
Abstract
Traditional pharmacological treatments for depression have a delayed therapeutic onset, ranging from several weeks to months, and there is a high percentage of individuals who never respond to treatment. In contrast, ketamine produces rapid-onset antidepressant, anti-suicidal, and anti-anhedonic actions following a single administration to patients with depression. Proposed mechanisms of the antidepressant action of ketamine include N-methyl-d-aspartate receptor (NMDAR) modulation, gamma aminobutyric acid (GABA)-ergic interneuron disinhibition, and direct actions of its hydroxynorketamine (HNK) metabolites. Downstream actions include activation of the mechanistic target of rapamycin (mTOR), deactivation of glycogen synthase kinase-3 and eukaryotic elongation factor 2 (eEF2), enhanced brain-derived neurotrophic factor (BDNF) signaling, and activation of α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors (AMPARs). These putative mechanisms of ketamine action are not mutually exclusive and may complement each other to induce potentiation of excitatory synapses in affective-regulating brain circuits, which results in amelioration of depression symptoms. We review these proposed mechanisms of ketamine action in the context of how such mechanisms are informing the development of novel putative rapid-acting antidepressant drugs. Such drugs that have undergone pre-clinical, and in some cases clinical, testing include the muscarinic acetylcholine receptor antagonist scopolamine, GluN2B-NMDAR antagonists (i.e., CP-101,606, MK-0657), (2R,6R)-HNK, NMDAR glycine site modulators (i.e., 4-chlorokynurenine, pro-drug of the glycineB NMDAR antagonist 7-chlorokynurenic acid), NMDAR agonists [i.e., GLYX-13 (rapastinel)], metabotropic glutamate receptor 2/3 (mGluR2/3) antagonists, GABAA receptor modulators, and drugs acting on various serotonin receptor subtypes. These ongoing studies suggest that the future acute treatment of depression will typically occur within hours, rather than months, of treatment initiation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Multiple mechanisms have been proposed to explain the rapid antidepressant actions of ketamine and other drugs. |
Proposed mechanisms underlying rapid antidepressant action are not mutually exclusive but may act in a complementary manner, resulting in rapid changes in synaptic plasticity, and sustained strengthening of excitatory synapses in limbic brain regions. |
There are a number of pre-clinically validated targets beyond N-methyl-d-aspartate receptor inhibition that provide hope for the development of novel rapid-acting antidepressants. |
1 Introduction
Major depressive disorder is a mental illness afflicting approximately 16% of the world population, characterized by depressed mood, lack of engagement in pleasurable activities, disturbances in activity levels, loss of concentration, and suicidal ideation [1]. Currently available interventions including monoamine-based pharmacotherapies and psycho-behavioral therapies require several weeks to months for beneficial effects to occur [2] and there is a high percent of patients with depression taking standard treatment who remain treatment resistant [3]. In addition to treatment resistance associated with the use of the currently available antidepressants, these treatments are often accompanied by undesirable side effects [4,5,6]. Therefore, there is an urgent need for better antidepressant medications, with a faster onset of action, which will be also effective in patients who do not respond to classical antidepressants.
1.1 Towards the Development of Rapid-Acting Antidepressants
In contrast to the delayed therapeutic effects of monoamine-based antidepressants, there is evidence for designing therapeutics with rapid-acting antidepressant actions. For instance, electroconvulsive therapy (ECT) often exerts a more rapid antidepressant action in major depression when compared with monoamine-acting antidepressants [7], with remission rates typically ranging from 50 to 75% [8,9,10]. For severe major depression cases and suicidal patients, ECT can induce a relatively rapid relief of symptoms [11]; however, any acute effects are transient [e.g., 12]. An average of six ECT applications over 2 weeks induces a sustained reduction in depressed mood symptom severity [13, 14].
Acute sleep deprivation is a well-documented, highly effective, fast-onset (within 24–48 h) antidepressant, which rapidly relieves depressed mood [15]. This initial improvement rapidly reverses following subsequent sleep cycles [15, 16]; however, the exact mechanisms underlying this rapid and transient effect of sleep deprivation are currently unknown. These established non-pharmacological interventions comprise a proof of principle for the more rapid relief of depressive symptoms and suggest the feasibility of designing rapid-acting antidepressant medications [17].
More recently, it has been found that ketamine, an anesthetic drug first commercially available for human use in 1970 [18, 19], exerts robust, rapid (within 2 h following administration), and sustained (7 days on average) antidepressant actions in patients with major depression, following a single administration (typically intravenous) at a sub-anesthetic dose [20,21,22,23,24]. Meta-analyses have confirmed and further supported the significance of both the antidepressant [25,26,27,28] and anti-suicidal [29] actions of ketamine compared with placebo controls. This finding revolutionized and established the concept of rapid-acting antidepressant medications. Nevertheless, the widespread clinical use of ketamine for the treatment of major depression is restricted to certain subgroups (e.g., treatment-resistant depression, suicidal ideation) and it requires close monitoring when it is administered, due to its side effects, including dissociation, psychotomimetic properties, and abuse potential [30, 31]. Consequently, alternative medications that share the robust antidepressant actions of ketamine, but lack its side effects, are urgently needed.
Following these promising findings with ketamine and based on the hypothesized mechanism of action of this prototype rapid-acting antidepressant medication, pre-clinical and clinical studies have assessed alternative, putative rapid-acting antidepressant medications including subunit-specific N-methyl-d-aspartate receptor (NMDAR) antagonists, the muscarinic acetylcholine receptor (mAChR) antagonist scopolamine, group II metabotropic glutamate receptor (mGluR2/3) antagonists, glycineB-NMDAR modulators, gamma aminobutyric acid (GABA)A receptor modulators (positive and negative allosteric modulators), and serotonin 2C (5-HT2C) receptor antagonists (see Table 1). These compounds/drugs have shown efficacy in several animal tests predictive of rapid-acting antidepressant actions (see Sect. 1.2).
1.2 Animal Tests Predictive of Rapid-Acting Antidepressant Efficacy
Tests in mice and rats predictive of antidepressant activity of different drugs have been widely used for mechanism-of-action studies and drug development purposes [32]. A valid model of antidepressant efficacy is expected to have predictive sensitivity in regards to the time course of antidepressant actions in humans. Classical monoamine-based antidepressants require long-term (weeks if not months) administration to exert their effects in patients with depression [2], thus these drugs should have a similar slow-onset time frame of action in animal tests with predictive validity [32]. Such validated tests (as reviewed by Ramaker and Dulawa [32]) include the forced-swim test assessed at 24 h following drug administration (i.e., 24-h forced-swim test), rather than the typical 1-h or 30-min time point, novelty-suppressed feeding, novelty-induced hypophagia, reversal of learned helplessness, tests of stress-induced social avoidance and anhedonia (i.e., long-term corticosterone administration, chronic mild stress, and chronic social defeat stress), as well as reversal of the hyperlocomotor effects following olfactory bulbectomy (Table 1). We note that the only drug repeatedly demonstrated to have rapid antidepressant efficacy in human clinical trials is ketamine, along with the non-pharmacological options ECT and sleep deprivation. Other putative rapid-acting antidepressant agents identified in preclinical models have not yet demonstrated a rapid clinical effect (within 72 h of administration), thus their rapid-onset antidepressant efficacy has yet to be validated in humans. For a description of animal tests predictive of rapid antidepressant efficacy, see Table 1.
