
Abstract
Despite the established efficacy of chimeric antigen receptor (CAR) T-cell therapy in hematologic malignancies, translating CAR T therapy to solid tumors has remained investigational. Glioblastoma, the most aggressive and lethal form of primary brain tumor, has recently been among the malignancies being trialed clinically with CAR T cells. Glioblastoma in particular holds several unique features that have hindered clinical translation, including its vast intertumoral and intratumoral heterogeneity, associated immunosuppressive environment, and lack of clear experimental models to predict response and analyze resistant phenotypes. Here, we review the history of CAR T therapy development, its current progress in treating glioblastoma, as well as the current challenges and future directions in establishing CAR T therapy as a viable alternative to the current standard of care. Tremendous efforts are currently ongoing to identify novel CAR targets and target combinations for glioblastoma, to modify T cells to enhance their efficacy and to enable them to resist tumor-mediated immunosuppression, and to utilize adjunct therapies such as lymphodepletion, checkpoint inhibition, and bi-specific engagers to improve CAR T persistence. Furthermore, new preclinical models of CAR T therapy are being developed that better reflect the clinical features seen in human trials. Current clinical trials that rapidly incorporate key preclinical findings to patient translation are emerging.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Treatment with chimeric antigen receptor (CAR) T cells, which are modified immune cells that target specific molecules unique to cancer, has been very successful in hematologic malignancies and has been trialed clinically in glioblastoma in phase I studies. |
Many challenges in glioblastoma CAR T treatment remain, including addressing tumor heterogeneity, immunosuppressive environments, and the need for more clinically relevant experimental models. |
Current efforts aim to identify novel target antigens and antigen combinations, modify T cells to enhance CAR T efficacy, and utilize therapeutic adjuncts to complement CAR T treatment. Clinical trials are rapidly incorporating this emerging preclinical data. |
1 Introduction
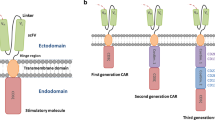
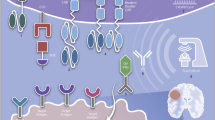
With recent advances, chimeric antigen receptor (CAR) T cells have become a promising new modality for patients with previously refractory cancers. CAR T cells represent a new mode of immunotherapy wherein T cells, generally from an autologous source, are genetically modified to express a synthetic receptor that targets a tumor-specific or tumor-associated antigen (Fig. 1) [1].

Summary of chimeric antigen receptor (CAR) T-cell generation. (1) T cells are isolated from patient leukopheresis. (2) T cells are modified via viral vectors or through novel mechanisms such as CRISPR/Cas9 to express the specific CAR of interest. (3) T cells are expanded ex vivo in order to generate sufficient CAR T-cell product for effective anti-tumor response. (4) CAR T cells are subsequently reintroduced to the patient for tumor-specific targeting. Common mechanisms of resistance include antigen-negative escape and T-cell exhaustion
In 2017, the US FDA approved two CAR T-based therapies, tisagenlecleucel (Kymriah; Novartis) and axicabtagene ciloleucel (Yescarta; Kite Pharma/Gilead Sciences), both for CD19-positive B-cell malignancies [2]. The FDA approved these drugs based on impressive results from two respective phase II trials. In the ELIANA trial, tisagenlecleucel showed complete remission rates of 81% in patients with relapsed or refractory (R/R) acute B-cell lymphoblastic leukemia (ALL) [3]. Similarly, the ZUMA-1 trial for axicabtagene ciloleucel showed an overall response rate of 82% and complete response rate of 58% for patients with R/R, aggressive non-Hodgkin lymphoma (NHL) [4]. These remarkable response rates, in patients with few remaining therapeutic options, have raised expectations for CAR T cells in other cancers.
We review the development of CAR T therapy in hematologic malignancy and recent attempts for clinical application in solid tumors with a focus on glioblastoma. We also discuss the unique challenges and potential avenues of development for CAR T treatment in glioblastoma, which include addressing intertumoral and intratumoral antigen heterogeneity, tumor antigen loss in response to therapy, adaptive immunosuppressive changes in the tumor microenvironment, and T-cell exhaustion.
1.1 The Development of Chimeric Antigen Receptor (CAR) T Cells
Almost three decades of research have taken CAR T cells from conception to FDA approval. Gross et al. first described a CAR in 1989, reporting their synthetic fusion of an antibody’s variable region with a T-cell receptor (TCR) signal transduction domain [5]. Using this construct, the authors could direct the specificity of T-cell cytolysis toward a desired antigen, without dependence on major histocompatibility complex (MHC) presentation. The essential components of a ‘first-generation’ CAR, analogous to the initial design by Gross et al., are now taken to include an extracellular antibody fragment, a linking domain, a transmembrane domain, and a CD3-ζ intracellular signaling component. Unfortunately, first-generation CARs proved to have limited efficacy against in vivo tumor targets, perhaps because they lacked the physiologic second signal required for full T-cell activation [6]. The earliest clinical trials evaluating first-generation CARs targeted the folate receptor in ovarian carcinoma, carbonic anhydrase receptor (CAIX) in renal cell carcinoma, GD2 in neuroblastoma, and CD20 in NHL [7,8,9,10,11]. There were no clinical responses in these studies except one partial response in the neuroblastoma study, in a patient with more limited disease than the other enrollees. All four studies raised concerns about persistence with first-generation CARs, with detectable CAR T cells declining in most patients within several days and becoming completely absent within several weeks. It was understood that first-generation CAR T cells were missing key ‘second-signal’ pathways, particularly co-stimulation by receptors such as CD28 and CD137, which led to CAR T defects such as minimal interleukin (IL)-2 stimulation on antigen engagement [9]. The renal, ovarian, and lymphoma trials attempted to work around this deficiency by coadministering IL-2, but clinical responses remained poor. The modest clinical results in these studies made clear the need for improved CAR T designs.
The second-generation CARs added domains for co-stimulatory signals, in particular CD28 and 4-1BB, in the intracellular chain alongside CD3-ζ. The first clinical trials with second-generation CARs, against CD19 in B-cell malignancies, produced striking complete remissions [12, 13]. It became clear by in vitro and in vivo experiments that CD28 and 4-1BB co-stimulatory signals enhance proliferation, persistence, and cytokine production of activated T cells [14,15,16]. The CD28 signal effectively amplifies the normal TCR signal cascade, leading to amplified cytokine production, proliferation, and an effector phenotype [14, 15]. The 4-1BB domain generally leads to a less brisk initial increase in cytokine production, but it enhances long-term persistence, increases central memory differentiation, and ameliorates activation-induced T-cell exhaustion [13, 15,16,17]. Included in the second-generation CARs are the current two FDA-approved drugs. Tisagenlecleucel utilizes the 4-1BB domain, while axicabtagene ciloleucel utilizes the CD28 co-stimulatory domain. The preclinical and clinical trial pipeline includes third- and later-generation CARs, the development of which have been reviewed in detail elsewhere [18]. The third-generation CARs add both CD28 and 4-1BB to the intracellular chain, and the fourth-generation constructs allow the secretion of certain anti-tumor proteins, such as cytokines, in response to T-cell activation [19].
