
Abstract
Alzheimer’s disease (AD), the most common cause of age-dependent dementia, is one of the most significant healthcare problems worldwide. Aggravating this situation, drugs that are currently US Food and Drug Administration (FDA)-approved for AD treatment do not prevent or delay disease progression. Therefore, developing effective therapies for AD patients is of critical urgency. Human genetic and clinical studies over the past three decades have indicated that abnormal generation or accumulation of amyloid-β (Aβ) peptides is a likely culprit in AD pathogenesis. Aβ is generated from amyloid precursor protein (APP) via proteolytic cleavage by β-site APP cleaving enzyme 1 (BACE1) (memapsin 2, β-secretase, Asp 2 protease) and γ-secretase. Mice deficient in BACE1 show abrogated production of Aβ. Therefore, pharmacological inhibition of BACE1 is being intensively pursued as a therapeutic approach to treat AD patients. Recent setbacks in clinical trials with BACE1 inhibitors have highlighted the critical importance of understanding how to properly inhibit BACE1 to treat AD patients. This review summarizes the recent studies on the role of BACE1 in synaptic functions as well as our views on BACE1 inhibition as an effective AD treatment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
β-Site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitors are potential disease-modifying drugs for Alzheimer’s disease (AD) treatment as they prevent the production of synaptotoxic amyloid-β (Aβ). |
Gradually increased deletion of BACE1 in an adult AD mouse model (5xFAD) reverses amyloid deposition and improves cognitive functions. |
Inhibition of BACE1 in human patients may obtain better results if an appropriate dose is determined in the future. |
1 Introduction
Alzheimer’s disease (AD) is the most common form of dementia and is characterized by widespread neurodegeneration that progresses throughout the neocortex and limbic system [1, 2]. AD patients experience progressive cognitive impairments and memory loss. More than 5 million people in the USA alone suffer from AD, with about 35 million having AD worldwide. It has been estimated that by 2030 there will be 74.7 million people with dementia, and the cost of caring for these individuals could rise to ~ US$2 trillion [3]. Provision of effective treatments for AD patients is therefore an urgent task. AD is a polygenic and neurodegenerative disease defined by the presence of extracellular amyloid plaques and intracellular neurofibrillary tangles, accompanied by neuroinflammation and loss of neurotransmitters in patients’ brains [4, 5]. Aberrant neural network activity, dysfunction, and loss of synapses as well as degeneration of specific neuronal populations are the main underlying causes of cognitive decline in AD.
1.1 Amyloid-β (Aβ) and the Amyloid Hypothesis
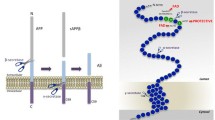
Amyloid-β (Aβ) peptides are heterogeneous peptides of 38–43 amino acids, which are excised from an integral membrane protein amyloid precursor protein (APP) by the β- and γ-secretase enzymes; Aβ is the main aggregated component of amyloid plaques in the brain (Fig. 1) [6]. Among the various Aβ peptides, Aβ1-40 is the most common form. Although Aβ1-42 is less abundant, it is more prone to form Aβ oligomers, and is viewed as a highly toxic component [7]. Genetic mutations in APP or presenilin-1 and -2 (PS1 and PS2), which are components of γ-secretase, facilitate production of Aβ1-42 near the lipid bilayer [8]. Aβ can be released into extracellular spaces either through the secretory pathway or directly into interstitial fluids or synaptic clefts [9]. At physiological levels, Aβ is suggested to regulate synaptic plasticity [7, 10, 11]. On the other hand, large numbers of studies suggest that Aβ at pathological levels causes synaptic toxicity [12,13,14,15], supporting a need to reduce Aβ levels to optimize synaptic functions, as highlighted in the amyloid cascade hypothesis [6, 16, 17].

The amyloidogenic pathway of APP. The amyloidogenic pathway involves the sequential cleavage of APP by β-secretase, which releases the soluble ectodomain sAPPβ. The remaining C99 fragment of APP is then cleaved by γ-secretase, resulting in the formation of the Aβ peptide. Due to its high propensity to aggregate, Aβ peptide oligomerizes, accumulates, and forms amyloid senile plaques, in turn leading to the documented alterations in Alzheimer’s disease. Aβ amyloid-β, AICD amyloid precursor protein intracellular domain, APP amyloid precursor protein, sAPPβ soluble peptide APPβ
Genetic studies identified 13 familial mutations in APP within or near the Aβ region [18]; these APP mutations are linked to either an increased risk for AD or atypical early onset, which is attributable to increased production of Aβ aggregates. Among these mutations, an increase in total Aβ production can result from a mutation in APP near the β-Site APP cleaving enzyme 1 (BACE1) (memapsin 2, β-secretase, Asp 2 protease) cleavage site by favoring BACE1 cleavage [19,20,21]. Many familial mutations in PS1 or PS2 appear to favor production of Aβ1-42 over smaller Aβ forms by altering γ-secretase activity [22]. On the other hand, excessive accumulation of Aβ monomers can result from failures to effectively clear Aβ [23,24,25]. In essence, abnormal accumulation of Aβ initiates a series of self-assembly steps that ultimately result in the formation of insoluble Aβ aggregates, which are known to impair many neuronal functions [26, 27]. These insoluble Aβ aggregates deposit extracellularly between neurons or near synapses, forming amyloid plaques [27].
As illustrated by the amyloid hypothesis [6, 16, 17], amyloid deposits likely occur prior to the formation of neurofibrillary tangles, which result from intraneuronal aggregation of the microtubule binding protein tau due to altered post-translational modification such as hyperphosphorylation and/or acetylation [28,29,30]. It has been reported that genetic mutations in the tau-coding gene can pathologically induce tau aggregation [31,32,33,34], suggesting that the formation of neurofibrillary tangles can be independent of toxic Aβ components. Although prevailing studies suggest that Aβ toxicity is an upstream event by catalyzing the formation of neurofibrillary tangles [35,36,37], neurofibrillary tangles appear to be more associated with neuronal and synaptic losses, which are two typical neuropathologies leading to memory impairment, confusion, personality changes, and cognitive decline over time in AD [38,39,40,41,42]. Hence, either Aβ or tau oligomers can damage the neuronal communication via damaging synaptic connections [27, 43, 44] (Fig. 2). The prevention of synaptic loss or preservation of synaptic connections should be a viable strategy to stop Alzheimer’s patients from developing memory problems.

Inhibition of synaptic transmission by amyloid plaques and Aβ peptides by physically interfering in signal transfer. This leads to initial synaptic failure, preceding any significant neuronal degeneration, and this Aβ-induced dysfunction of synaptic plasticity then appears to contribute to early memory loss. Aβ amyloid-β
2 Targeting β-Site Amyloid Precursor Protein Cleaving Enzyme 1 (BACE1)-Mediated Aβ Reduction as an Alzheimer’s Disease (AD) Therapy
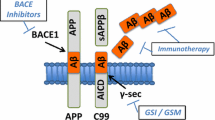
Although it is clear that AD is a disease of synaptic failure, the exact mechanisms causing neuronal dysfunctions and survival involve multiple aspects and remain to be determined. While aggregated Aβ is regarded as a catastrophic factor in AD pathogenesis, altered functions related to apolipoprotein E4 (ApoE4) [45, 46], tau [30, 47], α-synuclein [48, 49], and TDP-43 (transactive response DNA binding protein 43 kDa) [50,51,52] are likely co-morbidity factors. Other pathological factors such as inflammatory response, ion homeostasis disruption, oxidative stress, and decreased levels of neurotransmitters have also been identified and are considered to be sequential events of Aβ aggregation [53,54,55,56]. These factors will contribute to AD synaptic dysfunction and pathogenesis, but are beyond the scope of this review. Therefore, a plethora of strategies are being employed to implement checks towards the initiation and progression of AD (Fig. 3).