2 Ketamine: The Prototype Rapid-Acting Antidepressant
Among other actions, ketamine is a non-competitive NMDAR antagonist at the phencyclidine-binding site of the receptor [33, 34]. Although it was initially developed as an anesthetic drug [18], to be used as an alternative to phencyclidine, it was subsequently reported to exert robust antidepressant actions in patients with depression by Krystal and colleagues [20], who found that a single 40-min intravenous infusion of a sub-anesthetic dose of ketamine (0.5 mg/kg), which produced transient dissociative effects, resulted in significant improvement in depression symptoms within hours after administration of the drug (see Table 2). A robust antidepressant effect of ketamine occurred within 4 h post-administration [20], a time point well after the dissociative effects were no longer present. Following this finding, other clinical studies have replicated and further extended the findings of the efficacy of ketamine into patients with treatment-resistant depression, using the same dose and intravenous route of administration [e.g., 21, 24, 35]. Importantly, there is also significant separation between the antidepressant response of ketamine (64% of patients achieved a ≥ 50% reduction in the Montgomery–Åsberg Depression Scale scores) and placebo (28% of patients) when a psychoactive drug, i.e., midazolam, was used as an active placebo to counteract the functional un-blinding role of the dissociative side effects of ketamine [36]. In addition to its effects of relieving mood in patients with major unipolar depression, ketamine also exerts robust antidepressant effects in treatment-resistant bipolar depression, with a comparable response rate [37, 38] (Table 2). Such antidepressant effects may last 1–2 weeks [e.g., 21, 24, 35].
Nevertheless, individuals who positively respond to ketamine usually relapse. However, there is considerable variability in the time of relapse among patients receiving a single infusion of ketamine [e.g., 21, 24, 35], possibly influenced by the individual’s underlying genetics, as well as environmental influences, previous prescription history, and other factors. Relapse to depression following ketamine treatment might be related to the reversal of the synaptic effects of ketamine [39]; see Sects. 4 and 5.
In addition to its antidepressant actions, a single infusion of ketamine induces anti-anhedonic effects [40] and reduces suicidal ideation [29] in patients with depression with an effect commencing within a few hours of administration and lasting for up to 1 week, similar to its antidepressant time course. Importantly, the anti-suicidal actions of ketamine are partially independent of its antidepressant effects [29, 41], although further studies are required to confirm this conclusion. The clinical findings regarding the antidepressant actions of ketamine are also supported by animal tests that predict rapid-onset antidepressant action, including the 24-h forced-swim test [42,43,44], learned helplessness [42, 43, 45,46,47,48,49,50,51], novelty-suppressed feeding [49, 52,53,54,55], and novelty-induced hypophagia [56, 57]. Ketamine also reverses anhedonia and other maladaptive behaviors following chronic mild stress [43, 55] and chronic social defeat stress [45, 58,59,60,61]; also see Table 1. Only a few animal studies have been published to date assessing the effects of ketamine on endophenotypes of suicidal behavior (as discussed in [62]).
Ketamine is manufactured as a racemic mixture containing equal portions of its two enantiomers, the (S)- and (R)-ketamine. In a randomized double-blind placebo-controlled trial, intravenous infusion of the (S)-ketamine enantiomer (40-min infusion, 0.2 and 0.4 mg/kg) exerted antidepressant responses 1-day post-administration, which was sustained for 3 days, and in some patients lasted for up to 2 weeks following a single infusion [35]. In addition, a randomized controlled clinical trial indicated dose-dependent antidepressant actions of intranasally administered (S)-ketamine (administration regimen: 28–84 mg, twice a week for a total of 2 weeks) in patients with treatment-resistant depression receiving oral classical antidepressant treatment [63]. Pre-clinical rodent studies have also indicated rapid-acting antidepressant behavioral actions of (S)-ketamine following chronic social defeat stress, where the drug reduced behavioral despair (i.e., it decreased immobility time), and reversed anhedonia (i.e., it restored sucrose preference) induced by chronic stress [64]. Moreover, (S)-ketamine decreased escape failures in the learned helplessness paradigm [45] and reduced high behavioral despair in the forced-swim test following repeated corticosterone administration [65] in rodents. These behavioral actions of (S)-ketamine in rodent tests require higher doses compared with the (R)-ketamine enantiomer [45, 64, 65], indicating that (R)-ketamine is a more potent antidepressant compared with the (S)-ketamine enantiomer [66], at least in rodents. The (S)-ketamine enantiomer is currently in phase III clinical trials for the treatment of treatment-resistant depression and suicidal ideation after receiving the US Food and Drug Administration Fast Track Designation (Clinical Trial ID: NCT02417064). (R)-ketamine has yet to be tested in clinical trials for the treatment of major depression.
Although the antidepressant actions of ketamine are unique and it is the prototype rapid-acting antidepressant with multiple clinical trials supporting its robust effects in patients with major depression, its widespread use for the treatment of major depression is limited because of its significant adverse effects, as discussed earlier; also see [30, 67,68,69,70]. Therefore, other potent and effective antidepressant alternatives to ketamine that lack its undesirable side effects have become a focus in the search for rapid-acting treatments for major depression.
3 Scopolamine as a Rapid-Acting Antidepressant
Scopolamine is a non-selective mAChR antagonist, although it was reported to have more selective antagonist activity on mAChR subtypes 1 and 2 (M1 and M2, respectively) [71]. The first preclinical evidence indicating the antidepressant action of this drug came from Browne in 1979, where administration of scopolamine reduced behavioral despair of rodents in the acute forced-swim test [72]; a finding that was replicated in subsequent rodent studies also using the acute forced-swim test model [73]. However, this test does not predict fast-onset antidepressant actions (Table 1). Although studies assessing the effects of anticholinergic agents, such as biperiden, were performed in the early 1980s [74], the first human trial (not placebo controlled) assessing the effects of scopolamine in patients with major depression came from Gillin et al. 1991 in [75]. Administration of scopolamine for two consecutive nights induced a small, but significant, antidepressant effect 24 h following the second intramuscular injection [75].
Following these positive results, Furey and Drevets conducted a double-blind placebo-controlled trial in patients with depression, published in 2006 [76]. This study showed that 15-min intravenous infusions of scopolamine separated by 3–4 days between each administration session resulted in significant decreases in depression scores after three sessions (thus three scopolamine infusions in total), compared with placebo; significant clinical responses were observed in the evaluation after the first scopolamine infusion, 3–4 days after the first treatment. This effect of scopolamine was replicated in a second double-blind placebo-controlled trial conducted by the same investigators using the same study design [77]. The antidepressant effects of scopolamine were reported to appear following just a single 15-min intravenous infusion in a subsequent placebo-controlled clinical trial, with a greater effect in women than men [78] (see Table 2). It is worth noting that other anticholinergic drugs, including the M1 muscarinic antagonist biperiden, showed inconsistent results for the treatment of major depression in human trials, indicating that the effects of scopolamine might be mAchR subunit specific, or via another target [79, 80]. We note that in contrast to ketamine and ECT, there have been no studies published to date testing scopolamine in patients with treatment-resistant depression.