1.2 CAR T Cells in Solid Tumors
Trials of CAR T cells in solid tumors followed soon after those in hematologic cancers [17]. More than 100 clinical trials of CAR T cells in solid tumors have so far been initiated, most using a second- or third-generation of CARs. To our knowledge, at least 20 studies, have now published results [18]. The response rates, across solid tumor types, have been lower than those achieved in bone marrow-derived tumors. Nonetheless, there have been partial and complete responses in various trials that prove the principle that CAR T cells can be effective for solid tumors. The most positive trials to date have included the targeting of GD2 in neuroblastoma (3 of 11 with complete remissions), human epidermal growth factor receptor 2 (HER2) in sarcoma (4 of 17 with stable disease), and prostate-specific membrane antigen (PSMA) in prostate cancer (partial response in two patients and minimal response in one of five patients) [19,20,21,22]. One interesting study, using Epstein–Barr virus (EBV)-specific T cells expressing a first-generation CAR against GD2, achieved complete responses in four of a cohort of eight EBV+ patients with imaging-evaluable neuroblastoma [23]. This therapy was closer in mechanism to a second-generation CAR because the viral specificity of the native TCR allowed co-stimulatory activation both in vitro and likely in vivo. Tumor samples from patients with complete responses notably showed no PCR evidence of the CAR gene, suggesting an indirect therapeutic response may have occurred. Trials with less successful results have included the targeting of carcinoembryonic antigen (CEA) in hepatic metastases (stable disease in one of seven patients, progression or no response in the remainder) [24]. Overall, the clinical success in solid tumor trials has been less dramatic than for hematologic malignancies, but the complete and partial responses observed to date do suggest therapeutic activity.
1.3 CAR T Trials in Glioblastoma
Glioblastoma represents the highest grade of primary gliomas (WHO Grade IV), which holds a median survival of approximately 15 months with the standard of care of radiation and temozolomide [25]. Furthermore, the addition of temozolomide seems to only benefit a subset of patients who have an epigenetic silencing of O6-methylguanine-DNA methyltransferase (MGMT) [26]. Since the establishment of this standard of care therapy in 2004, the only other therapy that has prolonged survival in the newly diagnosed setting has been the use of tumor-treating fields, an external cap worn by patients that generates pulsating electrical fields and is believed to inhibit cellular proliferation, which has demonstrated an increase in survival to 20.9 months, compared with 16 months in those who had completed concomitant temozolomide and radiation [27, 28]. Furthermore, the bioavailability of targeted inhibitors commonly used in other cancers has been limited by the blood–brain barrier, which does not allow passive diffusion of molecules and in many cases drives active efflux [29]. With the limited success of standard treatments, and few recent advances, it is crucial to identify new strategies that can benefit patients with GBM.
The completed CAR T trials for glioblastoma have also showed signs of promise, while demonstrating the need for continued development, as manifested by ongoing trials (Table 1). To date, one CAR T trial targeting HER2, two targeting IL-13Rα2, and two targeting epidermal growth factor receptor (EGFR) vIII have been published [30,31,32,33]. All of these CAR targets are membrane bound, and when expressed on tumor cells are more highly expressed in tumor tissue than in normal brain [30,31,32,33]. HER2, commonly associated with breast cancer, is not normally expressed in the brain, but has been associated with certain forms of glioblastoma. IL-13Rα2 has low expression in the brain and is overexpressed in a subset of glioblastoma [34]. EGFRvIII is notable as a variant mutation, not normally seen in human tissues, of wild-type (WT) EGFR with a 2–7 exon deletion [35] that leads to a conformational change enabling tumor-specific targeting. Both studies of EGFRvIII utilized a CAR T target that was specific to EGFRvIII, with little to no activity to WT EGFR [32, 33].
One case report of a patient with multifocal recurrent glioblastoma multiforme (GBM) who received anti-IL-13Rα2 CAR T therapy demonstrated complete regression of all tumors, with recurrence after 7.5 months. This case was also notable for intracavitary and intraventricular delivery of CAR T, which may have several advantages, including the reduced need for CAR T-cell trafficking to the site of interest, as well as a reduced risk of systemic response from peripheral infusion [36]. When the tumor eventually recurred, preliminary data suggested decreased expression of IL-13Rα2 [31]. The anti-HER2 trial had one patient, of a total of 17, who demonstrated a partial response lasting 9.2 months, and three patients had stable disease for over 2 years of follow-up. This study was notable in that it consisted of both adult (n = 10) and pediatric (n = 7) patients with HER2+ glioblastoma. While the authors cited that pediatric patients have a better prognosis than adults [37], the study did not see any age-related survival benefit upon multivariate analysis. The anti-HER2 trial also demonstrated the challenge in using conventional imaging modalities to monitor response to CAR T therapy. Several patients had an increase in peritumoral edema in the weeks following CAR T infusion, but all of them survived more than 6 additional months, suggesting that perhaps some of those radiographic effects were not true progression [30]. The effect of immunotherapy on the blood–brain barrier may lead to increased contrast enhancement and create the false impression of tumor progression. At our institution, the trial of anti-EGFRvIII CAR T cells did not observe marked regression in tumor volume by serial magnetic resonance imaging (MRI), but it did demonstrate changes in tumor histology in post-infusion surgical specimens. In particular, several patients had decreased EGFRvIII antigen expression following infusion, as well as increases in anti-inflammatory adaptive responses such as regulatory T-cell (Treg) content and staining for programmed death-1 (PD-1) and programmed death-ligand 1 (PD-L1) [33]. The second study targeting EGFRvIII, by Goff et al., was unique among GBM CAR T trials for its use of lymphodepleting chemotherapy prior to infusion. There were no objective responses by MRI in this study, and the median progression-free survival (PFS) was 1.3 months. One patient had a PFS of 12.5 months and remained alive after 59 months of follow-up [32]. Collectively, these pilot studies included dozens of patients and observed radiographic objective responses in only two patients, one from Brown et al. when directed against IL-13Rα2, and the other from Ahmed et al. when directed against HER2 [30, 31]. However, these responses against an historically inveterate tumor suggest that CAR T has the potential to develop into an effective therapy for at least a subset of patients. New approaches are required that enhance the potency of CAR T and address the deficiencies of this treatment for GBM.
1.4 CAR T Safety Profile
Regarding safety, CAR T cells are theoretically more precise and potentially less toxic than conventional systemic chemotherapy. However, there are several serious adverse events associated with their use, including cytokine release syndrome (CRS) [38], neurotoxicity, and on-target off-tumor toxicity. Both CRS and neurotoxicity are thought to be related to the massive cytokine release that occurs when T cells are activated in the setting of a high tumor burden. CRS has proven quite common with anti-CD19 therapy in B-cell malignancies, occurring in 19–43% of patients, although treatments such as systemic corticosteroids and anti-IL-6 monoclonal antibodies are often rapidly effective in reversing these reactions [39]. In solid tumors, to our knowledge, two fatal adverse events have occurred. One case occurred during anti-HER2 CAR T therapy for colorectal cancer [40]. The patient developed pulmonary edema about 15 min after infusion of the CAR T cells. The dose the patient received was the highest planned in the first escalation cohort, suggesting that dose-dependent toxicity may have been a factor. Moreover, the selection of antigen may have played a role as HER2 is expressed at low levels in the lung. This may have led to non-specific binding and could have triggered the massive release of cytokines seen in this case. A second case occurred in a trial of anti-EGFRvIII CAR, and, similarly, the patient received the highest dose in the cohort and developed pulmonary edema shortly after transfusion [32]. Generally however, there appears to be a meaningful margin of safety for CAR T cells, as evidenced by the multiple trials that have shown clinical benefit with few adverse events [19, 20, 23, 31]. The anti-EGFRvIII trial in GBM at our institution noted a range of cytokine increases following transfusion, and while some patients did manifest mild systemic symptoms such as fever, there were no serious signs of CRS [33]. Approaches for making the therapies safer include the engineering of suicide genes, such as the herpes thymidine kinase, into the T cells. Another strategy is the expression of a fragment of EGFR on the T-cell surface, allowing the elimination of the product via monoclonal antibody [39]. The safety profile of CAR T will likely improve with the continued engineering of new safeguards.