Current therapeutic approaches in Alzheimer’s disease include (1) inhibition of β- and γ-secretases; (2) improving Aβ clearance; and (3) amelioration of inflammation and synaptic dysregulation. Aβ amyloid-β, APP amyloid precursor protein, BACE1 β-site amyloid precursor protein cleaving enzyme 1, CSF cerebrospinal fluid, MRI magnetic resonance imaging, PET positron emission tomography
Growth of amyloid plaques, both in size and density, is age-dependent; this is consistent with the fact that aging is the most important non-genetic risk factor for AD. Aβ oligomerizes and aggregates into small clusters or a nidus outside neurons, prior to the formation of dense core senile plaques [57]. Activation of microglia has been shown to migrate towards such growing Aβ plaques [58,59,60], where reactive astrocytes can also be found concurrently [61,62,63]. Dystrophic neurites, including those labeled by an antibody specific to a tubular protein named reticulon-3 (RTN3), can be detected when small Aβ plaques begin to form [64]. Hence, the formation of amyloid plaques, especially the core plaques, induces a cascade of changes including gliosis and neuritic dystrophy.
With a strong body of evidence supporting the amyloid hypothesis, AD therapy is actively focused on lowering Aβ levels to improve or reverse synaptic failures in patients. BACE1 inhibition emerges as one of the most attractive approaches for the following reasons. First, it has been shown that germline deletion of BACE1 abrogates Aβ production and ameliorates cognitive/behavioral deficiencies, as observed in transgenic mice overexpressing human APP with familial AD mutations [65,66,67], indicating that inhibition of BACE1 has a direct effect. Second, a rare human mutation at the BACE1 cleavage site of APP [A673T] results in a 40% decrease in Aβ production in vitro, a significantly reduced propensity for Aβ to aggregate, a five- to seven-fold reduced risk of developing AD, and greater resilience to cognitive dysfunction in elderly individuals [68, 69], implying that BACE1 cleavage alone appears to be beneficial in the human brain. Third, BACE1-containing vesicles have also been identified near active zones [43, 70, 71], and inhibition of BACE1 directly reduces Aβ-mediated impairments in synaptic transmission [14, 43, 72, 73]. Fourth, deletion of BACE1 in mice appears to have a minor impact on mouse growth or overall functions [43, 65, 74,75,76,77,78], perhaps related to the fact that most BACE1 substrates are also shed by α-secretase. Fifth, direct inhibition of γ-secretase, another strategy to reduce Aβ generation, is now recognized to be more challenging due to the indispensable physiological roles of γ-secretase [79, 80], leading investigators and many companies to focus on BACE1 inhibitors, which act upstream of γ-secretase in Aβ generation. Thus, BACE1 is recognized as a better-positioned target for treating AD patients.
2.1 BACE1 Inhibition as a Target for AD Therapy
BACE1 (memapsin 2, β-secretase, Asp 2 protease) is a type I membrane-bound aspartyl protease with the bilobal domain assembled by a pair of DTG/DSG motifs facing the extracytoplasmic side [81, 82]. A short C-terminal tail appears to mediate cellular trafficking of BACE1 [83]. The crystal structure of BACE1, first described by Tang and colleagues [82], reveals that the proteolytic pocket of BACE1 is relatively large and less hydrophobic, and can accommodate up to 11 residues [85,86,87]. This renders it more challenging for the development of small-molecular inhibitors using the high throughput screening approach [84, 88]. Structure-based designs of BACE1 inhibitors have been successful in developing highly potent and specific inhibitors in the peptidomemic structure [89,90,91,92,93,94]. Allosteric modulation of BACE1 activity, i.e., by targeting the distal exosite with an antibody, has also been explored [85, 87, 95,96,97]. The practical BACE1 inhibitors should be small in size and cross the blood–brain barrier easily as BACE1 is richly expressed in the brain, predominantly in neurons, and is readily detected in presynaptic hippocampal mossy fiber terminals [70], whereas its homolog BACE2 (64% amino acid sequence homology) is more sparsely expressed [98]. By crossing the blood–brain barrier, BACE1 inhibitory drugs can effectively reduce production of Aβ in neurons and in the brain overall.
Over the past 20 years, efforts to design and develop selective, cell-permeable, orally bioavailable, and brain-penetrant BACE1 inhibitors have undergone multiple challenging phases [88, 99, 100]. Following breakthroughs in the use of ‘fragment-based drug discovery’ for BACE1 inhibitors [84, 91, 101, 102], small-molecular BACE1 inhibitors were innovatively developed and are currently being tested in animals and in human trials. Five of these drugs being investigated clinical trials deserve a closer look (Sects. 2.1.1–2.1.5) [43, 84, 88, 99, 103].
2.1.1 Verubecestat (MK-8931)
Verubecestat (MK-8931) is the first small-molecular BACE1 inhibitor with oral availability and blood–brain barrier permeability [104] because of its physico-chemical properties such as high cellular permeability (Papp = 28.6 × 10−6 cm•s−1) and excellent aqueous solubility at neutral pH (1.6 mM in pH 7.4 buffer) [105]. Verubecestat forms interactions between the amidine moiety of verubecestat and the BACE1 catalytic dyad through hydrogen bonding. Inhibition of BACE1 by this drug is potent, although it is more potent for BACE2 inhibition (inhibition constant [Ki] = 2.2 nM for human BACE1 [hBACE1], Ki = 3.4 nM for mouse BACE1 (mBACE1), Ki = 0.34 nM for human BACE2 (hBACE2) but Ki is over 100,000 nM for human cathepsin D (hCatD)) [105]. Long-term treatment of verubecestat in animals can strongly reduce Aβ40, Aβ42, and soluble peptide APPβ Soluble Amyloid Precursor Protein (sAPPβ) in cerebrospinal fluid (CSF) and the brain. Preclinical tests of verubecestat in rats and monkeys exhibited no mechanism-based adverse effects, such as reduced nerve myelination, neurodegeneration, altered glucose homeostasis, or hepatotoxicity, which are seen in BACE1-null mice [105]. In the initial phase I trial (ClinicalTrials.gov identifier NCT01496170) that tested single doses up to 450 mg and multiple doses from 12 to 150 mg/day, verubecestat was well-tolerated and safe while a reduction of the CSF Aβ concentration in 32 participants reached ~ 90%. Another trial further confirmed tolerability to single and multiple doses. The phase II/III EPOCH trials (NCT01739348) focused on evaluation of conventional cognitive and functional outcomes. Therefore, participants in the trial had measurable cognitive deficits but were not entirely functionally impaired, as clinically confirmed by a positron emission tomography (PET) scan with the newly US Food and Drug Administration (FDA)-approved amyloid tracer flutemetamol [106]. Among the 1958 individuals recruited with moderate or mild AD, 80% were Caucasian and 63% had at least one ApoE4 allele; their Mini-Mental State Examination (MMSE) scores were between 15 and 28. They received either 12 or 40 mg of study drug or placebo for 18 months. Most participants either had taken or currently took a cholinesterase inhibitor and/or memantine. Although a dramatic reduction of Aβ40, Aβ42, and sAPPβ in CSF of up to 80% was detected and a small reduction in plaque load was confirmed by amyloid PET in participants taking the drug, the clinical trial was terminated in February 2018, with verubecestat exhibiting no improvement in cognitive function in AD patients cited as the reason [107]. The decline on the cognitive scale was at the same rate in both groups. Trial results also showed no impact of this drug on either total Tau (T-Tau) and phosphorylated Tau (P-Tau) levels in the treated group. Intriguingly, this drug appears to cause a small decrease in hippocampal and total brain volume but not delirium or brain edema. Trial participants did report adverse effects, mostly related to anxiety, falls, insomnia, and an average weight loss of 3.5 lb (1.6 kg). Rashes were almost twice as common in the verubecestat group than with placebo. Changes in hair color were frequent and this side effect is related to the inhibition of BACE2 for its control of hair pigmentation [108]. Unexpectedly, the administration of verubecestat in people with prodromal AD (APECS trial) resulted in a worsening of cognitive symptoms (NCT01953601). Hence, the inefficiency of the drug in improving cognitive function becomes complicated, perhaps being concealed by a secondary neuropsychiatric effect. Since adults older than 70 years often have co-morbidities such as α-synuclein, TDP-43, or vascular pathology, this could cloud the cognitive outcomes. The results of this drug trial suggest that BACE1 inhibitors need to be given several years before the onset of AD symptoms.