In rodents, scopolamine administration resulted in rapid antidepressant actions in the 24-h forced-swim test [81, 82], learned helplessness [83, 84], and novelty-suppressed feeding paradigm [81, 82] and attenuated chronic stress-induced deficits in sucrose preference [81] (see Table 1). These antidepressant effects in animal models were shown to be mediated by the effects of scopolamine to block the M1 subtype of mAChRs [81, 85]; however, some evidence for M2 mAChR blockade as the mediator of these effects of scopolamine in animal models also exists [86].
4 Mechanisms Underlying Fast/Rapid-Onset Antidepressants Actions
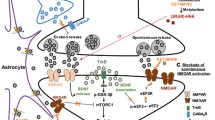
Consensus neurobiological mechanisms underlying the ability of ketamine and other drugs to exert rapid antidepressant actions are complex and have not been fully elucidated. The sustained effects of these drugs appear at time points well beyond when the drugs are eliminated from the brain [87], indicating that they act rapidly to induce long-lasting synaptic plasticity changes that maintain persistent antidepressant actions (see Fig. 1). Here, we review proposed mechanisms of the antidepressant action of ketamine in the context of describing shared, convergent, and also distinct mechanisms with putative novel rapid-acting antidepressants.

Proposed mechanisms of action of ketamine and other putative rapid-acting antidepressants. a Disinhibition hypothesis: ketamine or scopolamine selectively block N-methyl-d-aspartate receptors (NMDARs) or muscarinic acetylcholine receptors (mAChRs), respectively, expressed on gamma aminobutyric acid (GABA)ergic inhibitory interneurons, causing a decrease in interneuron activity, which leads to a disinhibition of pyramidal neurons and enhanced glutamatergic firing. b Negative modulators of GABA A receptors (GABA A R-NAMs) directly act to reduce pyramidal neuron inhibition. Evoked release of glutamate binds to and activates post-synaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR). c Role of ketamine metabolites: ketamine exerts NMDAR inhibition-independent antidepressant actions via the action of its metabolite, (2R,6R)-hydroxynorketamine (HNK), which acts to promote glutamate release (unpublished data) and AMPAR-mediated synaptic potentiation. d Antagonists of the group II metabotropic glutamate receptors (mGluR 2/3 ) disinhibit the tonic blockade of presynaptic glutamate release, thus enhancing synaptic glutamatergic neurotransmission and thus inducing AMPAR activation. AMPAR activation results in enhanced brain-derived neurotrophic factor (BDNF) release, activation of the tropomyosin receptor kinase B (TrkB) receptor, and a subsequent promotion of protein synthesis via the activation of the mechanistic target of rapamycin complex 1 of NMDARs (mTORC1 complex). e Inhibition of extra-synaptic NMDARs: ketamine selectively blocks extra-synaptic GluN2B-containing NMDARs, which are tonically activated by low levels of ambient glutamate regulated by the glutamate transporter 1 [excitatory amino acid transporter 2 (EAAT2)] located on astrocytes. Inhibition of the extra-synaptic GluN2B-NMDARs de-suppresses mTORC1 function, which in turn induces protein synthesis. f Blockade of spontaneous NMDAR activation: ketamine blocks NMDAR-mediated spontaneous neurotransmission (miniature excitatory postsynaptic currents—mEPSC), which results in the inhibition of eukaryotic elongation factor 2 kinase (eEF2K) activity, thus preventing phosphorylation of its eEF2 substrate. This effect subsequently leads to an enhancement of BDNF translation and ultimately protein synthesis. g GLYX-13-induced partial activation of NMDARs: activation of the NMDARs is hypothesized to activate mTORC1 and thus to induce protein synthesis. h Inhibition of NMDAR-dependent burst firing activity of lateral habenula (LHb) neurons: ketamine is proposed to decrease excessive NMDAR-dependent burst firing of LHb neurons linked to depressive symptomatology. All hypotheses propose sustained changes in synaptic plasticity, leading to strengthening of excitatory synapses, being necessary for antidepressant responses. GSK glycogen synthase kinase
4.1 N-Methyl-d-Aspartate Receptor (NMDAR) Modulation
4.1.1 NMDAR Inhibition
NMDARs are glutamatergic ion channel receptors, co-activated by glutamate and glycine, and are composed of four different subunits that may be derived from seven different subunit genes: GluN1, GluN2A-D, and GluN3A-B [88]. The first study to report that NMDAR blockers decrease behavioral despair in mice was conducted by Trullas and Skolnick in 1990, who showed that the non-competitive NMDAR channel blocker MK-801 (dizocilpine) and a competitive NMDAR inhibitor, AP-7, decrease immobility time in the forced-swim test acutely following administration [89]. At the time, long-term, but not short-term, administration of different antidepressant treatments, including tricyclic antidepressants, monoamine oxidase inhibitors, and ‘atypical agents’ had been shown to alter radioligand binding to NMDARs [90, 91]. These findings led to the hypothesis that NMDAR inhibition could be a key target for designing rapid-acting antidepressants. Indeed, it has long been thought that ketamine exerts its rapid antidepressant behavioral actions through its effects on blocking the NMDAR [92].
Evidence from pre-clinical work supports the hypothesis that NMDAR inhibition might induce rapid-onset antidepressant behavioral actions. In particular, a single administration of the GluN2B-acting NMDAR antagonist Ro 25-6981 decreases behavioral despair in the 24-h forced-swim test [42, 47, 93, 94], novelty-suppressed feeding test [42, 47, 55], and novelty-induced hypophagia [56] and reverses anhedonia following chronic mild stress [55]. Similarly, ifenprodil, another GluN2B-NMDAR antagonist, reverses chronic unpredictable stress-induced sucrose preference deficits and behavioral despair in rodents following a single administration [95]. Recently, it has been shown that ketamine might induce its acute (1-hour) antidepressant actions via blockade of NMDAR-dependent burst activity in the lateral habenula neurons [96]; however, it remains to be determined whether this action of ketamine is responsible for the long-lasting antidepressant actions of the drug.
Moreover, inhibiting the NMDAR at the co-agonist glycineB-binding site also exerts antidepressant actions in rodent tests predictive of rapid antidepressant activity. In particular, Zhu et al. [97] demonstrated that peripheral administration of 7-chlorokynurenic acid, a glycineB antagonist, exerted rapid antidepressant actions in the novelty-suppressed feeding and the learned helplessness paradigms following a single injection. Chronic stress-induced anhedonia was also reversed by administration of 7-chlorokynurenic acid, as it reversed decreased sucrose preference [97, 98]. Similar to these findings, 4-chlorokynurenine, a brain-penetrant pro-drug of 7-chlorokynurenic acid, induced fast-onset antidepressant behavioral actions in several animal tests including the 24-h forced-swim test, learned helplessness, and novelty-suppressed feeding paradigms [46]. Neither 7-chlorokynurenic acid nor 4-chlorokynurenine administration was associated with ketamine-like psychostimulant effects, sensory dissociation, or abuse liability in rodents [46, 97, 98]. 4-Chlorokynurenine is currently in a phase II clinical trial for the treatment of major depression (Clinical Trial ID: NCT02484456).