2 Challenges and Future Directions in CAR T Immunotherapy for Glioblastoma
2.1 Antigen Targeting
2.1.1 Glioblastoma Heterogeneity
In contrast to hematologic malignancies such as leukemia and lymphoma that typically have a common antigenic target, glioblastoma is exceptional in its vast intertumoral heterogeneity. The spectrum of glioblastoma encompasses a wide variety of genetic mutations and prevents the application of a single CAR T strategy to all patients. For instance, in the targets tested in a clinical trial, EGFRvIII was found to be highly expressed in 11% of tumors, as defined by a transcript allelic fraction of > 10% [41]. HER2 expression in glioblastoma was reported to be a similarly low proportion at 15.4%, as measured by immunohistochemistry [42]. IL-13Rα2 is a promising target, with expression in one study at a rate of 44% of tumors by microarray and 47% by quantitative polymerase chain reaction (qPCR) [43]. Due to the varied expression of these targets, the clinical trial design includes strict screening criteria that excludes many patients from eligibility (Table 1).
In addition to the intertumoral heterogeneity within single tumors, there is significant intratumoral variability of potential antigenic targets. Single-cell transcriptomic profiling has demonstrated that glioblastoma represents a diversity of cell types within a single tumor [44], which further complicates CAR T targeting. For example, all of the established subtypes of GBM (classical, mesenchymal, neural, and proneural) [45] can be observed in the transcriptomic profiles from single cells from the same tumor. EGFRvIII has been found to be variably expressed in different regions of the same tumor [46], with variations in expression before and after standard-of-care treatment [47]. Another analysis on the single-cell level has revealed varied degrees of EGFR amplification with subclonal populations of EGFR mutants [48]. This intratumoral heterogeneity in gene expression complicates the analysis of CAR T efficacy in glioblastoma patient samples as it may be difficult to discern efficacy versus sampling variability.
More recent work has expanded on the single-cell characterization of glioblastoma, and has compared the heterogeneity of cellular states in glioblastoma to neural development, with states resembling oligodendrocyte progenitors, neuronal progenitors, astrocyte-like cells, and mesenchymal-like cells [49]. Intriguingly, many cells exhibit mixed states by single-cell RNA analysis, suggesting that these cells have plasticity to transition from one cell state to another. This was further supported with genetic barcoding of a mouse model of glioblastoma, demonstrating the generation of multiple cell types [49]. This ability of glioblastoma to state transition further complicates treatment strategies. To address these issues, tumor targeting through combination therapies has become a strategy to overcome the challenges posed by intertumoral and intratumoral variability. Bivalent CAR T therapy utilizing IL-13Rα2 and HER2 was previously shown to reduce antigen escape in a murine glioblastoma model [50]. Using a trivalent strategy, a single CAR T-cell product that targeted HER2, IL-13Rα2, and Erythropoietin-producing hepatocellular carcinoma A2 (EphA2) was found to have increased interferon (IFN)-γ and IL-2 expression compared with monovalent and bivalent constructs, and mice had increased survival at 60 days with patient-derived xenografts expressing these antigens [51]. Analysis of 47 patient-derived tumor samples suggested that treatment with this trivalent strategy would be capable of effectively targeting nearly all of the cells analyzed. Future directions would include developing novel CAR T targets as well as novel combinations of CAR T antigens to reduce antigen escape.
2.1.2 Emerging Preclinical CAR T Targets
Several emerging clinical targets are currently being studied in glioblastoma. B7-H3 is a transmembrane molecule overexpressed in many cancers, and it was found to be highly expressed in 8/34 tumor samples and moderately expressed in 11/34 tumor samples [52]. In vitro and xenograft studies of CAR T cells directed against B7-H3-expressing cell lines demonstrated T-cell activation and improved survival [52]. This has led to the development of a clinical trial with an anticipated start date of May 2020 (NCT04077866). Another study similarly demonstrated high expression in 76% of 46 specimens analyzed, with effective CAR T targeting both in vitro models and using in vivo models [53].
Natural killer group 2 member D (NKG2D) is another antigenic target that is overexpressed relative to normal tissues in multiple cancers, including glioblastoma. A key feature of NKG2D is that its expression is induced by chemotherapy and radiotherapy [54]. CAR T cells targeting NKG2D demonstrated synergy with radiotherapy in an immunocompetent, murine model using GL-261 cells [55]. Other studies similarly demonstrated efficient targeting of CAR T cells directed against NKG2D in human-derived glioblastoma cell and xenografts [56].
Carbonic anhydrase IX (CAIX) is expressed in hypoxic environments, a hallmark of glioblastoma. This becomes an intriguing target as hypoxia is associated with the treatment-resistant mesenchymal phenotype [57] and has been found to impair CAR T targeting in vitro [58]. CAIX-directed CAR T cells demonstrated in vitro and in vivo efficacy against the U251 glioblastoma cell line. Direct tumor injection minimized the off-target effects [59].
EphA2 is another target associated with glioblastoma [60]. EphA2 is expressed in multiple cancers and has been associated with malignant transformation [61]. In a study examining EphA2 as a potential CAR T target, EphA2 was found to be expressed in U87 and U373 cell lines, had varied but detectable expression in 5/5 primary GBM cell lines, and, importantly, showed low levels of expression in normal brain. CAR T cells directed against EphA2 demonstrated T-cell activation and improved survival in murine models compared with non-transduced controls [62].
Chondroitin sulfate proteoglycan 4 (CSPG4) is a cell surface membrane protein that was found to be highly expressed in 31 of 46 GBM specimens. CAR T-cell targeting of CSPG4 controlled the growth of CSPG4-expressing glioblastoma models in vitro and in vivo. A unique feature of CSPG4 was that microglia-generated tumor necrosis factor (TNF)-α induced CSPG4 expression, and high levels of CSPG4 were associated with high levels of microglia, as identified by Iba1. Notably, tumor escape via antigen loss was not observed in this study [63].
CD133 was initially found to be a marker for brain tumor initiating cells [64,65,66] that are able to persist despite treatment, and promote tumor resistance. CAR T-cell therapy directed against the epitope of CD133, AC133 leads to selective targeting of AC133+ GBM stem cells, however it also paradoxically led to the upregulation of CD57 on CAR T cells. CD57 represents a terminally differentiated marker of T cells and may limit CAR T efficacy [67].
2.1.3 T-Cell-Mediated Antigen Loss and Escape
A fundamental mechanism of resistance to CAR T therapies across various systems is tumor cell persistence with antigen loss or low levels of target antigen [68, 69]. In clinical studies of glioblastoma, antigen loss was observed in the treatment with IL-13Rα2-directed CAR T cells [70]. This finding was also replicated in preclinical studies that combined IL-13Rα2 with transgenic IL-15 expression [71]. Targeting of EGFRvIII has similarly been limited by antigen loss in recurrence following administration of EGFRvIII-targeted CAR T cells [33]. In EGFRvIII peptide vaccines, 82% of patients had lost EGFRvIII expression upon recurrence [72], suggesting that this might be a general mechanism of resistance against antigen-specific targeting.
One study examining CAR T therapy in a mouse model of acute lymphoblastic leukemia (ALL) recently identified trogocytosis as a mechanism of antigen escape [73]. Trogocytosis is the process wherein T cells extract target antigens from antigen-presenting cells and express them on their own surface. In this study, trogocytosis of CD19 from tumor cells decreased antigen burden to the point of tumor cell escape. Furthermore, fratricide of CAR T cells was observed due to trogocytotic T-cell surface expression of CD19, leading to CAR T-mediated killing of CD19-bearing T cells. CAR T combination rather than monotherapy was shown to abrogate this mechanism of resistance [73]. The degree to which this process occurs in solid tumors such as glioblastoma remain unclear, but may be an important consideration for future CAR T strategies as this mechanism of resistance further supports the notion that targeting multiple antigens with CAR T therapy could potentially be advantageous.