2.1.2 Lanabecestat (AZD3293)
Lanabecestat (AZD3293), a small-molecule, orally administered BACE1 inhibitor developed by AstraZeneca, was first extensively tested in primary cortical neurons, mice, guinea pigs, and dogs prior to clinical trials [109, 110]. The drug has a slow off-rate (estimated half-life [t½] of 9 h for hBACE1) [109], which may result in a prolonged reduction of Aβ. The inhibition potency of AZD3293 is as follows: Ki = 0.4 nM for hBACE1, Ki = 0.8 nM for hBACE2, and Ki = 3797 nM for hCatD) [109]. The phase I study, begun in 2014, was conducted in collaboration with Eli Lilly, and trial results demonstrated excellent safety, tolerability, and metabolic profiles in elderly healthy volunteers and in AD patients with mild cognitive impairment [111, 112]. A separate phase I study in Japan (NCT02005211) also revealed excellent pharmacokinetics for the drug. Like Merck’s verubecestat, lanabecestat also strongly decreased the CSF Aβ level in the treated group. Phase II/III clinical trials, AMARANTH (NCT02245737), AMARANTH-EXTENSION (NCT02972658), and DAYBREAK-ALZ, recruited over 1400 participants and were planned to last up to 54 months with variable doses; they attempted to measure efficacy and safety in humans by analyzing results such as amyloid PET scans, CSF Aβ levels, and CSF amyloid inclusion. The primary functional outcome from these rigorous trials was anticipated to be improved patient cognition in addition to decreasing CSF levels of Aβ40, total tau, and phosphorylated tau. Although these results have not yet been released, this trial was also discontinued in July 2018 because an interim futility analysis unfortunately showed that lanabecestat treatment would not meet the primary endpoints. Interestingly, this compound also causes depigmentation in the epidermis and hair [111]. Similar to that of verubecestat, the lesson learnt from this announcement is that the appearance of even mild symptoms may be too late in the disease continuum for a BACE1 inhibitor to be efficacious. Firm conclusions may be drawn when the actual data are released.
2.1.3 Atabecestat (JNJ-54861911)
Atabecestat (JNJ-54861911) has been developed by Shionogi as a brain-penetrable small-molecular BACE1 inhibitor. In collaboration with Janssen, this orally available BACE1 inhibitor has entered a phase I trial (NCT01978548). Atabecestat is an efficient BACE1 inhibitor with a Ki of 9.8 nM for hBACE1 [113]. Daily administration of atabecestat 5–150 mg in healthy elderly and young participants for up to 14 days showed significant and consistent reduction of Aβ (up to 90% in the 90 mg cohort) in both plasma and CSF [114]. Another phase I trial (NCT02360657) in Japan aimed to evaluate a 1-month course of 10 or 50 mg in 18 patients with brain Aβ deposits and low CSF Aβ42 levels; these participants were categorized as “asymptomatic at risk of AD” as they exhibited an earlier stage of AD pathophysiology than pre-dementia or prodromal AD. A recently published study of two similarly designed phase I trials including Caucasian and Japanese patients administered atabecestat 10 and 50 mg for 4 weeks demonstrated mean CSF Aβ1-40 reductions of 67% and up to 90%, respectively [114]. Although minor adverse effects such as headache and back pain were noted in the phase I trial (NCT02360657), it was deemed safe enough to advance to phase II trials. NCT02260674, a multicenter phase II trial, recruited 114 pre-dementia individuals to determine the tolerability and long-term safety of atabecestat, including double-blind treatment for 6 months. Atabecestat was found to successfully reduce both plasma and CSF levels of Aβ1-37, Aβ1-38, Aβ1-40, and Aβ1-42 in a dose-dependent fashion, while levels of sAPPα were conversely increased. Atabecestat 5, 25, and 50 mg doses could reduce CSF Aβ levels by 50, 80, and 90%, respectively, indicating high potency for BACE1 inhibition. In a separate trial called EARLY (NCT02569398) that was launched in 2015, participants are asymptomatic but at risk of developing Alzheimer’s dementia and were intended to receive drug or placebo once daily for up to 4.5 years with continuous monitoring of cognitive scales. Unfortunately, observation of elevated liver enzymes in two patients led Janssen to announce the discontinuation of this trial on 17 May 2018. The main reason for this was the unfavorable benefit–risk ratio associated with the potential risk to patients of severe liver injury. Severe liver toxicity has not yet been seen in BACE1-null mice, but several BACE1 inhibitors, such as LY2886721, have been found to cause abnormal liver function [115].
2.1.4 Elenbecestat (E2609)
Elenbecestat (E2609), originally developed by Eisai as a small-molecule inhibitor of BACE1, is currently in clinical trials co-developed with Biogen. During preclinical testing in rodents, guinea pigs, and non-human primates, elenbecestat was shown to strongly reduce CSF and plasma Aβ levels (abstract in 2012 Alzheimer’s Association International Conference) [116]. A phase I trial (NCT01294540) reported that a single dose of 50 mg in 73 healthy participants (either sex, from age 30 to 85 years in six separate cohorts) was well-tolerated and safe. A single oral ascending-dose study of 5–800 mg and a 14-day multiple oral ascending-dose study of 25–400 mg showed that elenbecestat could significantly reduce plasma or CSF Aβ levels by as much as 92%: plasma Aβ(1-X) relative to baseline was 52% at 5 mg and 92% at 800 mg [168]. Participants reported adverse effects such as headache and dizziness. In a separate trial, 50 healthy adult volunteers, receiving doses between 25 and 400 mg daily for 14 days, also showed a dose-dependent reduction in plasma and CSF Aβ(1-X) levels: 46.2% in the 25 mg, 61.9% in the 50 mg, 73.8% in the 100 mg, and 79.9% in the 200 mg cohorts. No alarming safety concerns were reported apart from relapse of orolabial herpes in the 200 mg cohort. In addition, by comparing different doses to placebo in mild cognitive impairment/prodromal patients or two doses in subjects with mild AD dementia, elenbecestat was found to delay clinical symptoms at the endpoint of the trial. A phase IIa trial (NCT02322021) concluded that of elenbecestat 50 mg/day was safe and consistently reduced CSF Aβ(1-X) by about 70%. Thus, a standard phase III trial called MISSION AD was initiated in November 2016 (NCT02956486) and MISSION AD2 was initiated in January 2017 (NCT03036280), enrolling a total of 1330 early AD subjects. A regimen of a daily dose of 50 mg of elenbecestat for 24 months is being used to compare baseline cognitive scores with those at the end of the trial. This trial will also monitor changes in the amyloid PET and tau PET signal, CSF biomarkers such as t-tau and p-tau, Aβ(1-42) and Aβ(1-X), hippocampal volume, and alteration in the synaptic connectivity on functional magnetic resonance imaging (fMRI) at the end of 24 months (NCT02956486). Encouragingly, and unlike earlier mentioned discontinued trials, Biogen announced in June 2018 that the 18-month-long phase II study had revealed less decline in functional cognition in addition to a significant reduction in Aβ levels, quantified by amyloid PET imaging, in patients with mild to moderate form of AD [165]. However, they did disclose adverse effects such as upper respiratory tract infection, abnormal dreams and nightmares, contact dermatitis, headache, diarrhea, and falls. There were no reports of hepatic toxicity. This news provides great relief as different BACE1 inhibitors are showing promise.