Nevertheless, there are also clinical and pre-clinical findings challenging the hypothesis that NMDAR inhibition is the primary mechanism of the rapid antidepressant efficacy of ketamine. For example, (R)-ketamine has a ~ 4-fold lower affinity/potency for blocking the NMDAR compared with (S)-ketamine, but is a more potent antidepressant than (S)-ketamine in the 24-hour forced-swim test, the learned helplessness and novelty-suppressed feeding tests, as well as in reversing anhedonia following chronic social defeats in rodents [45, 64, 65, 99, 100].
In line with this, a recent study demonstrated that the antidepressant actions of ketamine are mediated by its (2R,6R)-hydroxynorketamine (HNK) metabolite. In particular, when breakdown of ketamine to its (2S,6S;2R,6R)-HNK metabolites was inhibited, ketamine did not exert long-lasting antidepressant behavioral effects in mice. There is evidence for enhanced antidepressant behavioral responses of ketamine in female compared with male rodents [45, 52, 101]. Notably, it was recently demonstrated that higher brain levels of the (2S,6S;2R,6R)-HNK metabolite of ketamine in female mice might be associated with these enhanced antidepressant behavioral actions of ketamine in female mice compared with male mice [45]. (2S,6S;2R,6R)-HNK is the most prominent HNK metabolite present in the plasma and brain of mice [45, 102] and the plasma of treatment-resistant patients with depression [103] following a single ketamine administration. Similar to the preclinical findings, higher levels of this metabolite have been measured in the plasma of female patients compared with male patients following a single infusion of ketamine, but this difference was not associated with any significantly different antidepressant responses in these patients [103].
(2R,6R)-HNK itself exerts dose-dependent rapid antidepressant actions in mouse tests [45] (see Table 1). In particular, a single intraperitoneal administration of (2R,6R)-HNK reduced immobility time in the 24-h forced-swim test, decreased escape failures in the learned helplessness paradigm, reduced anhedonia measures following chronic corticosterone administration, and reversed social interaction deficits induced by chronic social defeat stress [45] (see Table 1). Consequently, (2R,6R)-HNK is sufficient to exert the rapid antidepressant actions of ketamine, at least in animal tests.
In line with these findings, Pham et al. identified antidepressant-relevant actions of (2R,6R)-HNK in the 24-h forced-swim test following peripheral (10 mg/kg, intraperitoneally) administration or direct administration into the medial prefrontal cortex (mPFC; 1 nmol per side) in mice [104]. However, Yang et al. [105] did not observe significant antidepressant behavioral actions using a single dose of (2R,6R)-HNK (i.e., 10 mg/kg) following chronic social defeat stress in mice and the same group reported no effects of (2R,6R)-HNK in the learned helplessness paradigm in rats [106], highlighting that further studies are required to establish the effective doses of this metabolite in different animal tests predictive of antidepressant efficacy. Importantly, (2R,6R)-HNK does not inhibit the NMDAR at antidepressant-relevant concentrations, as was demonstrated by a lack of MK-801 displacement-binding studies (inhibitory constant: K i > 100 µM; half-maximal inhibitory concentration: IC50 > 100 µM) and lack of functional activity on NMDARs localized in stratum radiatum interneurons in rat hippocampal slices or dissociated primary cell culture [45, 107,108,109]. However, it does result in modest inhibition of mEPSC–NMDAR responses in mouse hippocampal cell cultures at higher concentrations (i.e., 50 µM; [108]).
Human clinical findings also challenge the NMDAR inhibition hypothesis. Several other NMDAR antagonists lack the rapid, robust, or sustained antidepressant action of ketamine in humans [25]; also see Table 2. For instance, memantine, an NMDAR channel antagonist, which acts at the same site as ketamine, failed to show significant antidepressant actions in individuals with major depressive disorder in multiple studies [110,111,112]. Moreover, a single administration of AZD6765, a low-trapping NMDAR channel blocker (i.e., fast off rate when glutamate is no longer bound to the NMDAR) also acting at the same site as ketamine, exerted modest but transient (~ 110 min) antidepressant effects in patients with major depression, although this effect was not sustained [113]. However, in a subsequent study, a single infusion of AZD6765 failed to induce a significant change in Montgomery–Åsberg Depression Scale scores compared with placebo at 24 h post-administration [114]. Within the same paper, it was reported that patients receiving AZD6765 (three intravenous infusions per week) for a total period of 3 weeks displayed an improvement in depressed mood and symptom remission during the trial [114]. Finally, a follow-up, larger, four-country, 49-site placebo-controlled human trial comparing AZD6765 with placebo as an adjunctive treatment for depression in a total of 302 patients, failed to show a difference between AZD6765 and placebo [115]. Thus, this drug is no longer under development for the treatment of major depression.
It has also been hypothesized that GluN2B-selective NMDAR inhibition would exert rapid antidepressant actions. Intravenous administration of one such compound, CP-101,606 (traxoprodil), did not result in a rapid beneficial effect (first assessed at 2 days post-treatment); however, it did induce a significant antidepressant response 5 and 8 days following a single infusion [116]. This was a small study (n = 15 subjects/group); there have been no further studies and this drug is not currently in development for depression treatment. There is evidence that this drug may also possess moderate-to-high affinity at sigma-1 receptors [117, 118], which has been suggested as a target for antidepressant action [119,120,121]. Therefore, it is difficult to conclude that the delayed antidepressant efficacy of CP-101,606 is solely owing to the block of GluN2B-containing NMDARs. Another GluN2B-selective NMDAR antagonist, MK-0657 (CERC-301), resulted in modest improvement in the Hamilton Depression Rating Scale, but not the Montgomery–Åsberg Depression Scale, 5 days following a single oral administration [122]; however, a larger phase II follow-up study with MK-0657 failed (as reported in [123]).
4.1.2 NMDAR Activation
Another finding that appears to contrast with the NMDAR inhibition hypothesis underlying rapid antidepressant efficacy is the observation that positive modulation of the NMDAR also exerts rapid antidepressant actions. However, as discussed later in this review, rapid-acting antidepressants converge to enhance excitatory neurotransmission, and subsequently activate α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) and NMDAR receptors [124, 125]. GLYX-13 is a synthetic peptide described as a functional partial agonist of the NMDAR [57, 126]. In humans, a single intravenous infusion of GLYX-13 decreased depression symptoms within 2 h of administration and this effect persisted for ~ 7 days in a double-blind randomized clinical trial [127] (see Table 2). Moreover, a single administration of GLYX-13 induces rapid and sustained antidepressant actions in several animal tests, including the 24-h forced-swim test [57, 128], learned helplessness [57], novelty-suppressed feeding [128], novelty-induced hypophagia [57], and female urine sniffing test [128]. Moreover, it reversed anhedonia and other maladaptive behaviors following chronic mild stress [57] and chronic social defeat stress [58] (see Table 1).