2.2 Immunosuppressive Microenvironment
2.2.1 Immune Checkpoint Upregulation and Inhibition
The immunosuppressive environment of glioblastoma remains a challenge for CAR T therapy [74]. Glioblastoma is known to have, at baseline, low levels of T-cell infiltration and only moderate levels of tumor mutational burden. Complicating these observations is the routine use of the corticosteroid dexamethasone in patients, which is often used to reduce cerebral edema but is also known to suppress T-cell responses [75,76,77]. The degree to which these factors impair CAR T responses in human trials is currently an area of active investigation.
Novel mechanisms to enhance immunologic responses have been developed and utilized in various malignancies [78]. In particular, inhibition of immunosuppressive checkpoint molecules such as CTLA-4, PD-1, and PD-L1 has had significant promise in other solid tumors such as melanoma [79] and lung cancer [80]. In CNS metastatic disease, evidence of tumor regression following treatment with the PD-1 inhibitors pembrolizumab and nivolumab has been observed, suggesting evidence of blood–brain barrier permeability [81], Given this efficacy, the use of checkpoint inhibition has recently been clinically explored in glioblastoma [82,83,84]. One study did demonstrate a statistical survival benefit of neoadjuvant treatment compared with adjuvant treatment (13.7 months vs. 7.5 months; p = 0.04) in recurrent glioblastoma and was associated with an upregulation of T cell and IFNγ gene-related expression [82]. In a second study, patients were treated with neoadjuvant and adjuvant nivolumab and compared with the pretreated baseline, as well as to untreated controls. Elevated T-cell infiltration and chemokine expression was similarly seen with suggestion of an altered immune microenvironment, although no clear survival benefit was noted [83]. A third, longitudinal study compared the genomic profiles of responders and non-responders to treatment with nivolumab or pembrolizumab. The responsive group enriched for mitogen-activated protein kinase (MAPK) pathway alterations, while non-responders enriched for phosphatase and tensin homolog (PTEN) mutations. This work similarly demonstrated alterations in the T-cell clonal diversity and tumor microenvironment [84]. Taken together, these works support the notion that immune-mediated checkpoints alter both the T-cell and tumor microenvironment, and could potentially enhance the efficacy of CAR T cells.
The aforementioned PD-1 and PD-L1 upregulation after CAR T exposure described in one of the anti-EGFRvIII clinical trials [33] prompted preclinical studies investigating the role of checkpoint inhibition in glioblastoma. In murine and canine models of glioblastoma, the efficacy of the appropriate checkpoint inhibitor varied based on the CAR T antigenic target. IL-13Rα2 CAR T-cell efficacy was enhanced with CTLA-4 blockade, while EGFRvIII CAR T-cell efficacy was enhanced with PD-1 and T-cell immunoglobulin mucin-3 (TIM-3) blockade [85]. Similarly, anti-HER2 CAR T cells had enhanced activity with the addition PD-1 blockade against HER2+ U251 cells. This led to increased cytokine activity and efficacy [86]. These preclinical studies led to the development of a phase I clinical trial that combines PD-1 checkpoint inhibition with pembrolizumab and EGFRvIII targeting CAR T therapy (ClinicalTrials.gov NCT03726515). This study is currently ongoing to analyze the safety of combined checkpoint inhibition with EGFRvIII CAR T therapy. In addition, a randomized study utilizing an IL-13Rα2-directed CAR T in combination with either nivolumab alone or nivolumab and ipilimumab is currently registered on ClinicalTrials.gov (NCT04003649), with an anticipated start date of December 2019.
2.2.2 T-Cell Exhaustion in Glioblastoma
Another potential challenge of CAR T therapy is the finding that glioblastoma, even in the treatment-naïve setting, is associated with sequestration of T cells in the bone marrow, leading to T-cell dysfunction [87]. This was prospectively measured clinically as a decrease in the numbers of both CD4+ and CD8+ T cells in treatment-naïve GBM patients compared with age-matched controls. Splenic volume as measured retrospectively from abdominal computed tomography (CT) scans was also shown to be decreased in GBM patients compared with controls. There was a concomitant three- to fivefold expansion of naïve T cells in the bone marrow. This phenotype was also noted in two lines of murine glioma. This T-cell sequestration may contribute to the lack of robust immunologic response seen in glioblastoma.
Lymphodepletion is also a common strategy to improve CAR T efficacy in hematologic cancers. Induction chemotherapy with fludarabine and cyclophosphamide was found to improve CAR T expansion and persistence [88]. This has been applied to glioblastoma as multiple trials targeting EGFRvIII (NCT01454596, NCT02844062) have incorporated fludarabine and cyclophosphamide into their protocols. As an alternative to fludarabine and cyclophosphamide, lymphodepletion with temozolomide, which is commonly utilized as standard of care in glioblastoma, improved CAR proliferation and persistence and improved survival in a preclinical murine model [89]. This preclinical model led to incorporation of temozolomide prior to CAR T treatment, in a recent clinical trial (NCT02664363).
One recently reported strategy that has been explored to enhance in vivo CAR T expansion has been the use of a novel antigen vaccine strategy that leverages the native lymph node APC cells in order to facilitate CAR T expansion and activation [90]. In this study, a CAR T ligand is modified such that it traffics into the lymph node and becomes expressed on the surface upon antigen-presenting cells. Infusion of CAR T cells leads to enhanced activation and proliferation in an immunocompetent mouse model. Future work can leverage similar strategies in enhancing the native response of CAR T cells in solid tumors.
Many preclinical efforts have been made to enhance the efficacy of CAR T cells through T-cell modifications. Third-generation CAR T cells are emerging that contain multiple co-stimulatory factors such as both CD28 and OX40 in an EGFRvIII targeting CAR [91]. As PD-1 upregulation is a known factor for T-cell exhaustion, CD133-directed CAR T cells have been modified to have a disruption of PD-1 expression by CRISPR/Cas9. This modification led to improved proliferation and cytotoxicity in vitro, as well as improved tumor growth inhibition in a murine glioma model [92]. CRISPR/Cas9 disruption of diacylglycerol kinase (DGK) similarly resulted in enhanced CAR T function and improved resistance to immunosuppressive signaling to transforming growth factor (TGF)-β and prostaglandin E2 in an EGFRvIII-targeting model [93]. IL-7 is a potent cytokine that has been co-expressed with a CAR targeting EphA2, leading to increased efficacy at lower cell doses, with complete tumor elimination in a murine xenograft model using the U373 glioblastoma cell line [94]. IL-15 co-expression in IL-13Rα2-targeted CAR T cells similarly led to enhanced efficacy against U373-based xenografts via enhanced proliferation, cytolytic activity, and persistence. However, this strategy was also notable for the development of IL-13Rα2-negative variants [71]. Due to the inherent non-specificity of targets such as WT EGFR, mRNA CARs have been developed to limit off-target effects. In vitro testing against U87, T98G, LN18, and other non-glioma cell lines demonstrated similar cytolytic activity, but reduced cytokine expression of IFNγ and TNFα [95].
