF inhibitors (combined or not with MEK inhibitors) for the treatment of low- and high-grade gliomas. We also provide a summary of the toxicity of these agents and describe resistance mechanisms that may be circumvented by alternative genomic approaches. Although the efficacy of targeted therapy for management of BRAF V600E-mutant gliomas has mostly been assessed in small retrospective and phase 2 studies with heterogeneous populations, the data generated so far are a proof of concept that genomic-directed therapies improve outcomes of patients with refractory/relapsed glioma and underpin the need of comprehensive genomic assessments for these difficult-to-treat diseases. In the future, the role of targeted therapy in the first-line setting and of genomic-directed therapies to overcome resistance mechanisms should be assessed in well-designed clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Targeted therapy with BRAF and MEK inhibitors improves outcomes of patients with BRAF V600E-mutant gliomas refractory to standard treatments. |
The efficacy of targeted therapy in patients with BRAF V600E-mutant glioma underscores the importance of comprehensive genomic assessments for patients with central nervous system tumors. |
Genomic-directed strategies to overcome resistance mechanisms should be assessed in clinical trials. |
1 Introduction
Gliomas are a heterogeneous group of primary neoplasms of the central nervous system (CNS) that differ in clinical presentation, molecular characteristics, and prognosis. The 2021 World Health Organization (WHO) classification of CNS tumors has acknowledged the evolving understanding of the molecular characteristics of gliomas and has introduced new subtypes based on specific molecular alterations [1, 2]. However, management of patients with glioma has not changed substantially in the last two decades, as treatment continues to be based on gross tumor resection that can be followed by either radiation therapy, chemotherapy, or a combination of both [3, 4]. This is particularly concerning for patients with high-grade and refractory gliomas, for whom therapeutic options are limited and survival is dismal [5,6,7,8].
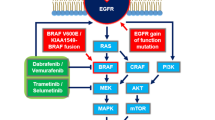
Identification of driver alterations has led to the development of targeted therapies that have resulted in improvements in recurrence-free survival, progression-free survival, and overall survival across several solid malignancies [9,10,11,12,13]. One such alteration occurs in the v-raf murine viral oncogene homolog B1 (BRAF). In physiological conditions, BRAF regulates the mitogen-activated protein kinase (MAPK) pathway, which is involved in the expression of genes related to cellular proliferation and survival. BRAF is also implicated in the development of adult tissues, including the CNS [14]. The most frequent alteration in BRAF is a point mutation characterized by substitution of valine for glutamic acid in codon 600 (V600E), which causes constitutive BRAF activation, independent of upstream RAS signaling [15]. BRAF mutations are frequent in melanomas [16] and also occur in colorectal cancers [17], non-small cell lung cancer [18], and thyroid carcinomas [19]. In gliomas, BRAF mutations have been identified across several histological subtypes and are more frequent in low-grade tumors [20]. However, the prognostic significance of BRAF mutations in gliomas is still debatable in the literature [21,22,23].
Dual inhibition of BRAF and mitogen-activated protein kinase kinase (MEK) is a standard of care for patients with BRAF V600-mutant melanomas, non-small cell lung cancers, and thyroid carcinomas [9,10,11, 13, 24, 25]. Targeted therapies, either with BRAF inhibitor monotherapy or in combination with a MEK inhibitor, have also been evaluated in patients with BRAF V600E-mutant recurrent or refractory gliomas in small retrospective series and prospective studies, with some patients benefiting from durable responses and extended survival [26,27,28,29,30,31]. However, there is no consensus on the best timing of initiation of these targeted therapies in patients with gliomas, and there is a concern regarding mechanisms of resistance and long-term toxicities, especially in patients with low-grade tumors [4, 32, 33].
In this review, we aim to discuss the role of BRAF in gliomagenesis and the prognostic implications of concurrent genomic alterations, as well as to describe the current landscape of the management of patients with BRAF V600E-mutant gliomas. We also discuss toxicities related to BRAF and MEK inhibition, and we summarize the evidence on mechanisms of resistance and possible approaches to overcome them.
2 BRAF V600 Mutations in Gliomas: Gliomagenesis, Concurrent Genomic Alterations, and Prognostic Implications
BRAF mutations are grouped in three classes according to their kinase activity. Class I BRAF mutations are characterized by strong activation and increased kinase activity of the MAPK pathway (approximately 500–700 fold as compared with wild-type BRAF). Class I BRAF mutations generate abnormal proteins that are constitutively activated without the need of dimerization and are sensitive to BRAF and MEK dual inhibition [34]. BRAF V600E is the most frequent class I mutation found in gliomas [35]. Class II mutations have lower kinase activity as compared with class I mutations, usually occur in the activation segment, and signal as RAS-independent dimers [34]. KIAA1549:BRAF, a BRAF fusion frequently found in pilocytic astrocytoma, functions as a class II BRAF mutation [36]. Class III BRAF mutations have lower kinase activity as compared to wild-type BRAF but may be implicated in tumorigenesis when they dimerize in the context of RAS upstream activation [34]. Class III mutations represent only 10% of BRAF mutations in gliomas, and are associated with NF1 loss-of-function mutations or EGFR amplification [35].
The frequency of BRAF mutations in gliomas varies according to histological subtypes and age of presentation. In the pediatric population, BRAF V600E mutation occurs in 7% of all glioma cases [37], but it can occur in 20% of pediatric low-grade gliomas (LGGs) [38]. In adults, BRAF V600E mutation occurs in only 4.6% of cases (mostly young adults), more frequently in epithelioid glioblastoma, pleomorphic xanthoastrocytoma, and anaplastic pleomorphic xanthoastrocytoma [37, 39]. However, it is possible that the frequency of BRAF V600E mutation in adult patients with glioma is underestimated, since BRAF testing is not routinely performed in all institutions. This is suggested by a single-center study in Japan in which all patients had access to comprehensive genomic profiling testing. In that cohort, 8% of patients with glioma had a BRAF V600E mutation [40]. In a study with 1320 central nervous system tumor samples, 96 BRAF mutations were detected (93 BRAF V600E), which were more frequently found in pleomorphic xanthoastrocytoma (63% of adult cases, 69% of pediatric cases), anaplastic pleomorphic xanthoastrocytoma (38% of adult cases, 100% of pediatric cases), and ganglioglioma (21% of adult cases, 18% of pediatric cases) [20]. Upon recurrence, BRAF V600E-mutant pediatric LGGs treated with either surgery, radiation, or chemotherapy continue to present BRAF V600E mutation in 98% of cases [41]. However, BRAF V600E-mutant glioblastoma subclones may not expand after exposure to radiation and alkylating chemotherapy, notwithstanding their theoretical proliferative advantage, and these tumors will not present BRAF V600E mutation at the time of recurrence [42]. Despite sharing the same point mutation, the prognosis of patients with BRAF V600E-mutant gliomas varies widely [39, 43], which implies that BRAF V600E is not the sole driver to influence tumorigenesis and clinical outcomes.