2.1.5 CNP520
CNP520 is an orally available small-molecule inhibitor of BACE1 from Novartis developed through optimizing the backbone of 3-amino-1,4-oxazine. This brain-penetrable compound has a good selectivity over BACE2 (Ki = 11 nM for hBACE1 and 10 nM for mBACE1 compared with Ki = 30 nM for hBACE2); its inhibitory activity toward capthesin D is relative low (Ki = 205,000 nM) [117, 118]. A recently published study on the pharmacodynamics of this drug showed that it markedly reduced Aβ levels both in the brain and CSF in rats and dogs, along with reducing Aβ deposition in APP transgenic mice [117]. Toxicology studies revealed good safety margins, with no signs of hair depigmentation, retina degeneration, liver toxicity, or cardiovascular adverse effects [117]. No phase I trial has been publicized, but a phase II trial is being conducted in collaboration with Amgen that is primarily aimed at delaying the start and progression of AD.
In one trial (NCT02576639), CNP520 was given to 124 healthy participants over 60 years old at a dose between 2 and 85 mg once daily for 13 weeks during the period of May 2017 to August 2017. A clear dose-dependent reduction of Aβ38, Aβ40, and Aβ42 levels in CSF and plasma in participants was observed. Similar to animal studies, no obvious hair depigmentation, retina degeneration, liver toxicity, or cardiovascular adverse effects were noted. A trial was launched by Novartis and Amgen called GENERATION to evaluate CNP520 for a longer period [117]. About 1340 participants in the age range of 60–75 years, presumably with no obvious cognitive impairments but with two ApoE4 alleles, were recruited through multiple centers. Measures include multiple parameters as mentioned earlier and include brain magnetic resonance imaging (MRI) scans conducted at the sixth month, twelfth month, and annually thereafter. This costly trial is part of the Alzheimer’s Prevention Initiative (API) and will be concluded in September 2024.
The noted difference of this GENERATION trial from the mentioned single-compound trials is that it combines the strengths of two Novartis compounds: CNP520 and CAD106. CAD106 is a second-generation anti-Aβ vaccine that recognizes Aβ1-6 and aims to clear amyloid plaques in AD patients [119, 120]. It has been shown to be safe and to illicit high Aβ-antibody titers in previous phase II trials (NCT00733863, NCT00795418, NCT00956410, NCT01023685, and NCT01097096). Modified phase II/III trials (GENERATION-S1 [NCT02565511] which will recruit ~ 1340 participants, and GENERATION-S2 [NCT03131453], which will recruit ~ 2000 cognitively unimpaired male and female participants with at least one ApoE4 allele) will last between 60 and 84 months and test the combination effect of CAD106 and CNP520 versus either single agent or placebo in those at risk of the onset of clinical symptoms of AD. The posited advantage of the combination trial is to not only reduce Aβ generation (via CNP520) but also to remove existing amyloid plaques (via CAD106). It is hoped that this combination may be beneficial in late-stage AD patients as BACE1 inhibition alone may be too late for these elderly AD patients.
3 Plausible Beneficial Effect of BACE1 Inhibitors
The beneficial effects of BACE1 inhibition, such as reduced Aβ levels, have most frequently been shown in animal studies [88, 93, 99, 121, 122]. A recent exciting study involved using a BACE1-conditional knockout mouse model. Reversal of pre-existing amyloid deposition in the 5xFAD AD mouse model by sequential deletion of BACE1 in adult mice was demonstrated [64]. Importantly, removal of amyloid plaques concomitantly improves Aβ-mediated synaptic dysfunction, gliosis, and neuritic dystrophy. In BACE1 inhibitor clinical trials (Table 1) targeting asymptomatic, early, or prodromal AD patients, one common observation is that these five inhibitors are potent in reducing Aβ in human CSF or plasma by as much as 90% during trials. While BACE1 inhibition should reduce Aβ generation in humans, three trials have failed to proceed, and this may have been due to various reasons: verubecestat may have a selectivity issue as it is a stronger BACE2 inhibitor while atabecestat causes intolerable liver toxicity. These failures should not discourage trials of BACE1 inhibitors as compounds with higher potency and selectivity and less toxicity may emerge in the future. Indeed, encouragingly, Allosterix, Amgen, Pfizer, Evotec AG, Johnson & Johnson, and AstraZeneca are continuingly developing BACE1 inhibitors, which are currently either in early stages of discovery or early-phase trials. If a potent BACE1 inhibitor can be safely used for decades in humans, the benefit of BACE1 inhibition for AD patients will likely be better appreciated, especially considering the recent results in BACE1 conditional knockout mice [64]. Since conditional deletion of BACE1 was demonstrated in relatively young mice (2–4 months, correlating to 20–26 years old in humans), inhibition of BACE1 activity at younger ages appears to make the most sense if the drug is safe enough. Hence, the goals in developing BACE1 inhibitors should include stringent safety profiling.
BACE1 inhibition may have other beneficial effects such as body weight reduction along with increased glucose metabolism and insulin sensitivity [123]. In a study with injury models of BACE-null mice, increased regeneration and re-innervation of peripheral axon and neuromuscular junctions was noted [124]. Therefore, BACE1 inhibition may benefit patients reporting chronic diabetes and/or nerve degeneration.
4 Potential Adverse Effects Associated with BACE1 Inhibitors
The FDA has recently proposed new guidelines that are aimed at lowering the clinical study goals of AD drugs for treating early-stage non-symptomatic patients, who have not yet shown functional disability or clinical abnormality [166]. During the discussion at the 2018 Clinical Trials on Alzheimer’s Disease (CTAD) conference, optimization of BACE inhibition to a safe dose was emphasized based on trial results that cognitive loss appears to be a dose-dependent event [167]. Thus, giving BACE1 inhibitors early may potentially delay the onset of AD, and possibly slow down its progression. However, as chemical compounds, BACE1 inhibitors could have both on-target and off-target adverse effects that could result in the cessation of a trial. In the reports of two recently terminated BACE1 inhibitor trials, beneficial effects on AD patients’ cognitive outcome were not reached [107, 117]. One scenario that may be accountable for the failure to improve cognitive function is related to mechanism-based adverse effects.