Bi-specific T-cell engagers (BiTEs), which are engineered bispecific antibodies used to direct T cells to targets of interest [96], have been recently combined with CAR T cells. In a preclinical mouse model, EGFRvIII-directed CAR T cells secreting BiTEs against EGFR were able to eliminate a heterogeneously expressing EGFRvIII tumor model [97]. Alternatively, antibodies targeting both WT EGFR overexpressing cells and EGFRvIII overexpressing cells have been developed and utilized in preclinical models [98, 99] to help overcome tumor heterogeneity.
T-cell selection into specific subtypes is another potential avenue to enhance CAR T efficacy in glioblastoma. Selection for CD4+ T cells was found to have greater anti-tumor immunity and persistence compared with CD8+ T cells in an IL-13Rα2-targeting intracranial glioma model [100]. This concept of T-cell subtype selection is currently being explored in a registered clinical trial comparing subtype-enriched CAR T-cell populations against HER2-positive glioblastoma (NCT03389230).
2.2.3 Routine Use of Dexamethasone for Cerebral Edema
Glioblastoma is commonly treated clinically with dexamethasone for symptoms of cerebral edema [101]. There are scarce data regarding the effect of dexamethasone on CAR T therapies; however, preclinical studies suggest that dexamethasone can meaningfully inhibit immune responses to malignancies. Dexamethasone was found to upregulate the CTLA-4 checkpoint receptor in activated T cells, as well as blocking CD28-mediated cell cycle [102]. A study of an intracranial tumor in mice found that high-dose dexamethasone abolished the survival benefit conferred by a locally delivered IL-2 immunotherapy [55]. However, doses comparable with those commonly used in humans had no significant effect [75]. Similarly, one study of CAR T cells directed to IL-13Rα2 in mice demonstrated no significant impairment in CAR T anti-tumor activity at doses up to 1 mg/kg [103]. Potential mechanisms for corticosteroid inhibition of CAR T function include reduced trafficking to the tumor and suppressed cytokine release. In rats with orthotopic gliomas, a reduction in intratumoral lymphocyte invasion was observed with dexamethasone [76]. Moreover, dexamethasone is a known potent inhibitor of IFNγ, which can lead to decreased T-cell activation [77]. Overall, the RANO Working Group recommends that patients enrolled in immunotherapy clinical trials be given the lowest tolerable dose of dexamethasone [104]. One approach has been to allow for low-dose corticosteroids, up to 6 mg/day, based on the aforementioned preclinical data that found low-dose corticosteroids compatible with immunotherapy. Future studies could potentially evaluate the degree to which dexamethasone affects CAR T activity in human glioblastoma, to ensure that patients have the greatest chance of meaningful therapeutic response.
2.2.4 Leukapheresis for the Generation of CAR T Cells versus Universal CAR T Cells
The most common strategy for generating CAR T cells involves leukapheresis and modification of patient-specific T cells through viral vector transduction. The time required to manufacture CAR T cells can be prohibitive to patients with glioblastoma, who often face rapid clinical worsening [25]. The need for patient-specific leukapheresis is also troublesome for the routine use of dexamethasone, which can suppress T-cell activity. In order to address these issues, universal CAR T cells have been explored [105] that are gene-edited to be deficient of TCR, human lymphocyte antigen (HLA) class I, and PD-1. Such a strategy could potentially be used to create a T cell devoid of alloreactivity and suitable for transfusion into any recipient. Such universal CAR T cells have been used in the treatment of infant B-cell ALL (B-ALL) [106], producing a complete molecular response. The universal CAR T strategy will potentially reduce the manufacturing time and the cost of CAR T cells, allowing patients to be treated earlier in their disease course.
3 Limitations in Glioblastoma CAR T Modeling
3.1 Cell Culture Systems
Unlike hematologic malignancies that can be modeled through growth in suspension, modeling solid tumors such as glioblastoma become challenging due to the three-dimensional nature and the complex tumor microenvironment. Current culture systems typically rely on clonal growth from dissociated cells in the form of either attached monolayer cultures or in cell suspension as neurospheres [107]. These systems may not accurately reflect the diverse cell types that have been observed clinically from glioblastoma tissue samples [44, 49, 108]. This clonal selection of cell cultures can complicate analysis of resistant and/or adaptive phenotypes as responses to therapy may vary based on media conditions [109]. While many CAR targets endogenously express the target antigen in established cell lines [55, 59, 62], some mutational drivers, such as EGFRvIII in particular, are difficult to maintain in culture [35, 110]. Other variables such as EGFR amplification have also been found to vary based on the amount of EGF exposure [111], and, similarly, EGFRvIII expression has been theorized to be dependent on a lack of EGF exposure [112]. The generation of cell lines that retain stable, endogenous expression of EGFRvIII [113] has mitigated some of the challenges with CAR T targeting. However, as the heterogeneity of EGFRvIII and other heterogeneous target antigens within tumors becomes increasingly clear, further work in modeling tumor heterogeneity may be necessary [44, 48, 114].
3.2 Mouse Models
The use of mice to study CAR T immunotherapy in GBM provides an important preclinical assessment of treatment safety and efficacy. The most common animal GBM model for CAR T research at our institution has been immunocompromised NSG mice [115] implanted with permanent human tumor lines. Using xenografts from established tumor lines has the advantage that tumors retain the histologic and genetic features of human tumors. For example, implanting U251 and U87 cells in Balb/c immunodeficient mice generates tumors that exhibit neovascularization, pleomorphism, and T-cell and macrophage infiltrates, similar to native human GBMs [116]. The consistency of tumors across animals is also an important consideration that can impact research expense and the number of mice required per study. Permanent cell lines have the advantage of generating consistent and reproducible tumors, while models employing spontaneous, chemical-induced, or viral-induced tumors can be more variable in tumor grade, histology, and prognosis [117]. For CAR T research, immunocompromised mice also have the specific advantage that the CAR T treatment product can be derived from human T cells and still engraft in the mouse, because there is little host-versus-graft rejection.
There are disadvantages to xenograft models in immunocompromised mice. First, GBM employs immunosuppressive mechanisms to avoid immune surveillance in natural hosts. The interactions of the tumor with an intact immune system might have effects that could influence the success or failure of a new therapy. For example, NSG mice fail to develop fully functional macrophages and Tregs [115, 118]. Both of these immune cell types are upregulated in the GBM microenvironment and are suggested to function in tumor immune evasion. Immune-competent animals also have an advantage in more realistic representations of the safety of immunotherapies. For example, immune-competent mice can develop CRS in response to CAR T treatment, while NSG mice do not develop this complication [75]. There is also the question of whether permanent tumor cell lines implanted into immunocompromised mice can recapitulate the true clonal diversity of human GBMs [119].
Another approach that has been utilized for CAR T studies is the generation of murine CAR T cells with a murine glioma line in order to maintain an immunocompetent model. This has been performed in CAR T cells targeting IL-13Rα2 [120] and EGFRvIII [98]. In both studies, these immunocompetent models became resistant to tumor rechallenging following initial CAR T exposure, suggesting a CAR T-cell memory phenotype. The study of IL-13Rα2 further suggested a proinflammatory phenotype with the observed increased presence of T cells and dendritic cells and decrease in myeloid-derived suppressor cells [120]. Another potential representation of the behavior of human GBMs would be the spontaneous generation of tumors in immunocompetent animals. However, this approach has the disadvantage of requiring a large number of animals and suffering poor interanimal comparability [117].
There are approaches seeking to combine the realism of immune-competent models with the consistency and relevance to human disease of patient-derived cell lines. Patient-derived orthotopic xenografts may better recapitulate tumor heterogeneity, as well as provide an assessment of therapeutic response tailored to the specific patient [119]. Implanting human tumor tissue into a mouse engrafted with a human immune system is an investigational method that could yield consistent and relevant tumors while maintaining realistic tumor-immune interactions. Immunosuppressed mice engrafted with human hematopoietic stem cells can form functional ‘humanized’ immune systems [121]. The interactions of tumor cells with infiltrating immune cells contributes to tumor heterogeneity [114]. Future work in GBM modeling could potentially incorporate these humanized mouse models in order to better recapitulate GBM heterogeneity and immune interactions in the animal tumor.