Preclinical data have demonstrated that BRAF V600E alone is insufficient to cause gliomagenesis. In a pilocytic astrocytoma model, neurospheres derived from human fetal cerebral cortex were infected with a lentivirus containing BRAF V600E. Although there was evidence of MAPK pathway activation in the infected cells, this did not result in significantly increased proliferation as compared to controls. These cells eventually stopped proliferating and demonstrated evidence of oncogene-induced senescence [44]. However, loss of function of p16Ink4a and p14Arf (products coded by CDKN2A) in association with BRAF V600E is sufficient to induce tumorigenesis, which implies that deletion of CDKN2A supresses oncogene-induced senescence in neural progenitors [45]. Similarly, activation of the PI3K/Akt/mTOR pathway may also overcome senescence, as evidenced in BRAF V600E-mutant gangliogliomas and dysembryoplastic neuroepithelial tumors [46].
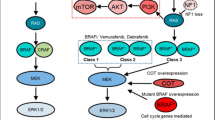
BRAF V600E mutation and CDKN2A homozygous deletion may be the initiating genomic events of a spectrum of gliomas with different histological characteristics and prognosis (Fig. 1). For example, in cases of epithelioid glioblastoma derived from anaplastic pleomorphic xanthoastrocytoma, the presence of BRAF V600E mutation and CDKN2A homozygous deletion has been evidenced in both low-grade and high-grade areas; however, differentiation to a more aggressive phenotype seems to be related to the overexpression of enhancer of zeste homolog 2 (EZH2), one of the proteins of the polycomb repressive complex 2, which catalyzes the trimethylation of H3K27. Overexpression of EZH2 has been observed in high-grade areas of epithelioid glioblastomas and of epithelioid differentiation in anaplastic pleomorphic xanthoastrocytomas carrying both BRAF V600E mutation and CDKN2A homozygous deletion, and it has been associated with worse survival [47]. Moreover, downregulation of both CDKN2A and MTAP (both localized at chromosome 9p21) has been observed in BRAF V600E-mutant pediatric high-grade gliomas (HGGs) [48].

Evolutionary pathways of a progenitor neural cell with BRAF V600E mutation and concurrent genomic alterations that lead to development of different gliomas. DNET: Dysembryoplastic neuroepithelial tumor; EZH: Enhancer of zeste homolog 2; TERTp: TERT promoter. Created with BioRender.com
The occurrence of TERT promoter mutation is associated with a higher histologic grade in BRAF V600E-mutant gliomas. Gabler et al [49]. developed a panel of glioma-derived cell lines to analyze the interplay between BRAF V600E and TERT promoter mutations. They observed that only cell lines with both BRAF V600E and TERT promoter mutation expressed TERT mRNA, and this finding was significantly more frequent in grades 3 and 4 gliomas as compared to LGGs (28% versus 3%, p = 0.003). Additionally, only BRAF V600E-mutant tumor cells with loss of CDKN2A and TERT promoter mutation developed stable and immortalized cell lines. These findings suggest that TERT promoter mutation and CDKN2A homozygous deletion have synergistic effects that lead to loss of oncogene-induced senescence caused by BRAF V600E mutation, and this results in a more aggressive phenotype.
The prognosis of patients with BRAF V600E-mutant gliomas is the result of a combination of histological grade and concurrent genomic alterations, as suggested by clinical observations. In a meta-analysis with survival data from 1308 patients with glioma, the presence of BRAF V600E mutation was associated with improved survival, but this benefit was restricted to young patients (< 35 years old) and with LGGs [21]. It is possible that some of these patients may have “molecularly defined glioblastoma,” i.e., a histologically low-grade tumor with the molecular characteristics of a glioblastoma (e.g., IDH wild-type, gain of chromosome 7/loss of chromosome 10); nonetheless, these patients still had good outcomes [50]. In contrast, the presence of BRAF V600E mutation in gangliogliomas, a WHO grade 1 tumor more frequent in children, adolescents, and young adults, is associated with shorter recurrence-free survival [23]. Parenthetically, the co-occurrence of H3K27M mutation with BRAF V600E mutation in patients with grade 1 gangliogliomas does not result in the adverse prognosis seen in patients with H3K27M-mutant diffuse midline gliomas [51]. In adults, BRAF V600E-mutant gliomas are less likely to have co-occurring IDH1/2, ATRX, and TP53 mutations, but homozygous deletion of CDKN2A or CDKN2B is a frequent co-occurring genomic event [22], and is also a marker of high-grade malignant astrocytomas in children [52]. Indeed, in patients with BRAF V600E-mutant pediatric LGGs, progression-free survival and overall survival were worse as compared with their BRAF wild-type counterparts (10 year progression-free survival for BRAF V600E-mutant versus BRAF wild type: 27% versus 60.2%; 10 year overall survival: 83.9% versus 92.1%), and patients with CDKN2A homozygous deletion and BRAF V600E mutation had worse outcomes than patients with CDKN2A homozygous deletion alone (10 year progression-free survival: 0% versus 45.9%, respectively) [53]. Therefore, a comprehensive genomic evaluation of BRAF V600E-mutant gliomas is necessary to prognosticate adequately and treat these tumors. This may be difficult in the context of lesions that are not amenable to surgical resection, but liquid biopsy may be an alternative to molecularly characterize these tumors [54].
3 Targeted Therapy in BRAF V600E-Mutant Gliomas: the Evidence
The efficacy of targeted therapies in other BRAF V600-mutant malignancies [9,10,11,12,13, 24, 25], including in patients with melanoma brain metastases [55], has led to the investigation of these agents for management of gliomas. In the preclinical setting, the BRAF inhibitor PLX-4720 resulted in increased survival in a murine astrocytoma model, and this was augmented when combined with palbociclib, a CDK4/6 inhibitor. Similar results were obtained in a human xenograft astrocytoma model with BRAF V600E mutation and CDKN2A deletion [45]. In a BRAF V600E-mutant pediatric glioma cell model, activity of the MAPK pathway was reduced by 59–63% with vemurafenib and by 72–74% with trametinib [56]. However, combined inhibition of BRAF and MEK results in more pronounced and prolonged inhibition of the MAPK pathway in glioma cell models than either agent alone, and this translated into more potent tumor growth inhibition in xenograft models while preventing ERK paradoxical reactivation and reducing the risk of development of cutaneous squamous cell carcinoma related to BRAF inhibitor monotherapy [57, 58].
In the clinical setting, activity of targeted therapy in patients with BRAF V600E-mutant gliomas has been demonstrated in several case reports [59,60,61,62,63,64], as well as in retrospective series. In a retrospective study with nine patients with BRAF V600E-mutant pediatric LGGs, the objective response rate (ORR) with dabrafenib monotherapy was 41.7%, disease control rate (DCR) was 100%, and progression-free survival was 26.1 months [32]. In another study, vemurafenib monotherapy led to treatment response in 57% (4/7) of patients with pediatric LGGs [65]. In a multiinstitutional retrospective series comprising 56 patients with pediatric LGGs (mostly pilocytic astrocytomas and gangliogliomas), dabrafenib monotherapy resulted in ≥ 25% tumor reduction in 80% of patients. Tumor responses were achieved at a median of 4 months and were sustained with a median time on treatment of 17.4 months. The presence of CDKN2A homozygous deletion did not seem to affect treatment response. Progressive disease was observed in eight patients, five of which achieved tumor control after adding a MEK inhibitor to dabrafenib therapy [30].