The antidepressant actions of GLYX-13 through its agonist activity at the NMDAR are in line with earlier evidence for antidepressant actions of other NMDAR glycineB-site agonists. Specifically, sarcosine, which is a co-agonist at the glycineB-binding site of the NMDAR [129], improved mood scores following 6-week administration in patients [130]. Similarly, the NMDAR glycine site partial agonist d-cycloserine also produced some antidepressant effects in human clinical trials [131, 132] and in the short-term 60-min forced-swim test in mice [133]. However, it should be noted that the high dose of d-cycloserine used in the study conducted by Heresco-Levy et al. [131] (i.e., 1 g/day) could have instead induced NMDAR inhibition [134]; therefore, further research is warranted to clarify whether d-cycloserine at the doses tested acted as an NMDAR partial agonist or antagonist. Finally, 1-aminocyclopropanecarboxylic acid, a high-affinity glycineB partial agonist, produces antidepressant actions for up to 6 h in the forced-swim test in mice [135].
Importantly, and in contrast to the side effects of ketamine, GLYX-13 does not induce sensory dissociation or display abuse liability properties in rodents [57, 136]. Furthermore, GLYX-13 administration prevents ketamine- and phencyclidine-induced memory deficits [137], consistent with its actions as a positive NMDAR modulator. These findings support that NMDAR inhibition is primarily involved in the side effects of ketamine, while activation of the NMDARs either through an enhanced synaptic glutamatergic neurotransmission, such in the case of ketamine, or direct activation of the receptor (i.e., GLYX-13) results in the antidepressant actions of these drugs [125]. GLYX-13 is currently in phase III clinical trials for the treatment of major depression (see Table 2; Clinical Trial ID: NCT02943577).
4.2 Enhancement of Synaptic Plasticity
4.2.1 Modulation of Excitatory Synaptic Neurotransmission
4.2.1.1 Inhibition of GABAergic Interneuron Activity
Hippocampal and cortical circuits comprise both excitatory glutamatergic pyramidal neurons and inhibitory GABAergic interneurons. GABAergic interneurons are critical for the balance between neural excitation and inhibition [138]. There are subgroups of interneurons that can be distinguished based on their dendritic and axonal morphology, electrophysiological properties (fast or low spiking), synaptic connections, and the genes expressed on these neurons, including calcium-binding proteins (e.g., parvalbumin) and co-transmitters (somatostatin) [139]. Parvalbumin-containing fast-spiking interneurons are primarily responsible for controlling the excitability of pyramidal neurons and the synchrony of their firing, via somatic and dendritic synapses [140,141,142]. High-frequency gamma power oscillations are hypothesized to be controlled by the activation/inactivation of parvalbumin-containing fast-spiking interneurons in the brain [143, 144]. In contrast to the parvalbumin-containing interneurons, somatostatin-expressing interneurons typically exhibit low-threshold spiking and project to distal dendritic pyramidal cells to control dendritic excitability [145, 146].
Although ketamine and scopolamine are expected to weaken excitatory neurotransmission via blocking NMDARs and mAChRs, respectively, both drugs, at low doses, induce a rapid increase in extracellular glutamate levels [85, 147, 148] and an enhancement of glutamate cycling [149] in the mPFC of rodents. One hypothesis that can explain this paradoxical effect is that ketamine and scopolamine exert the effects via receptor subtypes localized on inhibitory GABAergic interneurons, resulting in a reduction in their action potential firing of these neurons. This is predicted to decrease inhibition and increase pyramidal neuron discharge via disinhibition, thus enhancing excitatory glutamatergic neurotransmission [147]. This is also consistent with recent findings indicating that peripheral or intra-mPFC administration of (2R,6R)-HNK increases extracellular glutamate levels 24 h post-administration, an effect that was associated with its antidepressant behavioral actions in the 24-h forced-swim test [104]; however, this effect is not due to NMDAR inhibition [45, 107, 108].
NMDAR inhibition would typically lead to decreased neuronal excitability. However, MK-801, which acts on the ketamine/phencyclidine site, was shown to first reduce interneuron firing and subsequently enhance pyramidal neuron firing in awake rats [150]. This finding is proposed to explain how an antagonist of the NMDARs (i.e., ketamine) is able to induce an overall enhancement of excitatory neurotransmission. This effect might be owing to the fact that the glutamatergic excitatory synapses on GABAergic interneurons are disproportionally more sensitive to NMDAR blockers than pyramidal neurons [151, 152]. This might be because of their more depolarized resting potential, which results in a disproportionally larger NMDAR-mediated excitation, or the result of a unique NMDAR subunit composition that renders interneuron NMDARs more sensitive to ketamine [151, 152].
It is well documented that NMDAR antagonism induces an increase in gamma frequency oscillations. Indeed, MK-801 administration enhances cortical gamma electroencephalography (EEG) power in rats [153, 154], characterized by synchronized high-frequency firing as a result of pyramidal neuron disinhibition. Notably, non-selective and GluN2A-NMDAR antagonists, but not GluN2B-, or GluN2C-/2D selective subunit inhibitors increase gamma power in rats [155], suggesting NMDAR subunit-specific modulation of cortical EEG activity. However, there is also evidence that GluN2B-NMDAR inhibition increases gamma power specifically during rapid eye movement sleep state in rats, albeit at a lower magnitude compared with non-selective and GluN2A-NMDAR antagonists [156]. For ketamine, a critical role of GluN2D-NMDARs was reported to underlie the ability of the drug to induce high-frequency gamma oscillations because this increase was absent in mice lacking the GluN2D gene [157].
The antidepressant actions of scopolamine may also be mediated by its actions at interneurons. Activation of M1-mAChRs expressed on GABAergic interneurons was shown to result in rapid excitation of interneuron activity and thus reinforce an inhibitory input to the pyramidal neurons [158,159,160]. M1-mAChR knockdown specifically in somatostatin-positive GABAergic interneurons, but not parvalbumin interneurons or pyramidal neurons, prevents the antidepressant behavioral actions of scopolamine [82]. Together, these findings indicate that inhibition of M1-mAChR on GABAergic interneurons, resulting in disinhibition of pyramidal neurons and enhancement of glutamatergic transmission may be involved in the rapid antidepressant actions of scopolamine.