4 Assessment of Efficacy
Treatment-related changes, even with conventional chemoradiation, may lead to increased peritumoral edema and vascular leakage. On conventional CT and MRI with contrast, these changes can cause tumors to transiently appear larger, a phenomenon known as ‘pseudo-progression’ [122]. Immunotherapies can similarly create inflammatory changes that affect the blood–brain barrier and lead to increased contrast enhancement [123]. These effects confound the use of simple tumor volume on conventional imaging to measure treatment response. While cases of dramatic complete response may be evident on plain MRI, there may be cases of partial response where immune activity is occurring in the tumor. It is important to identify cases where the CAR T product is present and active in the tumor, as that activity could then be correlated to clinical outcomes such as survival. The methods for measuring CAR T activity in clinical trial patients are evolving as these studies take place, but rapidly emerging strategies to date include advanced imaging modalities, tissue analysis following CAR T therapy, and analysis of peripheral blood samples.
Advanced radiographic methods are being developed to more accurately describe both CAR T-cell trafficking into the tumor, as well as tumor biologic response. One appealing method in development is the tagging of CAR T cells with an MRI-sensitive probe [124]. The accurate assessment of T-cell biodistribution in solid tumors with these methods could provide a rapid surrogate for clinical response in tumors where volume changes on conventional imaging are less reliable. On the tumor side, MRI spectroscopy and perfusion imaging can yield metrics that may be associated with tumor histology. While still being validated for use with immunotherapy, the measurement of relative cerebral blood volume (rCBV) is particularly promising. The measurement of rCBV assesses microvascular volume, as apart from microvascular permeability, which is measured by contrast enhancement. According to a study comparing radiographic with pathologic tumor grading, the rCBV metric is almost perfectly predictive of tumor grade, with a particularly strong association with the mitotic index [125]. The measurement of rCBV could therefore provide a non-invasive means of assessing tumor response to immunotherapy.
When patients undergo surgery following CAR T treatment, tissue samples can be informative in assessing response to therapy. Reductions of target antigen expression following CAR T treatment can be suggestive of treatment response. However, this method is complicated by the association of chemoradiation with reductions in antigen expression [47] and by the significant sampling variability that might occur in heterogeneous tumors [126]. Furthermore, detection of target antigen by qPCR or RNA-based methods may not accurately reflect the target antigen at the protein level as there is evidence in CD19-directed therapy that CAR T targeting can lead to antigen loss without significant changes to RNA levels [73]. The EGFRvIII targeting trial at our institution found significant reductions of antigen expression in a subset of patients, but the fact that patients received conventional treatments between surgeries prior to CAR T made interpretation of that finding difficult [28]. Within tissue analysis, particularly important are the efforts to directly detect CAR T cells in the tumor tissue. Molecular techniques such as qPCR or immunohistochemistry directed to the CAR T cell can be used to detect whether the therapeutic product has tracked into the tumor. A relative enrichment of CAR T cells present in the tumor compared with normal tissue may provide supporting evidence for a specific tumor-directed therapeutic response. Moreover, even in cases where the tumor volume reduction is slight, the presence of CAR T cells in the tumor would be encouraging because future techniques such as CAR T cells secreting immunostimulatory molecules rely on the T cells co-locating with the tumor.
Peripheral blood analysis can potentially investigate the efficacy of CAR T therapy less invasively than reoperation. One approach includes the measurement of CAR T-cell levels in the blood to detect expansion of this subpopulation relative to the overall T-cell population. An expansion of CAR T cells could indicate antigen recognition and response. In addition, the persistence of CAR T cells in the periphery over time is an important predictor of clinical response in other cancers, making it a natural metric for GBM CAR T trials also [127]. Measuring tumor markers, such as circulating tumor DNA (ctDNA), is another appealing strategy. However, relative to other tumors, ctDNA has been found to be detected at low levels in glioblastoma, therefore further validation of the sensitivity of such an approach may be necessary [128]. Future work may define tumor markers in GBM that are more reliable in tracking immunotherapeutic response [129].
Future work aimed at analyzing tumor responses may ultimately rely on a multimodal approach that integrates several different studies and modalities. Due to the inherent heterogeneity of glioblastoma, the enhanced assessment of CAR T response may help define the patient subpopulations that respond best to each therapy.
5 Conclusions
While CAR T immunotherapy has revolutionized the treatment of hematologic malignancies, the role of CAR T therapy in solid tumors still remains in its infancy. Several early studies of CAR T therapy in glioblastoma have demonstrated a favorable safety profile, with several anecdotal incidences of CAR T efficacy, including two patients with objective radiographic responses [30, 31], evidence of antigen loss and CAR T engraftment [33], and long-term survivorship (59 months) following CAR T treatment [32]. However, it is becoming increasingly clear that glioblastoma in particular holds several unique challenges, including (1) vast intertumoral and intratumoral heterogeneity, which complicates antigen targeting; (2) baseline, adaptive, and iatrogenic immunosuppression, which attenuates CAR T responses; and (3) limitations in CAR T modeling and analysis, which hinders the prediction and evaluation of CAR T response. Multiple different strategies are currently being explored to address all of these issues. Ongoing work is aimed at identifying novel target antigens and antigen combinations, enhancing T-cell efficacy through further modification and selection, and utilizing immunotherapeutic adjuncts such as lymphodepletion, checkpoint inhibition, and bi-specific engagers to overcome CAR T resistance. Translating and integrating much of this promising preclinical work into the clinical realm may finally truly bring the promise of CAR T immunotherapy towards combating glioblastoma.
References
Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–28.
Yip A, Webster RM. The market for chimeric antigen receptor T cell therapies. Nat Rev Drug Discov. 2018;17:161–2.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378:439–48.
Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20:31–42.
Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. 1989;86:10024–8.
Brocker T. Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood. 2000;96:1999–2001.
Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2006;12:6106–15.
Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther J Am Soc Gene Ther. 2013;21:904–12.
Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther J Am Soc Gene Ther. 2007;15:825–33.
Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21:215–23.
Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–71.
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–20.
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33.
Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–1004.
Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther J Am Soc Gene Ther. 2009;17:1453–64.
van der Stegen SJC, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14:499–509.
Yan L, Liu B. Critical factors in chimeric antigen receptor-modified T-cell (CAR-T) therapy for solid tumors. OncoTargets Ther. 2019;12:193–204.
Li J, Li W, Huang K, Zhang Y, Kupfer G, Zhao Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: lessons learned and strategies for moving forward. J Hematol Oncol J Hematol Oncol. 2018;11:22.
Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol Off J Am Soc Clin Oncol. 2015;33:1688–96.
Junghans RP, Ma Q, Rathore R, Gomes EM, Bais AJ, Lo ASY, et al. Phase I trial of anti-PSMA designer CAR-T cells in prostate cancer: possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. Prostate. 2016;76:1257–70.
Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6.
Newick K, O’Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med. 2017;68:139–52.
Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–70.
Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res Off J Am Assoc Cancer Res. 2015;21:3149–59.
Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96.
Hegi ME, Diserens A-C, Gorlia T, Hamou M-F, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003.
Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA, et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA. 2015;314:2535–43.
Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA. 2017;318:2306–16.