Previous studies demonstrate that germline BACE1 deficiency can impair hippocampal activity-dependent long-term potentiation (LTP) at mossy fibers to CA3 while enhancing long-term depression (LTD) [125, 126]. Recent examinations of mice with deletion of BACE1 in the adult reveal a similar reduction in LTP [61] and altered mossy fiber structures [127], suggesting that chronic usage of BACE1 inhibitors in adults for a long period may potentially impair synaptic transmission and plasticity. The mechanistic reason for such an impairment remains to be determined. One possibility is that Aβ at physiological concentrations may actually be required for optimal synaptic function [128], and a high dose of chronic BACE1 inhibition perhaps diminishes Aβ levels to far below the normal physiological range, which may cause abnormal neuronal functions [43]. Neuregulin-1 (Nrg1) is known to regulate synaptic functions [129], and it is a validated substrate of BACE1 [130]. Such reduced cleavage of Nrg1 was readily seen when about 50% of BACE1 was deleted in adult mice [61]. Reduced Nrg1 signaling function, due to reduced release of the signaling Nrg1 N-terminal fragment, in neurons may contribute to impaired synaptic function upon BACE1 deletion. On the other hand, BACE1 is continually required for axon guidance and organization in the adult via cleaving of the CHI1 molecule [127], and altered axonal structures may impact proper formations of synapses. Further extensive studies investigating and comparing wild-type animals chronically treated with BACE1 inhibitors for the effect on synaptic transmission and plasticity are required to decipher the underlying molecular mechanisms. A quick study has indeed shown that long-term administration of high doses of two different BACE inhibitors (SCH1682496 and LY2811376), administered twice daily for 16 days, lead to detrimental effects on dendritic spine turnover and resulted in significant reductions in dendritic spine density at the end of the treatment period [131]. One interpretation is that significant inhibition of BACE1 in humans may compromise cognitive functions, and this may partially be responsible for the failure of these clinical trials.
Since BACE1 is a protease, drastic inhibition of BACE1 will reduce the cleavage of many physiological substrates for BACE1 (see examples in Table 2), and this potentially interferes with multiple physiological events [43, 97, 132, 133]. For example, chronic blockade of BACE1 using LY2811376 in mice showed impairments in muscle spindles [134]. This appears to be related to a reduction of the muscle spindle pool and impaired coordination of movement, probably arising from abrogated cleavage of the immunoglobulin domain-containing β1 isoform of Nrg1. Another interesting example is the observed retinal toxicity that caused immediate termination of clinical trials of the first BACE1 inhibitors [135]. It was discovered that BACE1 inhibitors prevented the normal turnover of photoreceptors in the eye, causing lipofuscinosis via blocking of cathepsin D, which is also an aspartyl protease and can be an off-site target of BACE1 inhibition [136]. An accumulation of lipofuscin age pigment is also observed in retinal pigment epithelial cells that have been treated with BACE1 small interfering RNA (siRNA) or with a BACE1 inhibitor, which reflects the perturbation of lysosomal function. The retina of the BACE1-deficient mouse is thinner than in wild-type mice and displays defects in vascularization [135, 137]. These defects are associated with impaired shedding and signaling of the vascular endothelial growth factor receptor 1 (VEGFR1) and a different chemical class of BACE1 inhibitor, LY2811376, appears to cause the same phenotype.
Therefore, it is important to investigate the effects of long-term BACE1 inhibition in animal AD models as well as in the normal adult brain in order to understand the mechanism of action and to determine whether the predicted positive effects of Aβ reduction will outweigh any deleterious effects on synapses and cognitive function. One way to mitigate adverse effects of BACE inhibition would be to find a dose range that suppresses Aβ accumulation while allowing sufficient processing of other substrates.
5 Conclusion
Extensive preclinical trials have demonstrated the efficacy of BACE1 inhibitors in lowering Aβ levels in the brain and in rescuing cognitive deficits in AD animal models, and current clinical trials have confirmed at least that BACE1 inhibition will significantly lower Aβ levels. The benefits of BACE1 inhibition for improving cognitive functions in AD patients have not yet been fully realized at this stage, partly due to timing of administration and perhaps also complications related to the physiological function of BACE1. BACE1 gene knockout experiments have offered insight into the physiological functions of BACE1 and warned of the risks associated with total eradication of its activity. BACE1 clearly plays an important role in the development of the central and peripheral nervous systems, and it is required for the maintenance and repair of these systems, particularly in response to injury and inflammation. Thus, it is imperative that safe therapies based on BACE1 inhibition should demonstrate high specificity toward BACE1 activity and aim to reduce BACE1 to manageable levels, but not to suppress it too dramatically. Early intervention should be recommended, as PET imaging studies have demonstrated that Aβ starts accumulating in the brain long before the first signs of cognitive impairment are observed, and 20–30 years before a clinical AD diagnosis is established and irreparable neuronal loss has occurred [138]. It will be beneficial to monitor early changes in biomarkers that indicate changes in BACE1 levels. Some of the newly identified BACE1 substrates may provide novel biomarkers to aid in quantification of BACE1 activity and to control the effects, positive and negative, of BACE1 inhibitor therapy.
The path leading to BACE1 inhibition to reduce AD pathology and improve cognition in AD patients has not been straight. However, this therapeutic strategy should not be discontinued because of these setbacks. BACE1 inhibition together with other strategies, including immunotherapy designed to clear brain amyloid plaques or neurofibrillary tangles, is likely to be more effective in improving cognition in AD. It is hoped that a combination therapy will be developed for treating AD patients in the near future.
References
Dubois B, et al. Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12(3):292–323.
Corriveau RA, et al. Alzheimer’s Disease-Related Dementias Summit 2016: national research priorities. Neurology. 2017;89(23):2381–91.
Hung SY, Fu WM. Drug candidates in clinical trials for Alzheimer’s disease. J Biomed Sci. 2017;24(1):47.
Efthymiou AG, Goate AM. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. 2017;12(1):43.
Dos Santos Picanco LC, et al. Alzheimer’s disease: a review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Curr Med Chem. 2018;25(26):3141–59.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608.
Storey E, Cappai R. The amyloid precursor protein of Alzheimer’s disease and the Abeta peptide. Neuropathol Appl Neurobiol. 1999;25(2):81–97.
De Strooper B, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol. 2010;6(2):99–107.
Cirrito JR, et al. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58(1):42–51.
Turner PR, et al. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol. 2003;70(1):1–32.
Berry BJ, et al. Physiological Abeta concentrations produce a more biomimetic representation of the Alzheimer’s disease phenotype in iPSC derived human neurons. ACS Chem Neurosci. 2018;9(7):1693–701.
Tu S, et al. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol Neurodegener. 2014;9:48.
Larson ME, Lesne SE. Soluble Abeta oligomer production and toxicity. J Neurochem. 2012;120(Suppl 1):125–39.
Yan R, et al. Inhibiting BACE1 to reverse synaptic dysfunctions in Alzheimer’s disease. Neurosci Biobehav Rev. 2016;65:326–40.
Rajmohan R, Reddy PH. Amyloid-Beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. J Alzheimers Dis. 2017;57(4):975–99.
Haass C, Selkoe DJ. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell. 1993;75(6):1039–42.
Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120(3):885–90.
Selkoe DJ, Podlisny MB. Deciphering the genetic basis of Alzheimer’s disease. Annu Rev Genom Hum Genet. 2002;3:67–99.
Li S, et al. Swedish mutant APP-based BACE1 binding site peptide reduces APP beta-cleavage and cerebral Abeta levels in Alzheimer’s mice. Sci Rep. 2015;5:11322.
Zhang S, et al. BACE1 cleavage site selection critical for amyloidogenesis and Alzheimer’s pathogenesis. J Neurosci. 2017;37(29):6915–25.
Ben Halima S, et al. Specific inhibition of beta-secretase processing of the Alzheimer disease amyloid precursor protein. Cell Rep. 2016;14(9):2127–41.
Chau DM, et al. Familial Alzheimer disease presenilin-1 mutations alter the active site conformation of gamma-secretase. J Biol Chem. 2012;287(21):17288–96.
Selkoe DJ. Clearing the brain’s amyloid cobwebs. Neuron. 2001;32(2):177–80.