In support of the disinhibition hypothesis, administration of negative allosteric modulators of GABAA receptors (GABA-NAMs), that is compounds acting as partial inverse agonists at the benzodiazepine-binding site of the GABAA receptor, exerts rapid antidepressant actions in several animal tests (see Table 1; Fig. 1), presumably through a disinhibition of excitatory glutamatergic neurotransmission [161]. Specifically, administration of GABA-NAMs that are selective for receptors containing alpha5 subunits, such as L-655,781 or MRK-016, reversed social interaction and sucrose preference deficits following several chronic stress paradigms 24 h after administration in rats [162]. The effects of a single injection were long lasting since decreases in behavioral despair in the forced-swim test and restoration of sucrose preference deficits could be seen up to 7 days post-treatment [163]. In mice, MRK-016 administration rapidly reversed chronic restraint stress-induced decreases in female urine sniffing preference (a measure of hedonic behavior in male mice) and it decreased immobility time in the 24-h forced-swim test [164]. More recently, the alpha5-selective GABA-NAM, RY-080, was shown to reduce behavioral despair in the acute forced-swim paradigm [165]. Importantly, in contrast to ketamine, alpha5-selective GABA-NAMs lack the sensory dissociation and abuse liability at antidepressant-relevant doses in animal models [163, 164] and in humans [166]. Rodent expression of the alpha5 subunit is largely limited to the hippocampus and PFC [167], with humans also showing preferential expression in these regions [168]. Although these findings are promising, the effects of these agents are yet to be assessed in patients with major depression.
In contrast to the evidence supporting a role of inhibition of GABAergic interneuron activity to underlie the rapid antidepressant actions of ketamine and scopolamine, there is also some pre-clinical evidence against this hypothesis. In particular, GABAergic synaptic cortical disinhibition induced via a global reduction in GABAA receptor function, increased baseline behavioral despair in the forced-swim test and latency for food consumption in the novelty-suppressed feeding test in mice [169,170,171], indicative of a depressive-related phenotype. In addition, disinhibition of somatostatin-positive GABAergic interneuron activity in mice, resulting in an enhanced interneuron excitability, and thus inhibition of pyramidal neuron activity induced antidepressant-related behavioral responses in mice (i.e., reduced latency to feed in the novelty-suppressed feeding test and decreased escape failures in the learned helplessness paradigm) [172]; however, long-term deletion of GABAA receptors could result in compensatory changes that might contribute to the observed behavioral effects.
There is evidence of GABAergic neurotransmission deficits in patients with depression. Such subjects have been shown to manifest decreased cerebrospinal fluid GABA levels and reductions in parahippocampal and lateral temporal GABAA receptor density, as measured by positron emission tomography tracer [11C]-flumazenil [173,174,175]. Long-term treatment with classical monoamine-based antidepressants restores decreased GABA levels in the occipital cortex of patients with depression [176]. Therefore, GABAA receptor-positive allosteric modulators might be able to restore these GABAergic neurotransmission deficits in patients with depression. Clinically, a report combining data from two previously published studies revealed enhanced antidepressant actions of selective serotonin reuptake inhibitors (SSRIs) when these were administered in conjunction with the positive GABAA receptor allosteric modulator eszopiclone (Lunesta®), a benzodiazepine site partial agonist, compared with the placebo-SSRI monotherapy groups [177]. However, there was no difference in the remission rates, and the response rate advantage of the combined administration of the drugs was lost when insomnia parameters were excluded from the depression score analysis, and there was no evidence for a faster acting antidepressant response of the combined SSRI and eszopiclone group [177]. Positive modulation of GABAA receptors through an increase in glycolytic byproduct methylglyoxal [178], delivered via inhibition of the cytosolic enzyme lactoylglutathione lyase (GLO1), induces fast-onset antidepressant actions in animal tests [179]. In particular, a 5-day administration of two GLO1 inhibitors, S-bromobenzylglutathione cyclopentyl diester (pBBG) and methyl-gerfelin, reduced behavioral despair in the mouse forced-swim test and tail suspension test and it also blocked maladaptive phenotypes induced by chronic mild stress and ameliorated olfactory bulbectomy-induced locomotor hyperactivity [179]; see Table 1. Additionally, administration of an alpha5-containing GABAAR positive allosteric modulator reduced behavioral deficits following chronic unpredictable mild stress, when injected 30 min prior to testing [180]. Further studies will be required to understand the exact impact of altered GABA levels to the symptoms of depression, as well as the effects of GABAA receptor agonists, antagonists, and inverse agonists on GABAA receptor function and excitatory-inhibitory balance.
4.2.1.2 Inhibition of Pre-synaptic Metabotropic Glutamate Receptors
The metabotropic glutamate receptor family is comprised of eight different receptor subtypes (mGluR1–mGluR8), which are divided into three main groups (group I: mGluR1 and mGluR5; group II: mGluR2 and mGluR3; group III: mGluR4, mGluR6, mGluR7, and mGluR8). Pre-clinical studies have reported the efficacy of group II metabotropic glutamate receptor (mGluR2/3) antagonists in reducing behavioral despair in the acute forced-swim test at 30–60 min following drug administration [181,182,183] and decreasing escape failures in the learned helplessness paradigm [184]. Group II metabotropic glutamate receptors are expressed at hippocampal, synaptic, mossy fiber-CA3 pyramidal cells and at excitatory synapses in the PFC [185]. mGluR2 receptors are primarily localized peri-synaptically in close proximity to the pre-synaptic terminals [186, 187], where they act as auto-receptors to decrease synaptic glutamate transmission when activated, presumably serving as a homeostatic mechanism to prevent excessive glutamate release [188, 189]. In contrast, mGluR3 receptors are primarily localized to glial cells [190] and their activation inhibits astrocyte growth [191] and increases glutamate transporter proteins [192], thus indirectly decreasing extracellular glutamate levels.
Group II metabotropic glutamate receptor inhibition has been shown to elicit rapid antidepressant actions in preclinical studies, similar to ketamine. In particular, a single administration of an mGluR2/3 antagonist reduced immobility time in the 24-h forced-swim test [193], decreased time delay until food consumption in the novelty-suppressed feeding test [194, 195], rapidly reversed chronic stress-induced decreases in sucrose preference, which was sustained for at least 10 days [196], and reversed chronic corticosterone-induced behavioral deficits [197] in rodents (see Table 1). In addition, mGluR2/3 blockade reversed the decrease in sucrose preference produced by chronic social defeat stress in mice [198]. While a large (n = 310 patients) clinical trial of a negative allosteric modulator of mGluR2 (RG1578; decoglurant) failed to demonstrate antidepressant responses compared with placebo (see abstract: [199]) (see Table 2), no measure of target engagement was included in this trial and therefore additional studies are needed to determine the potential of mGluR2/3 antagonists in the treatment of treatment-resistant depression.
4.2.2 Inhibition of Long-Term Depression and Induction/Enhancement of Long-Term Potentiation
There are two main forms of activity-dependent synaptic plasticity: long-term depression (LTD), characterized by a persistent decrease in synaptic efficacy, and long-term potentiation (LTP) [200], characterized by a persistent increase in synaptic efficacy [201]. N-Methyl-d-aspartate receptor activation is typically required for the induction of both LTP and LTD, although NMDAR-independent mechanisms of triggering LTP have been also described [202]. Chronic stress was shown to result in the enhancement of LTD and a reduction in LTP, leading to synaptic hypo-function, neuronal loss, as well as decreased spine density and length/number of dendritic branches at some, but not all, synapses in the PFC and hippocampus of rodents [55, 203,204,205,206,207,208,209]. Induction of LTP (and inhibition of LTD) has been implicated in antidepressant behavioral responses [210], including the ability of SSRIs to strengthen excitatory synapses [211].