Harder BG, Blomquist MR, Wang J, Kim AJ, Woodworth GF, Winkles JA, et al. Developments in blood–brain barrier penetrance and drug repurposing for improved treatment of glioblastoma. Front Oncol. 2018;8:462.
Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3:1094–101.
Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375:2561–9.
Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother (Hagerstown, MD). 1997;2019(42):126–35.
O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984.
Debinski W, Gibo DM. Molecular expression analysis of restrictive receptor for interleukin 13, a brain tumor-associated cancer/testis antigen. Mol Med. 2000;6:440–9.
Gan HK, Cvrljevic AN, Johns TG. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J. 2013;280:5350–70.
Sridhar P, Petrocca F. Regional delivery of chimeric antigen receptor (CAR) T-cells for cancer therapy. Cancers. 2017;9(7):E92.
Cohen KJ, Pollack IF, Zhou T, Buxton A, Holmes EJ, Burger PC, et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children’s Oncology Group. Neuro-Oncol. 2011;13:317–23.
Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24:739.
Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011.
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51.
Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77.
Koka V, Potti A, Forseen SE, Pervez H, Fraiman GN, Koch M, et al. Role of Her-2/neu overexpression and clinical determinants of early mortality in glioblastoma multiforme. Am J Clin Oncol. 2003;26:332–5.
Jarboe JS, Johnson KR, Choi Y, Lonser RR, Park JK. Expression of interleukin-13 receptor alpha2 in glioblastoma multiforme: implications for targeted therapies. Cancer Res. 2007;67:7983–6.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401.
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110.
Del Vecchio CA, Giacomini CP, Vogel H, Jensen KC, Florio T, Merlo A, et al. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene. 2013;32:2670–81.
Felsberg J, Hentschel B, Kaulich K, Gramatzki D, Zacher A, Malzkorn B, et al. Epidermal growth factor receptor variant III (EGFRvIII) positivity in EGFR-amplified glioblastomas: prognostic role and comparison between primary and recurrent tumors. Clin Cancer Res Off J Am Assoc Cancer Res. 2017;23:6846–55.
Francis JM, Zhang C-Z, Maire CL, Jung J, Manzo VE, Adalsteinsson VA, et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014;4:956–71.
Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019;178(4):835–849.e21.
Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Investig. 2016;126:3036–52.
Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncol. 2018;20:506–18.
Tang X, Zhao S, Zhang Y, Wang Y, Zhang Z, Yang M, et al. B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Mol Ther Oncolytics. 2019;14:279–87.
Nehama D, Di Ianni N, Musio S, Du H, Patané M, Pollo B, et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neurospheres. EBioMedicine. 2019;47:33–43.
Weiss T, Schneider H, Silginer M, Steinle A, Pruschy M, Polić B, et al. NKG2D-dependent antitumor effects of chemotherapy and radiotherapy against glioblastoma. Clin Cancer Res. 2018;24:882–95.
Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res. 2018;78:1031–43.
Yang D, Sun B, Dai H, Li W, Shi L, Zhang P, et al. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J Immunother Cancer. 2019;7:171.
Tejero R, Huang Y, Katsyv I, Kluge M, Lin J-Y, Tome-Garcia J, et al. Gene signatures of quiescent glioblastoma cells reveal mesenchymal shift and interactions with niche microenvironment. EBioMedicine. 2019;42:252–69.
Berahovich R, Liu X, Zhou H, Tsadik E, Xu S, Golubovskaya V, et al. Hypoxia selectively impairs CAR-T cells in vitro. Cancers. 2019;11:E602.
Cui J, Zhang Q, Song Q, Wang H, Dmitriev P, Sun M, et al. Targeting hypoxia downstream signaling protein, CAIX, for CAR-T cell therapy against glioblastoma. Neuro-Oncol. 2019;21:1436–46.
Hatano M, Eguchi J, Tatsumi T, Kuwashima N, Dusak JE, Kinch MS, et al. EphA2 as a glioma-associated antigen: a novel target for glioma vaccines. Neoplasia N Y N. 2005;7:717–22.
Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001;61:2301–6.
Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21:629–37.
Pellegatta S, Savoldo B, Ianni ND, Corbetta C, Chen Y, Patané M, et al. Constitutive and TNFα-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: implications for CAR-T cell therapy. Sci Transl Med. 2018;10:eaao2731.
Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, et al. CD133+ and CD133-glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–5.
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8.
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401.
Zhu X, Prasad S, Gaedicke S, Hettich M, Firat E, Niedermann G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget. 2015;6:171–84.
Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. 2019;10:128.
Brown CE, Mackall CL. CAR T cell therapy: inroads to response and resistance. Nat Rev Immunol. 2019;19:73–4.
Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang W-C, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2015;21:4062–72.
Krenciute G, Prinzing BL, Yi Z, Wu M-F, Liu H, Dotti G, et al. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. 2017;5:571–81.
Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:4722–9.
Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568:112–6.
See AP, Parker JJ, Waziri A. The role of regulatory T cells and microglia in glioblastoma-associated immunosuppression. J Neurooncol. 2015;123:405–12.
Lesniak MS, Gabikian P, Tyler BM, Pardoll DM, Brem H. Dexamethasone mediated inhibition of local IL-2 immunotherapy is dose dependent in experimental brain tumors. J Neurooncol. 2004;70:23–8.
Badie B, Schartner JM, Paul J, Bartley BA, Vorpahl J, Preston JK. Dexamethasone-induced abolition of the inflammatory response in an experimental glioma model: a flow cytometry study. J Neurosurg. 2000;93:634–9.
Arya SK, Wong-Staal F, Gallo RC. Dexamethasone-mediated inhibition of human T cell growth factor and gamma-interferon messenger RNA. J Immunol (Baltim, MD). 1950;1984(133):273–6.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54.
Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–44.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8.
Abid H, Watthanasuntorn K, Shah O, Gnanajothy R. Efficacy of pembrolizumab and nivolumab in crossing the blood brain barrier. Cureus. 2019;11(4):e4446.
Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477–86.
Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25:470–6.
Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25:462–9.
Yin Y, Boesteanu AC, Binder ZA, Xu C, Reid RA, Rodriguez JL, et al. Checkpoint blockade reverses anergy in IL-13Rα2 humanized scFv-based CAR T cells to treat murine and canine gliomas. Mol Ther Oncolytics. 2018;11:20–38.
Shen L, Li H, Bin S, Li P, Chen J, Gu H, et al. The efficacy of third generation anti-HER2 chimeric antigen receptor T cells in combination with PD1 blockade against malignant glioblastoma cells. Oncol Rep. 2019. https://doi.org/10.3892/or.2019.7263.
Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. 2018;24:1459–68.
Turtle CJ, Berger C, Sommermeyer D, Hanafi L-A, Pender B, Robinson EM, et al. Anti-CD19 chimeric antigen receptor-modified T cell therapy for B cell non-Hodgkin lymphoma and chronic lymphocytic leukemia: fludarabine and cyclophosphamide lymphodepletion improves in vivo expansion and persistence of CAR-T cells and clinical outcomes. Blood. 2015;126:184.
Suryadevara CM, Desai R, Abel ML, Riccione KA, Batich KA, Shen SH, et al. Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. Oncoimmunology. 2018;7:e1434464.
Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ, et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. 2019;365:162–8.
Sahin A, Sanchez C, Bullain S, Waterman P, Weissleder R, Carter BS. Development of third generation anti-EGFRvIII chimeric T cells and EGFRvIII-expressing artificial antigen presenting cells for adoptive cell therapy for glioma. PLoS One. 2018;13(7):e0199414.
Hu B, Zou Y, Zhang L, Tang J, Niedermann G, Firat E, et al. Nucleofection with plasmid DNA for CRISPR/Cas9-mediated inactivation of programmed cell death protein 1 in CD133-specific CAR T cells. Hum Gene Ther. 2019;30:446–58.