Miners JS, et al. Abeta-degrading enzymes: potential for treatment of Alzheimer disease. J Neuropathol Exp Neurol. 2011;70(11):944–59.
Saido T, Leissring MA. Proteolytic degradation of amyloid beta-protein. Cold Spring Harb Perspect Med. 2012;2(6):a006379.
Willen K, et al. Abeta accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Mol Neurodegener. 2017;12(1):61.
Arbel-Ornath M, et al. Soluble oligomeric amyloid-beta induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol Neurodegener. 2017;12(1):27.
Mietelska-Porowska A, et al. Tau protein modifications and interactions: their role in function and dysfunction. Int J Mol Sci. 2014;15(3):4671–713.
Simic G, et al. Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules. 2016;6(1):6.
Iqbal K, et al. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010;7(8):656–64.
Tacik P, et al. Genetic disorders with tau pathology: a review of the literature and report of two patients with tauopathy and positive family histories. Neurodegener Dis. 2016;16(1–2):12–21.
Goedert M. Tau gene mutations and their effects. Mov Disord. 2005;20(Suppl 12):S45–52.
Stanford PM, et al. Mutations in the tau gene that cause an increase in three repeat tau and frontotemporal dementia. Brain. 2003;126(Pt 4):814–26.
Sohn PD, et al. Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol Neurodegener. 2016;11(1):47.
Gotz J, et al. An update on the toxicity of Abeta in Alzheimer’s disease. Neuropsychiatr Dis Treat. 2008;4(6):1033–42.
Gotz J, et al. Formation of neurofibrillary tangles in P301 l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491–5.
Gotz J, et al. Amyloid-induced neurofibrillary tangle formation in Alzheimer’s disease: insight from transgenic mouse and tissue-culture models. Int J Dev Neurosci. 2004;22(7):453–65.
Akram A, et al. Stereologic estimates of total spinophilin-immunoreactive spine number in area 9 and the CA1 field: relationship with the progression of Alzheimer’s disease. Neurobiol Aging. 2008;29(9):1296–307.
Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol Aging. 1990;11(1):29–37.
Masliah E, et al. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci Lett. 1989;103(2):234–9.
Bussiere T, et al. Stereologic analysis of neurofibrillary tangle formation in prefrontal cortex area 9 in aging and Alzheimer’s disease. Neuroscience. 2003;117(3):577–92.
Audrain M, et al. Alzheimer’s disease-like APP processing in wild-type mice identifies synaptic defects as initial steps of disease progression. Mol Neurodegener. 2016;11:5.
Das B, Yan R. Role of BACE1 in Alzheimer’s synaptic function. Transl Neurodegener. 2017;6:23.
Wang Y, et al. The release and trans-synaptic transmission of tau via exosomes. Mol Neurodegener. 2017;12(1):5.
Huynh TV, et al. Apolipoprotein E and Alzheimer’s disease: the influence of apolipoprotein E on amyloid-beta and other amyloidogenic proteins. J Lipid Res. 2017;58(5):824–36.
Engstrom AK, et al. Gene-environment interaction between lead and Apolipoprotein E4 causes cognitive behavior deficits in mice. Mol Neurodegener. 2017;12(1):14.
Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63(3):287–303.
Hashimoto M, Masliah E. Alpha-synuclein in Lewy body disease and Alzheimer’s disease. Brain Pathol. 1999;9(4):707–20.
Wirths O, Bayer TA. Alpha-synuclein, Abeta and Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(1):103–8.
Wilhite R, et al. Platelet phosphorylated TDP-43: an exploratory study for a peripheral surrogate biomarker development for Alzheimer’s disease. Future Sci OA. 2017;3(4):FSO238.
James BD, et al. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain. 2016;139(11):2983–93.
Josephs KA, et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014;127(6):811–24.
Su B, et al. Oxidative stress signaling in Alzheimer’s disease. Curr Alzheimer Res. 2008;5(6):525–32.
Green KN, LaFerla FM. Linking calcium to Abeta and Alzheimer’s disease. Neuron. 2008;59(2):190–4.
Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383–8.
Babic T. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;67(4):558.
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6.
Mrak RE. Microglia in Alzheimer brain: a neuropathological perspective. Int J Alzheimers Dis. 2012;2012:165021.
Koenigsknecht-Talboo J, et al. Rapid microglial response around amyloid pathology after systemic anti-Abeta antibody administration in PDAPP mice. J Neurosci. 2008;28(52):14156–64.
Condello C, Yuan P, Grutzendler J. Microglia-mediated neuroprotection, TREM2, and Alzheimer’s disease: evidence from optical imaging. Biol Psychiatry. 2018;83(4):377–87.
Kuchibhotla KV, et al. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science. 2009;323(5918):1211–5.
Chun H, Lee CJ. Reactive astrocytes in Alzheimer’s disease: a double-edged sword. Neurosci Res. 2018;126:44–52.
Frost GR, Li YM. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017;7(12):170228.
Hu X, et al. BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J Exp Med. 2018;215(3):927–40.
Dominguez D, et al. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem. 2005;280(35):30797–806.
Ohno M, et al. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis. 2007;26(1):134–45.
McConlogue L, et al. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J Biol Chem. 2007;282(36):26326–34.
Jonsson T, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488(7409):96–9.
Maloney JA, et al. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J Biol Chem. 2014;289(45):30990–1000.
Kandalepas PC, et al. The Alzheimer’s beta-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol. 2013;126(3):329–52.
Das B, Yan R. Role of BACE1 in Alzheimer’s synaptic function. Transl Neurodegener. 2017;6(1):23.
Ovsepian SV, et al. Synaptic vesicle cycle and amyloid β: biting the hand that feeds. Alzheimers Dement. 2018;14(4):502–13.
Shankar GM, Walsh DM. Alzheimer’s disease: synaptic dysfunction and Abeta. Mol Neurodegener. 2009;4:48.
Roberds SL, et al. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum Mol Genet. 2001;10(12):1317–24.
Luo Y, et al. BACE1 (beta-secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time. Neurobiol Dis. 2003;14(1):81–8.
Luo Y, et al. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci. 2001;4(3):231–2.
Vassar R. Beta-secretase (BACE) as a drug target for Alzheimer’s disease. Adv Drug Deliv Rev. 2002;54(12):1589–602.
Golde TE, Petrucelli L, Lewis J. Targeting Abeta and tau in Alzheimer’s disease, an early interim report. Exp Neurol. 2010;223(2):252–66.
Carroll CM, Li YM. Physiological and pathological roles of the gamma-secretase complex. Brain Res Bull. 2016;126(Pt 2):199–206.
De Strooper B. Lessons from a failed gamma-secretase Alzheimer trial. Cell. 2014;159(4):721–6.
Willem M, Lammich S, Haass C. Function, regulation and therapeutic properties of beta-secretase (BACE1). Semin Cell Dev Biol. 2009;20(2):175–82.
Tang J, et al. Study of memapsin 2 (beta-secretase) and strategy of inhibitor design. J Mol Neurosci. 2003;20(3):299–304.
Tan J, Evin G. Beta-site APP-cleaving enzyme 1 trafficking and Alzheimer’s disease pathogenesis. J Neurochem. 2012;120(6):869–80.
Yan R. Stepping closer to treating Alzheimer’s disease patients with BACE1 inhibitor drugs. Transl Neurodegener. 2016;5:13.
Turner RT 3rd, et al. Subsite specificity of memapsin 2 (beta-secretase): implications for inhibitor design. Biochemistry. 2001;40(34):10001–6.
Turner RT 3rd, et al. Structural locations and functional roles of new subsites S5, S6, and S7 in memapsin 2 (beta-secretase). Biochemistry. 2005;44(1):105–12.