These results were replicated in a phase 1/2a clinical trial in pediatric patients with refractory BRAF V600E-mutant LGGs. In 32 evaluable patients, ORR with dabrafenib monotherapy was 44%, median duration of response was 26 months, and disease control rate was 78% [66]. In another phase 1 study with 19 patients with pediatric LGGs, the ORR with vemurafenib monotherapy was 32% and some responses were maintained with 40 months of follow-up [67]. A phase 1 trial of trametinib alone or with dabrafenib in pediatric patients with refractory LGGs demonstrated higher ORR in patients receiving combination therapy (25% versus 15%), and progression-free survival was longer in patients receiving dual inhibition (36.9 versus 16.4 months) [68]. Finally, a randomized phase 2 study of dabrafenib and trametinib versus carboplatin and vincristine in patients with relapsed pediatric LGGs demonstrated superiority of targeted therapy with higher ORR (47% versus 11%) and superior median progression-free survival (20.1 versus 7.4 months) [69]. These results underpin the clinical efficacy of targeted therapy in patients with refractory pediatric LGGs.
Evidence for first-line treatment with targeted therapy in patients with pediatric LGG is scarce, with one retrospective study (including patients with different MAPK pathway alterations) demonstrating an ORR of 75% and DCR of 100% [70]. In another retrospective study with 19 patients with pediatric HGG treated with upfront targeted therapy (11 with BRAF and MEK inhibitor combination, and 8 with BRAF inhibitor monotherapy), the response rate (in 14 evaluable patients) was 57%. The estimated progression-free survival at 3 and 5 years was 65% and 44%, respectively, and overall survival at 3 and 5 years was 82%. These results compare favorably to historical controls of patients with BRAF V600E-mutant pediatric HGG treated with radiotherapy and conventional chemotherapy [71]. These studies suggest there is a survival advantage for using targeted therapy in the first-line setting, but confirmation of this hypothesis depends on results of ongoing prospective clinical trials.
In patients with relapsed or refractory pediatric HGGs, the combination of dabrafenib and trametinib was assessed in a single-arm phase 2 trial. Among 41 patients enrolled, ORR was 56.1%. Median progression-free survival was 9 months, median duration of response was 22.2 months, and median overall survival 32.8 months [72]. These results compare favorably to a retrospective cohort of patients with pediatric HGGs treated with dabrafenib alone (11 patients, ORR 36%, and median progression-free survival 10 months) [30].
In contrast to the pediatric population, data on the efficacy of targeted therapy in adults with BRAF V600E-mutant gliomas were very limited until recently. In a retrospective study of 28 adult patients with refractory or disseminated BRAF V600E-mutant gliomas with high-grade features, 13 patients were treated with BRAF inhibitor monotherapy, and 15 patients received a combination of BRAF and MEK inhibitors. In the whole cohort, tumor responses were achieved in 11 (39%) patients, and responding patients had a median reduction of tumor burden of 78%. The probability of response in patients treated with dual inhibition was not statistically different when compared with patients treated with BRAF inhibitor alone (27% versus 54%, p = 0.25). The authors observed that, despite being treated in the recurrent setting, responding patients achieved a median progression-free survival that was longer than achieved with standard first-line treatment (18 versus 7 months, p = 0.047). Additionally, tumor response was associated with improvement in performance status [31].
Adult patients with BRAF V600E-mutant gliomas have been evaluated in a few basket trials. The NCI-MATCH trial assessed the efficacy of dabrafenib and trametinib in patients with BRAF V600E-mutant solid tumors and included five patients with CNS tumors (one each of epithelioid glioblastoma, pilocytic astrocytoma, anaplastic astroblastoma, pleomorphic xanthoastrocytoma, and histiocytic sarcoma). Three of these patients were evaluable for response, and a partial response was seen in two of them, while the other patient had stable disease. One patient sustained response for approximately 15 months [29].
The activity of single-agent vemurafenib in patients with recurrent BRAF V600E-mutant gliomas was assessed in the VE-BASKET study, a multicohort, nonrandomized trial. In total, 24 patients with glioma were enrolled, of which 11 had malignant diffuse glioma (5 with anaplastic astrocytoma and 6 with glioblastoma) and 7 had pleomorphic xanthoastrocytoma. In the whole cohort, confirmed ORR was 25% (one partial response was observed in the malignant diffuse glioma subcohort, and one complete and two partial responses were observed in the pleomorphic xanthoastrocytoma subcohort). Despite utilizing RECIST criteria for assessing response, the responses rates reported in the VE-BASKET study would be similar if RANO criteria were applied, as all but two responders (both in the pleomorphic xanthoastrocytoma subcohort) had at least 50% reduction in the sum of the largest diameters of the target lesions. Median progression-free survival was 5.5 months, and median overall survival was 28.2 months. The later result was influenced by the outcomes of patients with pleomorphic xanthoastrocytoma (median overall survival not reached); patients with malignant diffuse glioma had a median overall survival of 11.9 months [26].
A small phase 2 trial assessed encorafenib and binimetinib in patients with recurrent BRAF V600E-mutant HGG. This trial enrolled only five patients, and it was closed because of slow accrual. Three patients achieved a radiological response (two complete responses) and another had stable disease [73]. The ROAR study prospectively evaluated the combination of dabrafenib and trametinib in patients with BRAF V600E-mutant tumors. This study enrolled 58 patients with recurrent glioma: 45 had HGGs (31 with glioblastoma), and 13 had LGGs. In the HGG cohort, ORR by RANO criteria was 33%, and median duration of response was 31.2 months. Median progression-free survival was 5.5 months and median overall survival was 17.6 months. In contrast, the ORR in the LGG cohort was 54%, and median duration of response, median progression-free survival, and median overall survival were not reached [27, 28].
It should be emphasized that patients with HGG treated with targeted therapy within the VE-BASKET and ROAR trials had been previously treated with multiple lines of therapy, and the results obtained in these studies compare favorably with lomustine and bevacizumab, an approved combination for treating patients with recurrent glioblastomas and grade 4 astrocytomas (median progression-free survival: 4.2 months; median overall survival: 9.1 months) [8, 26,27,28].
Table 1 summarizes the efficacy results of targeted therapy for patients with BRAF V600E-mutant gliomas across several studies.
Although with small numbers and with a heterogeneous population, results of both VE-BASKET and ROAR trials reveal the different biological behaviors of LGGs and HGGs when treated with targeted therapies. Similar to what is seen in the pediatric population [30], in whom BRAF V600E-mutant LGGs are more frequent, adult patients with low-grade BRAF V600E-mutant gliomas may achieve long-term control of disease with BRAF inhibitor alone; however, patients with HGG seem to require a more aggressive inhibition of the MAPK pathway, which is achieved by the combination of BRAF and MEK inhibitors [57]. Given the lack of randomized data, the decision of whether treating a patient with BRAF inhibitor alone or combined with a MEK inhibitor may rely on tumor grade, toxicity, and possible mechanisms of resistance (discussed in detail below).