Blocking the NMDAR with an acute application of ketamine (10 µM) on rat hippocampal slices prevents the induction of LTP [212], and can be predicted to inhibit induction of LTD. Administration of ketamine or (2R,6R)-HNK occluded the induction of LTP in the nucleus accumbens and ventral tegmental area 24 h post-injection in mice [213]. However, ketamine paradoxically enhances LTP in hippocampal brain slices taken 24 h after a single in-vivo administration in rodents [57, 212]. Therefore, ketamine appears to exert differential effects in vivo and in vitro in terms of LTP induction. One factor to consider in regard to this paradox is the lack of metabolism of ketamine to its HNK metabolites in vitro [214] and/or that enhanced glutamatergic neurotransmission (i.e., increased extracellular glutamate levels [147], increased glutamate cycling [149]) might not occur in slices. Another important factor is the relatively rapid (within 2 h post-injection) clearance of ketamine from the brain in vivo [45]. Finally, the degree of NMDAR inhibition in vivo produced by antidepressant-relevant concentrations of ketamine is uncertain, while in slices, concentrations of ketamine may block the NMDAR both on GABAergic interneurons, as well as on glutamatergic pyramidal neurons.
GLYX-13 administered in vivo was shown to reduce hippocampal LTD and to simultaneously enhance the magnitude of LTP induced in rat slices [215, 216], possibly via a preferential activation of GluN2B-containing NMDARs [215]. Importantly, GLYX-13 reverses chronic stress-induced reduction in NMDAR-dependent LTP [217], which is a mechanism hypothesized to mediate its antidepressant actions. Moreover, similar to ketamine, GLYX-13 enhances hippocampal LTP in brain slices taken 24 h and 7 days following a single in vivo injection in rodents [218].
Group II metabotropic glutamate receptor activation is required for the expression of hippocampal LTD and induces a bi-directional inhibition of LTP [188, 189]. In contrast to mGluR2 receptors, activation of mGluR3 receptors is critical for the expression of LTD, but it is not important for LTP [219]. It can therefore be hypothesized that inhibition of mGluR2/3 receptors might produce rapid antidepressant actions via blockade of the presynaptic mGluR2 autoreceptors, thereby promoting induction of LTP/synaptic strengthening via a disinhibition of excitatory neurotransmission. Furthermore, administration of mGluR2/3 antagonists in rodents increases PFC glutamate levels [220], which might be involved in the antidepressant actions of these drugs via the activation of the AMPA-preferring glutamate receptors (AMPARs), as described below.
4.3 Synaptogenesis
A convergent hypothesis for the actions of rapid-acting antidepressants is that administration of these agents leads to a restoration of synapse number following depression-induced synaptic loss and changes in neuronal morphology. There is evidence showing decreased clustering of neurons [221], reduced neuronal size and glial cell density [222,223,224,225], decreased cortical thickness [225], as well as decreased soma size of pyramidal neurons [226] in the PFC, anterior cingulate cortex and/or hippocampus in post-mortem brain tissue from patients with major depression. Moreover, evidence shows that the density of dendritic spines and the extent of dendritic branching in the PFC and hippocampus of chronically stressed rodents are reduced [227,228,229,230,231,232,233]. Although findings in post-mortem brain tissue have limitations owing to poor fixation and tissue condition, loss of synapses were also observed in the PFC of patients with depression using electron microscopy [234].
Long-term, but not short-term monoamine-based antidepressant treatment restores dendritic atrophy induced by chronic stress in rodents [235]. In contrast to the need for long-term administration of conventional antidepressants to induce neuronal remodeling, ketamine rapidly reverses the reduction in the number of dendritic synaptic connections following chronic stress in the PFC of rodents [42], consistent with a promotion of the formation of new synapses. This increase in mature spine density was observed 24 h following a single ketamine administration [42]. The same authors also showed that ketamine produced an increase in the amplitude and frequency of serotonin- and hypocretin-induced excitatory post-synaptic currents in the PFC of rats [42]. Recently, Cavalleri et al. [236] demonstrated that a 60-min exposure of mouse mesencephalic and human-induced pluripotent stem cells derived dopamine neurons to ketamine (1–10 μM) and (2R,6R)-HNK (0.5 μΜ) increased dendritic arborization and soma size of both these cell populations [236].
Similar to ketamine, scopolamine administration has been reported to rapidly increase synaptogenesis in conjunction with its antidepressant behavioral responses in rats [85]. Likewise, the mGluR2/3 antagonist MGS0039 also reverses chronic stress-induced decreases in spine density in the prelimbic area of the mPFC and hippocampus of mice [198]. GLYX-13 also rapidly increases spine number and function in layer 5 neurons of the mPFC [128]. In addition, electroconvulsive shock increases dendritic branching in the dentate gyrus [237] and 24 h of sleep deprivation increases the number of spines in the PFC, but not hippocampus, of rats [238]. Finally, sub-chronic administration of 5-HT2C receptor antagonists (RS102221 and SB242084) induces fast-onset (after 5 days of administration) antidepressant actions following chronic mild stress that was associated with a reversal of stress-induced dendritic atrophy in the mPFC of mice [239]. These findings suggest that increases in spine density and strengthening of neuronal connections are critical mechanisms underlying rapid antidepressant behavioral responses. Increases in synaptic spine densities were shown to be preceded by an activation of the mechanistic target of rapamycin (mTOR) [240], as described below.
5 Downstream Pathways Involved in Rapid Antidepressant Actions
5.1 Activation of α-Amino-3-Hydroxy-5-Methyl-4-Isoxazole-Propionic Acid Receptors (AMPAR)
Changes in AMPARs play a major role in the expression of plasticity at excitatory synapses [241]. Enhancement of synaptic glutamatergic neurotransmission is largely mediated by an increase in the number and/or conductance of post-synaptic AMPARs [242, 243]. The role of AMPAR-mediated plasticity in rapid antidepressant actions is a topic of great interest. Important considerations to understand the role of AMPARs in rapid antidepressant actions is to distinguish between changes in AMPAR activation occurring during the induction of the antidepressant response, and changes in AMPAR number or function in the expression of a persistent antidepressant response, when the drugs have been metabolized and are no longer in the system.
There is considerable evidence that AMPAR activation is required for the induction of a rapid antidepressant response. In rodent pre-clinical studies, administration of the AMPAR antagonist NBQX prevents the antidepressant actions of ketamine [45, 47, 64, 195, 244,245,246,247], GluN2B-NMDAR antagonists [47], 4-chlorokynurenic acid [46], GLYX-13 [57], scopolamine [85, 248], mGluR2/3 antagonists [244, 249], GABAAR-NAMs [164], and (2R,6R)-HNK [45]. Importantly, NBQX pre-treatment does not block the antidepressant actions of monoamine-based antidepressants [47, 250].