Jung I-Y, Kim Y-Y, Yu H-S, Lee M, Kim S, Lee J. CRISPR/Cas9-mediated knockout of DGK improves antitumor activities of human T cells. Cancer Res. 2018;78:4692–703.
Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017;7:1238–47.
Caruso HG, Torikai H, Zhang L, Maiti S, Dai J, Do K-A, et al. Redirecting T-cell specificity to EGFR using mRNA to self-limit expression of chimeric antigen receptor. J Immunother (Hagerstown, MD). 1997;2016(39):205–17.
Choi BD, Gedeon PC, Herndon JE, Archer GE, Reap EA, Sanchez-Perez L, et al. Human regulatory T cells kill tumor cells through granzyme-dependent cytotoxicity upon retargeting with a bispecific antibody. Cancer Immunol Res. 2013;1:163.
Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. 2019;37:1049–58.
Chen M, Sun R, Shi B, Wang Y, Di S, Luo H, et al. Antitumor efficacy of chimeric antigen receptor T cells against EGFRvIII-expressing glioblastoma in C57BL/6 mice. Biomed Pharmacother. 2019;113:108734.
Jiang H, Gao H, Kong J, Song B, Wang P, Shi B, et al. Selective targeting of glioblastoma with EGFRvIII/EGFR bitargeted chimeric antigen receptor T cell. Cancer Immunol Res. 2018;6:1314–26.
Wang D, Aguilar B, Starr R, Alizadeh D, Brito A, Sarkissian A, et al. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight. 2018;3:99048.
Hockey B, Leslie K, Williams D. Dexamethasone for intracranial neurosurgery and anaesthesia. J Clin Neurosci Off J Neurosurg Soc Australas. 2009;16:1389–93.
Giles AJ, Hutchinson M-KND, Sonnemann HM, Jung J, Fecci PE, Ratnam NM, et al. Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. J Immunother Cancer. 2018;6:51.
Brown CE, Aguilar B, Starr R, Yang X, Chang W-C, Weng L, et al. Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for Improved anti-tumor efficacy against glioblastoma. Mol Ther. 2018;26:31–44.
Okada H, Weller M, Huang R, Finocchiaro G, Gilbert MR, Wick W, et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. Lancet Oncol. 2015;16:e534–42.
Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23:2255–66.
Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017;9:eaaj2013.
Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403.
Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, et al. Single-Cell RNA-Seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 2017;21:1399–410.
Ledur PF, Onzi GR, Zong H, Lenz G. Culture conditions defining glioblastoma cells behavior: what is the impact for novel discoveries? Oncotarget. 2017;8:69185–97.
Stockhausen M-T, Broholm H, Villingshøj M, Kirchhoff M, Gerdes T, Kristoffersen K, et al. Maintenance of EGFR and EGFRvIII expressions in an in vivo and in vitro model of human glioblastoma multiforme. Exp Cell Res. 2011;317:1513–26.
Schulte A, Günther HS, Martens T, Zapf S, Riethdorf S, Wülfing C, et al. Glioblastoma stem-like cell lines with either maintenance or loss of high-level EGFR amplification, generated via modulation of ligand concentration. Clin Cancer Res Off J Am Assoc Cancer Res. 2012;18:1901–13.
Patanè M, Porrati P, Bottega E, Morosini S, Cantini G, Girgenti V, et al. Frequency of NFKBIA deletions is low in glioblastomas and skewed in glioblastoma neurospheres. Mol Cancer. 2013;12:160.
Stec WJ, Rosiak K, Siejka P, Peciak J, Popeda M, Banaszczyk M, et al. Cell line with endogenous EGFRvIII expression is a suitable model for research and drug development purposes. Oncotarget. 2016;7:31907–25.
Cassidy JW, Caldas C, Bruna A. Maintaining tumor heterogeneity in patient-derived tumor xenografts. Cancer Res. 2015;75:2963–8.
Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol (Baltim, MD). 1950;1995(154):180–91.
Candolfi M, Curtin JF, Nichols WS, Muhammad AG, King GD, Pluhar GE, et al. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J Neurooncol. 2007;85:133–48.
Oh T, Fakurnejad S, Sayegh ET, Clark AJ, Ivan ME, Sun MZ, et al. Immunocompetent murine models for the study of glioblastoma immunotherapy. J Transl Med. 2014;12:107.
Halkias J, Yen B, Taylor KT, Reinhartz O, Winoto A, Robey EA, et al. Conserved and divergent aspects of human T-cell development and migration in humanized mice. Immunol Cell Biol. 2015;93:716–26.
Patrizii M, Bartucci M, Pine SR, Sabaawy HE. Utility of glioblastoma patient-derived orthotopic xenografts in drug discovery and personalized therapy. Front Oncol. 2018;8:23.
Pituch KC, Miska J, Krenciute G, Panek WK, Li G, Rodriguez-Cruz T, et al. Adoptive transfer of IL13Rα2-specific chimeric antigen receptor T cells creates a pro-inflammatory environment in glioblastoma. Mol Ther J Am Soc Gene Ther. 2018;26:986–95.
Walsh NC, Kenney LL, Jangalwe S, Aryee K-E, Greiner DL, Brehm MA, et al. Humanized mouse models of clinical disease. Annu Rev Pathol. 2017;12:187–215.
Sanghera P, Perry J, Sahgal A, Symons S, Aviv R, Morrison M, et al. Pseudoprogression following chemoradiotherapy for glioblastoma multiforme. Can J Neurol Sci J Can Sci Neurol. 2010;37:36–42.
Ellingson BM, Chung C, Pope WB, Boxerman JL, Kaufmann TJ. Pseudoprogression, radionecrosis, inflammation or true tumor progression? Challenges associated with glioblastoma response assessment in an evolving therapeutic landscape. J Neurooncol. 2017;134:495–504.
Chapelin F, Capitini CM, Ahrens ET. Fluorine-19 MRI for detection and quantification of immune cell therapy for cancer. J Immunother Cancer. 2018;6(1):105.
Jain KK, Sahoo P, Tyagi R, Mehta A, Patir R, Vaishya S, et al. Prospective glioma grading using single-dose dynamic contrast-enhanced perfusion MRI. Clin Radiol. 2015;70:1128–35.
Nishikawa R, Sugiyama T, Narita Y, Furnari F, Cavenee WK, Matsutani M. Immunohistochemical analysis of the mutant epidermal growth factor, ΔEGFR, in glioblastoma. Brain Tumor Pathol. 2004;21:53–6.
Song D-G, Ye Q, Carpenito C, Poussin M, Wang L-P, Ji C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. 2011;71:4617–27.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24.
Nakamura S, Yokoyama K, Yusa N, Ogawa M, Takei T, Kobayashi A, et al. Circulating tumor DNA dynamically predicts response and/or relapse in patients with hematological malignancies. Int J Hematol. 2018;108:402–10.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflict of interest
Donald M. O’Rourke has received research grants related to the development of CAR T cells in glioblastoma (Novartis), and holds patents pending and patents filed for CAR T cells in glioblastoma (Novartis, University of Pennsylvania). Ryan D. Salinas and Joseph S. Durgin declare they have no conflicts of interest that might be relevant to the contents of this article.
Rights and permissions
About this article

Cite this article
Salinas, R.D., Durgin, J.S. & O’Rourke, D.M. Potential of Glioblastoma-Targeted Chimeric Antigen Receptor (CAR) T-Cell Therapy. CNS Drugs 34, 127–145 (2020). https://doi.org/10.1007/s40263-019-00687-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-019-00687-3