4 BRAF and MEK Inhibition-Related Toxicity
Data on short- and long-term toxicity of BRAF and MEK inhibitors are furnished largely from clinical trials in melanoma, in which these drugs were evaluated in phase 3 trials with long follow-up. Currently, there are three BRAF and MEK inhibitor combinations approved for metastatic melanoma based on randomized trials combining BRAF and MEK inhibitors in comparison to BRAF inhibitor monotherapy: dabrafenib and trametinib, encorafenib and binimetinib, and vemurafenib and cobimetinib [10, 11, 74, 75]. These trials reveal there are class effect adverse events (i.e., that are common to any BRAF inhibitor or to any combination of BRAF and MEK inhibitors) and drug-specific adverse events (Tables 2 and 3).
Monotherapy with a BRAF inhibitor is associated with increased rates of cutaneous toxicities as compared with dual BRAF and MEK inhibition, especially hyperkeratosis (6–40%) and cutaneous squamous cell carcinoma/keratoacanthoma (8–29%) [10, 11, 13, 74]. Dual inhibition significantly reduces the incidence of cutaneous toxicities, but it is more likely to cause increase of AST (13–22%) and ALT (13–23%) [10, 11, 13, 75]. Additionally, the combination of BRAF and MEK inhibitors increases the rate of visual adverse events, such as blurred vision (16%), serous retinopathy (20%), and retinal detachment (8%) [10, 11, 13, 74, 76]. Fortunately, most of these events are mild and are primarily managed by close monitoring or temporarily withholding the drugs. The discontinuation rate of BRAF and MEK inhibitors due to adverse events varies between 10% and 16%, similar to the discontinuation rates of BRAF inhibitor monotherapy [10, 11, 13, 74, 75].
Cardiac adverse events, such as decreased left ventricular ejection fraction, cardiac failure, and QT interval prolongation, have been closely monitored in clinical trials on patients with melanoma and, despite being more frequent with BRAF and MEK dual inhibition, occur at a low incidence and are mostly mild (e.g., the incidence of grade ≥ 3 decreased left ventricular ejection fraction varies between 1% and 2%) [10, 11, 74]. In comparison with BRAF inhibitor monotherapy, the combination of BRAF and MEK inhibitors is associated with increased risk of pulmonary embolism (2.2% versus 0.4%, RR 4.36, 95% CI 1.23–15.44, p = 0.02), hypertension (19.5% versus 14%, RR 1.49, 95% CI 1.12–1.48, p = 0.005), and decreased left ventricular ejection fraction (8.1% versus 2%, RR 3.72, 95% CI 1.74–7.95, p < 0.001), particularly in patients younger than 55 years (RR 26.50, 95% CI 3.58–196.10, p = 0.001) [77]. Currently, there are no specific recommendations on how to monitor and treat patients with decreased left ventricular ejection fraction related to BRAF and MEK inhibitors, but holding the drugs and consultation with cardiology is recommended [78].
Dabrafenib and trametinib is the most frequently studied combination in the treatment of BRAF V600E-mutant gliomas [27,28,29, 69, 72]. Pyrexia is a frequent adverse event of this combination (mostly associated with dabrafenib), and it occurs in 59% of patients with melanoma [10]. In patients with LGG treated within the ROAR study, pyrexia occurred in 62% of patients; however, in the HGG cohort, the frequency of pyrexia dropped to 24% [27]. This difference is likely due to more frequent use of steroids in patients with HGG. Classically, pyrexia is managed by holding dabrafenib, prescribing antipyretics, continuing trametinib, and resuming dabrafenib after resolution of pyrexia. However, in an adjuvant trial of dabrafenib and trametinib for fully resected stage III melanoma, pyrexia was managed by immediately holding both drugs and by resuming them after the patient remained asymptomatic for at least 24 hours. This approach reduced the rate of grade ≥ 3 pyrexia and resulted in comparable rates of recurrence-free survival as holding dabrafenib alone [79]. The rates of fatigue and nausea are comparable between the melanoma and glioma trials [10, 27].
Importantly, the rate of neurotoxicity in patients with glioma was not increased while on treatment with dabrafenib and trametinib. Intracranial hemorrhage was not reported in the ROAR study, and seizures occurred in four (8.9%) patients in the HGG glioma cohort [27, 28]. This is consistent with the rates of tumor-related epilepsy in patients with HGG [80]. Headaches were observed in 45% of patients treated with dabrafenib and trametinib, which is in line with what was reported in patients with metastatic melanoma [10, 27, 55].
Vemurafenib causes skin rashes in 67.5% and photosensitivity reactions in 37.8% of patients when given as monotherapy; when combined with cobimetinib, the rate of skin rashes increases to 72.5% and photosensitivity reactions to 47.8% [81]. In the glioma cohort of the VE-BASKET study, the frequency of photosensitivity with single-agent vemurafenib was 38% (nine patients) and of skin rashes was 29% (seven patients). There were no grade ≥ 3 photosensitivity reaction or skin rashes [26]. Most of the adverse events occurring with vemurafenib and cobimetinib are managed with dose reduction or interruption, and supportive care [81].
Encorafenib and binimetinib cause increase of creatine phosphokinase in 26% of patients. The same adverse event is observed in 35.2% of patients receiving vemurafenib and cobimetinib. Constipation, abdominal pain, and dizziness are also more frequent with encorafenib and binimetinib [74].
The data presented above were generated by studies in adult populations with melanoma or gliomas. In pediatric patients with LGG, the rate of pyrexia with single agent dabrafenib is 28% [66]. In a phase 1 trial, the rate of pyrexia in patients with pediatric LGGs receiving dabrafenib and trametinib was 50% [68]. In another phase 1 study investigating vemurafenib in 19 patients with pediatric LGGs, 10 patients developed grade 3 maculopapular rash; however, photosensitivity was observed in only one patient. Another patient developed cutaneous squamous cell carcinoma after four cycles of vemurafenib [67]. Decreased left ventricular ejection fraction with BRAF and MEK inhibition seems to occur at similar rates as seen in adult populations [68]. Taken together, the pattern of adverse events observed in the pediatric population seems to be consistent with that observed in adults, but larger trials with long-term follow-up are needed to adequately assess the safety of BRAF and MEK inhibitors in children.
5 Mechanisms of Resistance to BRAF and MEK Inhibition in Gliomas and Possible Solutions
Development of resistance to targeted therapy is a concern when treating patients with BRAF V600E-mutant gliomas. In patients treated with BRAF inhibitor monotherapy, reactivation of the MAPK pathway through ERK paradoxical activation is a well-described mechanism of resistance and can be prevented by concurrent MEK inhibition [58]. Data from clinical trials in patients with BRAF V600-mutant melanoma demonstrate that upfront dual inhibition of BRAF and MEK not only delays progression (10.5 versus 5.6 months) but also improves overall survival when compared with BRAF inhibitor alone (1 year overall survival: 79% versus 70%), and the overall survival benefit is superior with upfront combination despite introduction of MEK inhibitor following progression while on BRAF inhibitor monotherapy [82]. However, patients with BRAF V600E-mutant pediatric LGGs have prolonged control of disease with BRAF inhibition alone, and most patients who present disease progression can have their disease successfully controlled with addition of a MEK inhibitor [30, 83]. This suggests that gliomas have distinct mechanisms of resistance to BRAF inhibition when compared with melanomas.