Li X, et al. Predicting memapsin 2 (beta-secretase) hydrolytic activity. Protein Sci. 2010;19(11):2175–85.
Ghosh AK, Osswald HL. BACE1 (beta-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem Soc Rev. 2014;43(19):6765–813.
Ghosh AK, et al. Design, synthesis, and X-ray structural studies of BACE-1 inhibitors containing substituted 2-oxopiperazines as P1′-P2′ ligands. Bioorg Med Chem Lett. 2017;27(11):2432–8.
Yuan J, et al. Structure-based design of beta-site APP cleaving enzyme 1 (BACE1) inhibitors for the treatment of Alzheimer’s disease. J Med Chem. 2013;56(11):4156–80.
Stamford A, Strickland C. Inhibitors of BACE for treating Alzheimer’s disease: a fragment-based drug discovery story. Curr Opin Chem Biol. 2013;17(3):320–8.
Butini S, et al. Novel peptidomimetics as BACE-1 inhibitors: synthesis, molecular modeling, and biological studies. Bioorg Med Chem Lett. 2013;23(1):85–9.
Ghosh AK, Brindisi M, Tang J. Developing beta-secretase inhibitors for treatment of Alzheimer’s disease. J Neurochem. 2012;120(Suppl 1):71–83.
Ghosh AK, Gemma S, Tang J. beta-Secretase as a therapeutic target for Alzheimer’s disease. Neurotherapeutics. 2008;5(3):399–408.
Hong L, et al. Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science. 2000;290(5489):150–3.
Wang W, Liu Y, Lazarus RA. Allosteric inhibition of BACE1 by an exosite-binding antibody. Curr Opin Struct Biol. 2013;23(6):797–805.
Koelsch G. BACE1 Function and Inhibition: implications of intervention in the amyloid pathway of Alzheimer’s disease pathology. Molecules. 2017;22(10):1723.
Vassar R, et al. The beta-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J Neurosci. 2009;29(41):12787–94.
Yan R, Vassar R. Targeting the beta secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014;13(3):319–29.
Zhu K, et al. Consequences of pharmacological BACE inhibition on synaptic structure and function. Biol Psychiatry. 2018;84(7):478–87.
Oehlrich D, Prokopcova H, Gijsen HJ. The evolution of amidine-based brain penetrant BACE1 inhibitors. Bioorg Med Chem Lett. 2014;24(9):2033–45.
Das S, Chakraborty S, Basu S. Fragment-based designing for the generation of novel leads against BACE1. Cent Nerv Syst Agents Med Chem. 2015;15(1):52–64.
Munro KM, et al. Functions of the Alzheimer’s disease protease BACE1 at the synapse in the central nervous system. J Mol Neurosci. 2016;60(3):305–15.
Scott JD, et al. Discovery of the 3-imino-1,2,4-thiadiazinane 1,1-dioxide derivative verubecestat (MK-8931)–a β-site amyloid precursor protein cleaving enzyme 1 inhibitor for the treatment of Alzheimer’s disease. J Med Chem. 2016;59(23):10435–50.
Kennedy ME, et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS beta-amyloid in animal models and in Alzheimer’s disease patients. Sci Transl Med. 2016;8(363):363ra150.
Zwan MD, et al. Diagnostic impact of [18F]flutemetamol PET in early-onset dementia. Alzheimers Res Ther. 2017;9(1):2.
Egan MF, et al. Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2018;378(18):1691–703.
Shimshek DR, et al. Pharmacological BACE1 and BACE2 inhibition induces hair depigmentation by inhibiting PMEL17 processing in mice. Sci Rep. 2016;6:21917.
Eketjall S, et al. AZD3293: a novel, orally active BACE1 inhibitor with high potency and permeability and markedly slow off-rate kinetics. J Alzheimers Dis. 2016;50(4):1109–23.
Sims JR, et al. Development review of the BACE1 inhibitor lanabecestat (AZD3293/LY3314814). J Prev Alzheimers Dis. 2017;4(4):247–54.
Cebers G, et al. AZD3293: pharmacokinetic and pharmacodynamic effects in healthy subjects and patients with Alzheimer’s disease. J Alzheimers Dis. 2016;55(3):1039–53.
Sakamoto K, et al. BACE1 inhibitor lanabecestat (AZD3293) in a phase 1 study of healthy Japanese subjects: pharmacokinetics and effects on plasma and cerebrospinal fluid Abeta peptides. J Clin Pharmacol. 2017;57(11):1460–71.
Timmers M, et al. Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer’s disease: randomized, double-blind, placebo-controlled study. Alzheimers Res Ther. 2018;10(1):85.
Timmers M, et al. Profiling the dynamics of CSF and plasma Abeta reduction after treatment with JNJ-54861911, a potent oral BACE inhibitor. Alzheimers Dement (N Y). 2016;2(3):202–12.
Lahiri DK, et al. Lessons from a BACE1 inhibitor trial: off-site but not off base. Alzheimers Dement. 2014;10(5 Suppl):S411–9.
2012 AAIC conference in Vancouver, Canada, Eisai presented data to suggest that elenbecestat lowers Aβ concentration in the brain, CSF, and plasma of rats and guinea pigs, and that it lowers CSF and plasma Aβ in non-human primates information from news report https://www.alzforum.org/news/conference-coverage/wave-new-bace-inhibitors-heading-phase-2.
Neumann U, et al. The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer’s disease. EMBO Mol Med. 2018;10(11):e9316.
Dobrowolska Zakaria JA, Vassar RJ. A promising, novel, and unique BACE1 inhibitor emerges in the quest to prevent Alzheimer’s disease. EMBO Mol Med. 2018;10(11):e9717.
Vandenberghe R, et al. Active Abeta immunotherapy CAD106 in Alzheimer’s disease: a phase 2b study. Alzheimers Dement (N Y). 2017;3(1):10–22.
Farlow MR, et al. Long-term treatment with active Abeta immunotherapy with CAD106 in mild Alzheimer’s disease. Alzheimers Res Ther. 2015;7(1):23.
Wu G, et al. Characterization of plasma beta-secretase (BACE1) activity and soluble amyloid precursor proteins as potential biomarkers for Alzheimer’s disease. J Neurosci Res. 2012;90(12):2247–58.
Vassar R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimers Res Ther. 2014;6(9):89.
Meakin PJ, et al. Reduction in BACE1 decreases body weight, protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem J. 2012;441(1):285–96.
Farah MH, et al. Reduced BACE1 activity enhances clearance of myelin debris and regeneration of axons in the injured peripheral nervous system. J Neurosci. 2011;31(15):5744–54.
Wang H, Song L, Laird F, Wong PC, Lee HK. BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. J Neurosci. 2008;28(35):8677–81
Wang H, Megill A, Wong PC, Kirkwood A, Lee HK. Postsynaptic target specific synaptic dysfunctions in the CA3 area of BACE1 knockout mice. PLoS One. 2014;9(3):e92279.
Ou-Yang M-H, et al. Axonal organization defects in the hippocampus of adult conditional BACE1 knockout mice. Sci Transl Med. 2018;10(459):eaao5620.
Parihar MS, Brewer GJ. Amyloid-beta as a modulator of synaptic plasticity. J Alzheimers Dis. 2010;22(3):741–63.
Mei L, Nave KA. Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron. 2014;83(1):27–49.
Hu X, et al. Neurological dysfunctions associated with altered BACE1-dependent Neuregulin-1 signaling. J Neurochem. 2016;136(2):234–49.
Filser S, et al. Pharmacological inhibition of BACE1 impairs synaptic plasticity and cognitive functions. Biol Psychiatry. 2015;77(8):729–39.