Putative mechanisms of resistance to BRAF inhibition in LGG and HGG include alterations in genes that modulate receptor tyrosine kinase activity, such as CBL (an E3 ubiquitin-ligase) and ERFFI1 (ERBB receptor feedback inhibitor 1), NF1 loss-of-function missense mutations, activating mutations in MAP2K1, emergent mutations in PTEN and PIK3C2G, alterations in cell cycle regulators (such as BAP1 and ANKHD1) and TET2 alterations (a gene involved in epigenetic modulation of DNA). Additionally, resistance to BRAF inhibitors may emerge from a switch from BRAF to CRAF-mediated ERK activation, making it independent from BRAF V600E. Resistance may also emerge through changes in gene expression, as demonstrated by an RNA sequencing analysis that revealed enrichment of mesenchymal (TGFB1-high) and proneural (TGFB1-low) genotypes with different expressions of EGFR, YAP1 and KRAS. Those changes may occur at low variant allele frequencies, but this may be clinically significant in the context of low drug penetration through the blood–brain barrier [84]. Other mechanisms of resistance to BRAF inhibition include increased expression of AXL, a gene that activates the JAK/STAT, MAPK/ERK and PI3K/AKT pathways, increased EGFR expression, and elevated Wnt signaling [85]. The occurrence of in cis BRAF L514V mutation, which allows dimerization with BRAF V600E and results in decreased sensitivity to BRAF inhibitor monotherapy, has also been described as a resistance mechanism in BRAF V600E-mutant glioma [86], and this could potentially be counteracted by pan-RAF inhibition [87], although this has not been prospectively evaluated.
As mechanisms of resistance are identified, potential therapies to overcome resistance are being explored in the preclinical and clinical settings. Glioma cell lines treated with the BRAF inhibitor PLX-4720 present decreased expression of the protein tyrosine phosphatase PTPN9, a negative regulator of EGFR. As a result, those cell lines present hyperexpression of EGFR, making them resistant to BRAF inhibition. The combination of PLX-4720 and neratinib, an EGFR inhibitor, has been demonstrated to reduce tumor growth in BRAF V600E inhibitor-resistant xenograft models [88].
Inhibition of autophagy has been identified as a possible therapeutic approach to overcome different resistance mechanisms, including KRAS and NRAS activation, and EGFR hyperexpression [89]. In a preclinical model, the combination of vemurafenib and chloroquine (which functions as an autophagy inhibitor) resensitized resistant BRAF V600E-mutant glioma cells to BRAF inhibition [90]. In the clinical setting, this combination has led to extended tumor control in a patient with BRAF V600E-mutant pleomorphic xanthoastrocytoma who had disease progression on dabrafenib and trametinib [91]. A clinical trial combining dabrafenib, trametinib, and hydroxychloroquine for patients with recurrent BRAF-altered gliomas previously exposed to BRAF and MEK inhibitors is actively recruiting patients (NCT04201457).
Activation of mTOR pathway is a mechanism of resistance to MEK inhibitors, and combination of trametinib with the mTORC1/2 inhibitor sapanisertib has been investigated in glioma xenograft models. This combination effectively inhibited downstream signaling of both MAPK and mTOR pathways, as well as cell cycle regulator proteins such as CDK1, CDK2, CDK4, CDK5, and CDK6. Decrease of VEGF expression was also noted with trametinib and sapanisertib combination. However, upregulation of EGFR and class 1 histone deacetylase proteins has been identified as potential resistance mechanisms to this combination [92]. Similarly, the combination of dabrafenib, trametinib, and an HSP90 inhibitor resulted in inhibition of both MAPK and mTOR pathways. This combination resulted in cytotoxic effects in BRAF and MEK inhibitor-resistant glioma cells, and this effect was observed both in vitro and in vivo [93]. In another study with glioma xenograft models, MEK1 inhibition with selumetinib caused STAT3 activation, and combined inhibition with LLL12 (a STAT3 inhibitor) induced tumor complete responses[94].
Although multiple mechanisms of resistance have been described and possible therapeutic approaches to overcome resistance have been identified, these findings have not yet been translated to clinical practice. This is due to the rarity of BRAF V600E-mutant gliomas, and to the difficulty in acquiring tissue samples at the time of relapse. Additionally, trials with targeted therapy have only been conducted after relapse to standard first-line treatment, and it is not known what the impact of front-line targeted therapy in patients with BRAF V600E-mutant gliomas would be. A phase 2 trial (NCT03919071) is assessing the event-free survival in patients with newly diagnosed BRAF V600E-mutant glioma treated with dabrafenib and trametinib maintenance following radiation therapy, but this study is not expected to be completed until 2027.
6 Conclusions
The identification of BRAF V600E mutations in gliomas has provided new therapeutic approaches for patients who otherwise would have limited treatment options. Results from retrospective and prospective studies provide evidence that targeted therapy can lead to tumor shrinkage, extended disease control, and prolonged survival in patients with gliomas refractory to standard first-line therapies. The results of these trials also evidence that genomic-directed therapies may be effective for patients with glioma. However, there are questions that still need to be answered in well-conducted clinical trials. For example, it is unknown whether patients with LGG are best treated with a BRAF inhibitor alone or in combination with a MEK inhibitor upfront. Additionally, for this population with a better prognosis, long-term follow-up is necessary to assess potential late toxicities of targeted therapy. For patients with HGG, results of first-line targeted therapy are eagerly waited. Whenever possible, patients who develop progressive disease should have genomic studies to assess mechanisms of resistance that can be targeted by alternative genomic approaches that should be evaluated in prospective clinical trials. Finally, the effectiveness of targeted therapy in patients with BRAF V600E-mutant gliomas highlights the importance of a comprehensive genomic evaluation for patients affected with primary brain malignancies.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23(8):1231–51.
van der Meulen M, Ramos RC, Mason WP, Von Deimling A, Maas SLN. Opinion & Special Article: Glioma classification: how to interpret molecular markers in a diffuse glioma pathology report. Neurology. 2022 Sep 2.
Mohile NA, Messersmith H, Gatson NTN, Hottinger AF, Lassman AB, Morton J, et al. Therapy for diffuse astrocytic and oligodendroglial tumors in adults: ASCO-SNO Guideline. J Clin Oncol. 2022;24(3):358–83.
de Blank P, Bandopadhayay P, Haas-Kogan D, Fouladi M, Fangusaro J. Management of pediatric low-grade glioma. Curr Opin Pediatr. 2019;31(1):21–7.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96.
Minniti G, De Sanctis V, Muni R, Filippone F, Bozzao A, Valeriani M, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma in elderly patients. J Neurooncol. 2008;88(1):97–103.
Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA. 2017;318(23):2306–16.
Wick W, Gorlia T, Bendszus M, Taphoorn M, Sahm F, Harting I, et al. Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med. 2017;377(20):1954–63.
Dummer R, Hauschild A, Santinami M, Atkinson V, Mandalà M, Kirkwood JM, et al. Five-year analysis of adjuvant dabrafenib plus trametinib in stage III melanoma. N Engl J Med. 2020;383(12):1139–48.
Long GV, Flaherty KT, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2019;30(11):1848.
Dummer R, Flaherty KT, Robert C, Arance A, de Groot JWB, Garbe C, et al. COLUMBUS 5-year update: a randomized, open-label, phase III trial of encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF V600-mutant melanoma. J Clin Oncol. 2022;40(36):4178–88.
Tabernero J, Grothey A, Van Cutsem E, Yaeger R, Wasan H, Yoshino T, et al. Encorafenib plus cetuximab as a new standard of care for previously treated BRAF V600E-mutant metastatic colorectal cancer: updated survival results and subgroup analyses from the BEACON study. J Clin Oncol. 2021;39(4):273–84.
Ascierto PA, McArthur GA, Dreno B, Atkinson V, Liszkay G, Di Giacomo AM, et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17(9):1248–60.
Ye Q, Srivastava P, Al-Kuwari N, Chen X. Oncogenic BRAF(V600E) induces microglial proliferation through extracellular signal-regulated kinase and neuronal death through c-Jun N-terminal kinase. Neural Regen Res. 2023;18(7):1613–22.
Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017;2017.
Saginala K, Barsouk A, Aluru JS, Rawla P, Barsouk A. Epidemiology of melanoma. Med Sci. 2021;9(4).
Grothey A, Fakih M, Tabernero J. Management of BRAF-mutant metastatic colorectal cancer: a review of treatment options and evidence-based guidelines. Ann Oncol. 2021;32(8):959–67.
Leonetti A, Facchinetti F, Rossi G, Minari R, Conti A, Friboulet L, et al. BRAF in non-small cell lung cancer (NSCLC): pickaxing another brick in the wall. Cancer Treat Rev. 2018;66:82–94.
Scheffel RS, Dora JM, Maia AL. BRAF mutations in thyroid cancer. Curr Opin Oncol. 2022;34(1):9–18.
Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011;121(3):397–405.
Vuong HG, Altibi AMA, Duong UNP, Ngo HTT, Pham TQ, Fung KM, et al. BRAF mutation is associated with an improved survival in glioma-a systematic review and meta-analysis. Mol Neurobiol. 2018;55(5):3718–24.
Wang W, Wang M, Jiang H, Wang T, Da R. BRAF(non-V600E) more frequently co-occurs with IDH1/2 mutations in adult patients with gliomas than in patients harboring BRAF(V600E) but without a survival advantage. BMC Neurol. 2021;21(1):195.
Dahiya S, Haydon DH, Alvarado D, Gurnett CA, Gutmann DH, Leonard JR. BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol. 2013;125(6):901–10.
Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland A, et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18(10):1307–16.
Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol. 2018;36(1):7–13.
Kaley T, Touat M, Subbiah V, Hollebecque A, Rodon J, Lockhart AC, et al. BRAF inhibition in BRAF(V600)-mutant gliomas: results from the VE-BASKET study. J Clin Oncol. 2018;36(35):3477–84.
Wen PY, Stein A, van den Bent M, De Greve J, Wick A, de Vos F, et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutant low-grade and high-grade glioma (ROAR): a multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol. 2022;23(1):53–64.
Subbiah V, Kreitman RJ, Wainberg ZA, Gazzah A, Lassen U, Stein A, et al. Dabrafenib plus trametinib in BRAFV600E-mutated rare cancers: the phase 2 ROAR trial. Nat Med. 2023 Apr 14.
Salama AKS, Li S, Macrae ER, Park JI, Mitchell EP, Zwiebel JA, et al. Dabrafenib and trametinib in patients with tumors with BRAF(V600E) mutations: results of the NCI-MATCH trial subprotocol H. J Clin Oncol. 2020;38(33):3895–904.
Nobre L, Zapotocky M, Ramaswamy V, Ryall S, Bennett J, Alderete D, et al. Outcomes of BRAF V600E pediatric gliomas treated with targeted BRAF inhibition. JCO Precis Oncol. 2020;4.
Berzero G, Bellu L, Baldini C, Ducray F, Guyon D, Eoli M, et al. Sustained tumor control with MAPK inhibition in BRAF V600-mutant adult glial and glioneuronal tumors. Neurology. 2021;97(7):e673–83.
Perez JPM, Muchart J, Lopez VS, Capella MS, Salvador N, Jaume SP, et al. Targeted therapy for pediatric low-grade glioma. Childs Nerv Syst. 2021;37(8):2511–20.
Di Nunno V, Gatto L, Tosoni A, Bartolini S, Franceschi E. Implications of BRAF V600E mutation in gliomas: Molecular considerations, prognostic value and treatment evolution. Front Oncol. 2022;12:1067252.
Dankner M, Rose AAN, Rajkumar S, Siegel PM, Watson IR. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene. 2018;37(24):3183–99.
Schreck KC, Grossman SA, Pratilas CA. BRAF mutations and the utility of RAF and MEK inhibitors in primary brain tumors. Cancers. 2019;11(9).
Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68(21):8673–7.
Andrews LJ, Thornton ZA, Saincher SS, Yao IY, Dawson S, McGuinness LA, et al. Prevalence of BRAFV600 in glioma and use of BRAF inhibitors in patients with BRAFV600 mutation-positive glioma: systematic review. Neuro Oncol. 2022;24(4):528–40.
Ryall S, Zapotocky M, Fukuoka K, Nobre L, Guerreiro Stucklin A, Bennett J, et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell. 2020;37(4):569-83 e5.
Dono A, Vu J, Anapolsky M, Hines G, Takayasu T, Yan Y, et al. Additional genetic alterations in BRAF-mutant gliomas correlate with histologic diagnoses. J Neurooncol. 2020;149(3):463–72.
Omura T, Takahashi M, Ohno M, Miyakita Y, Yanagisawa S, Tamura Y, et al. Clinical application of comprehensive genomic profiling tests for diffuse gliomas. Cancers. 2022;14(10).
Lazow MA, Hoffman L, Schafer A, Osorio DS, Boue DR, Rush S, et al. Characterizing temporal genomic heterogeneity in pediatric low-grade gliomas. Acta Neuropathol Commun. 2020;8(1):182.
Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343(6167):189–93.
Ellison DW, Hawkins C, Jones DTW, Onar-Thomas A, Pfister SM, Reifenberger G, et al. cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF(V600E) mutation. Acta Neuropathol. 2019;137(4):683–7.
Raabe EH, Lim KS, Kim JM, Meeker A, Mao XG, Nikkhah G, et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res. 2011;17(11):3590–9.