Yan R. Physiological functions of the beta-site amyloid precursor protein cleaving enzyme 1 and 2. Front Mol Neurosci. 2017;10:97.
Coimbra JRM, et al. Highlights in BACE1 inhibitors for Alzheimer’s disease treatment. Front Chem. 2018;6:178.
May PC, et al. Robust central reduction of amyloid-beta in humans with an orally available, non-peptidic beta-secretase inhibitor. J Neurosci. 2011;31(46):16507–16.
Cai J, et al. beta-Secretase (BACE1) inhibition causes retinal pathology by vascular dysregulation and accumulation of age pigment. EMBO Mol Med. 2012;4(9):980–91.
Zuhl AM, et al. Chemoproteomic profiling reveals that cathepsin D off-target activity drives ocular toxicity of beta-secretase inhibitors. Nat Commun. 2016;7:13042.
Luna S, Cameron DJ, Ethell DW. Amyloid-beta and APP deficiencies cause severe cerebrovascular defects: important work for an old villain. PLoS ONE. 2013;8(9):e75052.
Dore V, et al. Cross-sectional and longitudinal analysis of the relationship between Abeta deposition, cortical thickness, and memory in cognitively unimpaired individuals and in Alzheimer disease. JAMA Neurol. 2013;70(7):903–11.
Ye N, et al. Clinical bioavailability of the novel BACE1 inhibitor lanabecestat (AZD3293): assessment of tablet formulations versus an oral solution and the impact of gastric pH on pharmacokinetics. Clin Pharmacol Drug Dev. 2018;7(3):233–43.
Albala B, et al. CSF amyloid lowering in human volunteers after 14 days’ oral administration of the novel BACE1 inhibitor E2609 [abstract no. S4-04-01]. Alzheimers Dement. 2012;8(4 Suppl):S743.
Kumar D, et al. Secretase inhibitors for the treatment of Alzheimer’s disease: long road ahead. Eur J Med Chem. 2018;148:436–52.
Ufer M, et al. Results from a first-in-human study with the BACE inhibitor CNP520 [abstract no. O1-10-06]. Alzheimers Dement. 2016;12(7):P200.
Weyer SW, et al. Comparative analysis of single and combined APP/APLP knockouts reveals reduced spine density in APP-KO mice that is prevented by APPsalpha expression. Acta Neuropathol Commun. 2014;2:36.
Hu X, et al. BACE1 regulates hippocampal astrogenesis via the Jagged1-Notch pathway. Cell Rep. 2013;4(1):40–9.
Kim D, Tsai LH. Bridging physiology and pathology in AD. Cell. 2009;137(6):997–1000.
Li Q, Sudhof TC. Cleavage of amyloid-beta precursor protein and amyloid-beta precursor-like protein by BACE 1. J Biol Chem. 2004;279(11):10542–50.
Eggert S, et al. The proteolytic processing of the amyloid precursor protein gene family members APLP-1 and APLP-2 involves alpha-, beta-, gamma-, and epsilon-like cleavages: modulation of APLP-1 processing by n-glycosylation. J Biol Chem. 2004;279(18):18146–56.
Heber S, et al. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000;20(21):7951–63.
Gautam V, et al. BACE1 activity regulates cell surface contactin-2 levels. Mol Neurodegener. 2014;9:4.
He W, et al. beta-Site amyloid precursor protein cleaving enzyme 1 (BACE1) regulates Notch signaling by controlling the cleavage of Jagged 1 (Jag1) and Jagged 2 (Jag2) proteins. J Biol Chem. 2014;289(30):20630–7.
Zhou L, et al. The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J Biol Chem. 2012;287(31):25927–40.
Hitt B, et al. beta-Site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. J Biol Chem. 2012;287(46):38408–25.
Kuhn PH, et al. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012;31(14):3157–68.
Kim W, et al. BACE1 elevation engendered by GGA3 deletion increases beta-amyloid pathology in association with APP elevation and decreased CHL1 processing in 5XFAD mice. Mol Neurodegener. 2018;13(1):6.
Bot N, et al. Processing of the synaptic cell adhesion molecule neurexin-3beta by Alzheimer disease alpha- and gamma-secretases. J Biol Chem. 2011;286(4):2762–73.
Fleck D, et al. Dual cleavage of neuregulin 1 type III by BACE1 and ADAM17 liberates its EGF-like domain and allows paracrine signaling. J Neurosci. 2013;33(18):7856–69.
Zhu K, et al. Beta-site amyloid precursor protein cleaving enzyme 1 inhibition impairs synaptic plasticity via seizure protein 6. Biol Psychiatry. 2018;83(5):428–37.
Gunnersen JM, et al. Sez-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron. 2007;56(4):621–39.
Herber J, et al. Click chemistry-mediated biotinylation reveals a function for the protease BACE1 in modulating the neuronal surface glycoproteome. Mol Cell Proteomics. 2018;17(8):1487–501.
Causevic M, et al. BACE1-cleavage of Sez6 and Sez6L is elevated in Niemann-Pick type C disease mouse brains. PLoS One. 2018;13(7):e0200344.
Wong HK, et al. beta Subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J Biol Chem. 2005;280(24):23009–17.
Kim DY, et al. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat Cell Biol. 2007;9(7):755–64.
Sachse CC, et al. BACE1 and presenilin/gamma-secretase regulate proteolytic processing of KCNE1 and 2, auxiliary subunits of voltage-gated potassium channels. FASEB J. 2013;27(6):2458–67.
Lehnert S, et al. Ion channel regulation by beta-secretase BACE1—enzymatic and non-enzymatic effects beyond Alzheimer’s disease. Channels (Austin). 2016;10(5):365–78.
Biogen. Phase II clinical study of elenbecestat demonstrates safety and tolerability in MCI and mild to moderate Alzheimer’s disease at 18-months [news release]. 2018 Jun 4. http://investors.biogen.com/news-releases/news-release-details/phase-ii-clinical-study-elenbecestat-demonstrates-safety-and. Accessed 24 Jan 2019.
NASDAQ. FDA sets the stage for earlier-stage Alzheimer’s treatments. 2018 Feb 16. https://www.nasdaq.com/article/fda-sets-the-stage-for-earlier-stage-alzheimers-treatments-cm922638. Accessed 24 Jan 2019.
Clinical Trials on Alzheimer’s Disease (CTAD). Is there a role for BACE inhibition in Alzheimer’s treatment? [press release]. http://www.ctad-alzheimer.com/files/files/BACE%20inhibitors%20press%20release%2025%20Oct%202018%20_0.pdf. Accessed 24 Jan 2019.
Eisai Global. Eisai presents first clinical data for BACE inhibitor E2609 at Alzheimer’s Association international conference 2012. 2012 Jul 19. https://www.eisai.com/news/news201247.html. Accessed 24 Jan 2019.
Acknowledgements
Due to space limitations, not all BACE1 inhibitor trials are cited, but many of these studies are discussed in reviews cited in this article.
Author information
Authors and Affiliations
Contributions
Both B. Das and R. Yan wrote this review.
Corresponding author
Ethics declarations
Funding
No funding was received for the review. R. Yan is supported by grants (MH103942, RF1AG058261, AG025493, NS074256, and AG046929) from the National Institutes of Health.
Conflict of interest
RY and BD declare no conflicts of interest or competing interests relevant to this review.
Availability of data and materials
The authors encourage free citation of the data or conclusions in this review article.
Rights and permissions
About this article

Cite this article
Das, B., Yan, R. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 33, 251–263 (2019). https://doi.org/10.1007/s40263-019-00613-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-019-00613-7