Huillard E, Hashizume R, Phillips JJ, Griveau A, Ihrie RA, Aoki Y, et al. Cooperative interactions of BRAFV600E kinase and CDKN2A locus deficiency in pediatric malignant astrocytoma as a basis for rational therapy. Proc Natl Acad Sci USA. 2012;109(22):8710–5.
Prabowo AS, Iyer AM, Veersema TJ, Anink JJ, Schouten-van Meeteren AY, Spliet WG, et al. BRAF V600E mutation is associated with mTOR signaling activation in glioneuronal tumors. Brain Pathol. 2014;24(1):52–66.
Wang J, Liu Z, Cui Y, Liu Y, Fang J, Xu L, et al. Evaluation of EZH2 expression, BRAF V600E mutation, and CDKN2A/B deletions in epithelioid glioblastoma and anaplastic pleomorphic xanthoastrocytoma. J Neurooncol. 2019;144(1):137–46.
Frazão L, do Carmo Martins M, Nunes VM, Pimentel J, Faria C, Miguéns J, et al. BRAF V600E mutation and 9p21: CDKN2A/B and MTAP co-deletions–markers in the clinical stratification of pediatric gliomas. BMC Cancer. 2018;18(1).
Gabler L, Lotsch D, Kirchhofer D, van Schoonhoven S, Schmidt HM, Mayr L, et al. TERT expression is susceptible to BRAF and ETS-factor inhibition in BRAF(V600E)/TERT promoter double-mutated glioma. Acta Neuropathol Commun. 2019;7(1):128.
Fukuoka K, Mamatjan Y, Ryall S, Komosa M, Bennett J, Zapotocky M, et al. BRAF V600E mutant oligodendroglioma-like tumors with chromosomal instability in adolescents and young adults. Brain Pathol. 2020;30(3):515–23.
Pages M, Beccaria K, Boddaert N, Saffroy R, Besnard A, Castel D, et al. Co-occurrence of histone H3 K27M and BRAF V600E mutations in paediatric midline grade I ganglioglioma. Brain Pathol. 2018;28(1):103–11.
Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MY, Yu M, et al. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res. 2010;70(2):512–9.
Lassaletta A, Zapotocky M, Mistry M, Ramaswamy V, Honnorat M, Krishnatry R, et al. Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol. 2017;35(25):2934–41.
Tan JY, Wijesinghe IVS, Alfarizal Kamarudin MN, Parhar I. Paediatric gliomas: BRAF and Histone H3 as biomarkers, therapy and perspective of liquid biopsies. Cancers. 2021;13(4).
Davies MA, Saiag P, Robert C, Grob J-J, Flaherty KT, Arance A, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18(7):863–73.
Usta D, Sigaud R, Buhl JL, Selt F, Marquardt V, Pauck D, et al. A cell-based MAPK reporter assay reveals synergistic MAPK pathway activity suppression by MAPK inhibitor combination in BRAF-driven pediatric low-grade glioma cells. Mol Cancer Ther. 2020;19(8):1736–50.
Zhang J, Yao TW, Hashizume R, Hariono S, Barkovich KJ, Fan QW, et al. Combined BRAF(V600E) and MEK blockade for BRAF(V600E)-mutant gliomas. J Neurooncol. 2017;131(3):495–505.
Grossauer S, Koeck K, Murphy NE, Meyers ID, Daynac M, Truffaux N, et al. Concurrent MEK targeted therapy prevents MAPK pathway reactivation during BRAFV600E targeted inhibition in a novel syngeneic murine glioma model. Oncotarget. 2016;7(46):75839–53.
Johanns TM, Ferguson CJ, Grierson PM, Dahiya S, Ansstas G. Rapid clinical and radiographic response with combined dabrafenib and trametinib in adults with BRAF-mutated high-grade glioma. J Natl Compr Canc Netw. 2018;16(1):4–10.
Costa IN, Reis J, Pinheiro J, Silva R, Fernandes C. Dabrafenib plus trametinib: an impressive response in an adult patient with BRAF V600E-mutated and isocitrate dehydrogenase (IDH) wild-type glioma. Cureus. 2022;14(8): e28156.
Fusco MJ, Pina Y, Macaulay RJ, Sahebjam S, Forsyth PA, Peguero E, et al. Durable progression-free survival with the use of BRAF and MEK inhibitors in four cases with BRAF V600E-mutated gliomas. Cancer Control. 2021;28:10732748211040012.
Toll SA, Tran HN, Cotter J, Judkins AR, Tamrazi B, Biegel JA, et al. Sustained response of three pediatric BRAF(V600E) mutated high-grade gliomas to combined BRAF and MEK inhibitor therapy. Oncotarget. 2019;10(4):551–7.
Schreck KC, Guajardo A, Lin DDM, Eberhart CG, Grossman SA. Concurrent BRAF/MEK inhibitors in BRAF V600-mutant high-grade primary brain tumors. J Natl Compr Canc Netw. 2018;16(4):343–7.
Smith-Cohn M, Davidson C, Colman H, Cohen AL. Challenges of targeting BRAF V600E mutations in adult primary brain tumor patients: a report of two cases. CNS Oncol. 2019;8(4):CNS48.
Del Bufalo F, Ceglie G, Cacchione A, Alessi I, Colafati GS, Carai A, et al. BRAF V600E inhibitor (vemurafenib) for BRAF V600E mutated low grade gliomas. Front Oncol. 2018;8:526.
Hargrave DR, Bouffet E, Tabori U, Broniscer A, Cohen KJ, Hansford JR, et al. Efficacy and safety of dabrafenib in pediatric patients with BRAF V600 mutation-positive relapsed or refractory low-grade glioma: results from a phase I/IIa study. Clin Cancer Res. 2019;25(24):7303–11.
Nicolaides T, Nazemi KJ, Crawford J, Kilburn L, Minturn J, Gajjar A, et al. Phase I study of vemurafenib in children with recurrent or progressive BRAF(V600E) mutant brain tumors: Pacific Pediatric Neuro-Oncology Consortium study (PNOC-002). Oncotarget. 2020;11(21):1942–52.
Bouffet E, Geoerger B, Moertel C, Whitlock JA, Aerts I, Hargrave D, et al. Efficacy and safety of trametinib monotherapy or in combination with dabrafenib in pediatric BRAF V600-mutant low-grade glioma. J Clin Oncol. 2023;41(3):664–74.
Eric Bouffet JH, Maria Luisa Garré, Junichi Hara, Ashley Plant-Fox, Isabelle Aerts, Franco Locatelli, Jasper Van der Lugt, Ludmila Papusha, Felix Sahm, Uri Tabori, Kenneth J. Cohen, Roger J. Packer, Olaf Witt, Lali Sandalic, Ana Bento Pereira da Silva, Mark W. Russo, and Darren R. Hargrave. Primary analysis of a phase II trial of dabrafenib plus trametinib (dab + tram) in BRAF V600-mutant pediatric low-grade glioma (pLGG). J Clin Oncol. 2022 40:17_suppl, LBA2002-LBA2002.
Leclair NK, Lambert W, Roche K, Gillan E, Gell JJ, Lau CC, et al. Early experience with targeted therapy as a first-line adjuvant treatment for pediatric low-grade glioma. Neurosurg Focus. 2022;53(6):E15.
Rosenberg T, Yeo KK, Mauguen A, Alexandrescu S, Prabhu SP, Tsai JW, et al. Upfront molecular targeted therapy for the treatment of BRAF-mutant pediatric high-grade glioma. Neuro Oncol. 2022;24(11):1964–75.
Darren R. Hargrave KT, Junichi Hara, Uwe R. Kordes, Santhosh A. Upadhyaya, Felix Sahm, Eric Bouffet, Roger J. Packer, Olaf Witt, Lali Sandalic, Agnieszka Kieloch, Mark W. Russo, and Kenneth J. Cohen. Dabrafenib + trametinib (dab + tram) in relapsed:refractory (r:r) BRAF V600–mutant pediatric high-grade glioma (pHGG)- Primary analysis of a phase II trial. 2022;DOI: https://doi.org/10.1200/JCO.2022.40.16_suppl.2009 J Clin Oncol. 40, no. 16_suppl (June 01, 2022) 2009-2009.
Karisa Schreck RS, L Burt Nabors, Ben Ellingson, Joy Fisher, Serena Desideri, Neeraja Danda, Michelle Rudek, Stuart Grossman, Xiaobu Ye. CTNI-60. Preliminary results of binimetinib and encorafenib in adults with recurrent BRAF V600E-mutated high-grade glioma. Neurooncology. 2022;24,7:vii86, https://doi.org/10.1093/neuonc/noac209325
Ascierto PA, Dummer R, Gogas HJ, Flaherty KT, Arance A, Mandala M, et al. Update on tolerability and overall survival in COLUMBUS: landmark analysis of a randomised phase 3 trial of encorafenib plus binimetinib vs vemurafenib or encorafenib in patients with BRAF V600-mutant melanoma. Eur J Cancer. 2020;126:33–44.
Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76.
Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19(5):603–15.
Mincu RI, Mahabadi AA, Michel L, Mrotzek SM, Schadendorf D, Rassaf T, et al. Cardiovascular adverse events associated with BRAF and MEK inhibitors: a systematic review and meta-analysis. JAMA Netw Open. 2019;2(8): e198890.
Glen C, Tan YY, Waterston A, Evans TRJ, Jones RJ, Petrie MC, et al. Mechanistic and clinical overview cardiovascular toxicity of BRAF and MEK inhibitors: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2022;4(1):1–18.
Atkinson V, Robert C, Grob JJ, Gogas H, Dutriaux C, Demidov L, et al. Improved pyrexia-related outcomes associated with an adapted pyrexia adverse event management algorithm in patients treated with adjuvant dabrafenib plus trametinib: primary results of COMBI-APlus. Eur J Cancer. 2022;163:79–87.
Li L, Fang S, Li G, Zhang K, Huang R, Wang Y, et al. Glioma-related epilepsy in patients with diffuse high-grade glioma after the 2016 WHO update: seizure characteristics, risk factors, and clinical outcomes. J Neurosurg. 2022;136(1):67–75.
Dreno B, Ribas A, Larkin J, Ascierto PA, Hauschild A, Thomas L, et al. Incidence, course, and management of toxicities associated with cobimetinib in combination with vemurafenib in the coBRIM study. Ann Oncol. 2017;28(5):1137–44.
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–703.
Touat M, Gratieux J, Condette Auliac S, Sejean K, Aldea S, Savatovsky J, et al. Vemurafenib and cobimetinib overcome resistance to vemurafenib in BRAF-mutant ganglioglioma. Neurology. 2018;91(11):523–5.
Schreck KC, Morin A, Zhao G, Allen AN, Flannery P, Glantz M, et al. Deconvoluting mechanisms of acquired resistance to RAF inhibitors in BRAF(V600E)-mutant human glioma. Clin Cancer Res. 2021;27(22):6197–208.
Yao TW, Zhang J, Prados M, Weiss WA, James CD, Nicolaides T. Acquired resistance to BRAF inhibition in BRAFV600E mutant gliomas. Oncotarget. 2017;8(1):583–95.
Wang J, Yao Z, Jonsson P, Allen AN, Qin ACR, Uddin S, et al. A secondary mutation in BRAF confers resistance to RAF inhibition in a BRAF(V600E)-mutant brain tumor. Cancer Discov. 2018;8(9):1130–41.
Jain P, Fierst TM, Han HJ, Smith TE, Vakil A, Storm PB, et al. CRAF gene fusions in pediatric low-grade gliomas define a distinct drug response based on dimerization profiles. Oncogene. 2017;36(45):6348–58.
Yao TW, Zhang J, Prados M, Weiss WA, James CD, Nicolaides T. EGFR blockade prevents glioma escape from BRAFV600E targeted therapy. Oncotarget. 2015;6(26):21993–2005.
Mulcahy Levy JM, Zahedi S, Griesinger AM, Morin A, Davies KD, Aisner DL, et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife. 2017;6.
Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014;4(7):773–80.
Pina Y, Fusco MJ, Macaulay RJ, Walko CM, Peguero E, Evernden BR, et al. Using personalized medicine in gliomas: a genomic approach to diagnosis and overcoming treatment resistance in a case with pleomorphic xanthoastrocytoma. J Neurol. 2020;267(3):783–90.
Maxwell MJ, Arnold A, Sweeney H, Chen L, Lih TM, Schnaubelt M, et al. Unbiased proteomic and phosphoproteomic analysis identifies response signatures and novel susceptibilities after combined MEK and mTOR inhibition in BRAF(V600E) mutant glioma. Mol Cell Proteomics. 2021;20: 100123.
Sasame J, Ikegaya N, Kawazu M, Natsumeda M, Hayashi T, Isoda M, et al. HSP90 inhibition overcomes resistance to molecular targeted therapy in BRAFV600E-mutant high-grade glioma. Clin Cancer Res. 2022;28(11):2425–39.
Bid HK, Kibler A, Phelps DA, Manap S, Xiao L, Lin J, et al. Development, characterization, and reversal of acquired resistance to the MEK1 inhibitor selumetinib (AZD6244) in an in vivo model of childhood astrocytoma. Clin Cancer Res. 2013;19(24):6716–29.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflict of Interest
Thiago P. Muniz is a prior recipient of the Novartis Oncology Young Canadian Investigator Award. Warren P. Mason has received honoraria from Bayer, Viatris, Inc., and GlaxoSmithKline, and is part of the advisory board for Novocure.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Code Availability
Not applicable.
Author Contributions
Thiago P. Muniz: conceptualization, methodology, data curation, writing—original draft, writing—review and editing. Warren P. Mason: conceptualization, writing—review and editing, supervision.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Muniz, T.P., Mason, W.P. BRAF Mutations in CNS Tumors—Prognostic Markers and Therapeutic Targets. CNS Drugs 37, 587–598 (2023). https://doi.org/10.1007/s40263-023-01016-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-023-01016-5