A marker of enhanced AMPAR activation during the induction of the antidepressant response is an increase in synchronous oscillatory high frequency EEG activity [248,249,253]. In fact, most of the agents exerting rapid antidepressant actions in clinical and/or pre-clinical studies, including ketamine [45, 114, 254], AZD6765 [114], mGluR2/3 antagonists [255], 4-chlorokynurenine (Zanos, P. and Gould, TD unpublished data), alpha5-selective GABA-NAMs [164], and (2R,6R)-HNK [45], were reported to enhance surface EEG power in the gamma frequency band (30–80 Hz) measured in the frontal cortex. Notably, NBQX pre-treatment prevented not only the increases in gamma activity, but also the persistent behavioral consequences of (2R,6R)-HNK [45] and alpha5-selective GABA-NAMs [164]. Because gamma activity strongly depends on the balance of synaptic excitation and inhibition [256], it is possible that NBQX prevents the antidepressant effects of ketamine and other putative rapid-acting antidepressants by disrupting gamma oscillations.
Gamma oscillatory activity is known to favor induction of activity-dependent synaptic plasticity [256]. One hypothesis for how gamma oscillations might induce a change in behavioral state is through an activity-dependent strengthening of abnormally weakened excitatory synaptic networks, particularly those associated with reward and affect regulation [124]. Acute rhythmic optogenetic activation of the PFC, in the absence of any pharmacological manipulation, exerts an acute antidepressant-like effect in the forced-swim test in mice [257] and a sustained ketamine-like antidepressant response in the forced-swim test 24 h after stimulation in rats [258]. These findings suggest that monitoring EEG activity may be of use to predict novel rapid antidepressant compounds and which patients are likely to respond to this treatment.
There is also considerable evidence that changes in AMPAR number and function underlie the expression of a persistent antidepressant response, long after the rapid-acting agent has been cleared. Similar to mechanisms underlying LTP induction, ketamine administration results in a rapid upregulation of cell surface expression of the GluA1 subunit of the AMPAR, in the mPFC and hippocampus of stress-naive rodents [57, 259], as well as an increase in the number of synaptoneurosomal GluA1-AMPAR subunits in the same brain regions 24 h post-injection [42, 45, 128]. Similar, albeit more slowly occuring, changes in AMPAR function are observed at stress-weakened synapses after long-term, but not short-term, SSRI administration in rodents [211].
Similar effects were reported in the PFC and/or hippocampus of rodents following administration of a selective GluN2B antagonist [42], GLYX-13 [128] or (2R,6R)-HNK [45]. Chronic stress-induced decreases in hippocampal GluA1 expression are also rapidly reversed following a single dose of a selective GluN2B antagonist [91] an alpha5-selective GABA-NAM [162]. An increase in AMPAR expression in specific synapses is likely to be indicative of a potentiation of AMPAR-mediated synaptic transmission following administration of rapid-acting antidepressants. Indeed, electrophysiological indicate that ketamine application enhances AMPAR-mediated synaptic transmission in pyramidal neurons of mPFC [260] and the CA3 region of hippocampus [261] of stress-naive rats.
In addition to the GluA1 subunit involvement, GluA2 subunit of the AMPAR (GluA2) surface expression is increased after 4–8 h of sleep deprivation in the cortex and hippocampus of rats [262, 263] and following a single administration of ketamine in the hippocampus of mice [43, 45]. Synaptoneurosomal GluA2 levels were also increased 24 h after (2R,6R)-HNK administration [45]. GluA2 subunits may be required for the induction of synaptic potentiation of ketamine because ketamine did not induce AMPAR-mediated synaptic potentiation of Schaffer collateral-CA1 synapses in hippocampal slices of mice lacking the GluA2 gene, and these mice did not manifest antidepressant responses to ketamine in the acute (30-min) forced-swim test [259].
There is evidence that rapid-acting antidepressants exert acute effects on AMPAR function. Application of ketamine to hippocampal slices induces AMPAR-mediated synaptic potentiation in the CA1 region [264], even in the absence of ongoing synaptic stimulation [43, 259]. Similar to ketamine, application of (2R,6R)-HNK increases AMPAR-mediated excitatory post-synaptic potentials recorded from the CA1 region of hippocampus [45] at a concentration that does not alter the NMDA-evoked responses in brain slices [45] or hippocampal cell cultures [107, 108]. However, there is no evidence indicating that these actions are a direct effect of ketamine or its metabolite on the AMPARs.
The involvement of AMPAR activation in the antidepressant actions of rapid-acting drugs is further supported by pre-clinical findings showing that positive allosteric modulators of AMPAR [265,266,267] and AMPAR agonists (including AMPA itself) [247, 268] decrease behavioral despair in the acute forced-swim test and the demonstration of an enhanced antidepressant potency (increased sucrose preference) of ketamine when administered in combination with AMPA in a putative rat model of depression [268]. Nevertheless, the effects of AMPAR agonists and positive modulators in better validated animal tests (as discussed earlier) have not been reported yet.
5.2 Brain-Derived Neurotrophic Factor (BDNF)
Brain-derived neurotrophic factor (BDNF) is a growth factor regulating functional neuronal connections and synaptic plasticity in the central nervous system [269,270,271,272]. It has long been postulated that BDNF signaling via its primary receptor, tropomyosin receptor kinase B (TrkB), is deficient in major depression and that elevation of BDNF-TrkB signaling contributes to antidepressant activity [44, 273,274,275,276,277,278,279,280]. For example, long-term administration of monoamine-acting antidepressants was reported to increase BDNF transcription in the hippocampus in rats [281]. In addition, long-term antidepressant treatment [282] and ECT [273, 283] reverse a deficit in serum BDNF levels in patients with major depression. Deletion of hippocampal BDNF attenuates antidepressant efficacy of classical antidepressants in rodent models [284, 285], and the BDNF receptor TrkB is required to exert antidepressant actions of typical antidepressants [286]. Moreover, systemic or intra-hippocampal administration of BDNF exerts antidepressant-like effects [287,288,289], while over-expression of BDNF in the hippocampus leads to resilience to chronic stress [290]. Activation of TrkB is necessary for these behavioral actions [291, 292]. It should be noted though that the effects on BDNF-TrkB signaling are region specific because there is evidence showing that enhanced BDNF-TrkB signaling in the mesolimbic dopaminergic system results in a depressive phenotype in rodents [293, 294].
Brain-derived neurotrophic factor signaling has been postulated to underlie the antidepressant actions of ketamine and scopolamine. In particular, ketamine did not exert antidepressant actions in mice with forebrain-specific Bdnf gene knockdown [43] and intra-mPFC infusion of a BDNF-neutralizing antibody abolished the antidepressant actions of ketamine [295]. In addition, mice expressing the human BDNFVal66Met (rs6265) single nucleotide polymorphism, which are characterized by deficits in BDNF processing and activity-dependent BDNF release [296], show attenuated responses to ketamine [297] and scopolamine [298]. Similar dependence on BDNF-TrkB signaling has been observed for GLYX-13 [299] and (2R,6R)-HNK (Duman RS, unpublished data). In line with these data, patients with major depression carrying the Val66Met rs6265 allele (both Val/Met and Met/Met) showed less robust antidepressant response to ketamine compared with individuals with homozygous Val/Val (~ 20–24% of Met carriers showed an improvement vs. 40% of Val/Val carriers) [300].
Although classical antidepressants require several weeks of administration to induce BDNF-related changes, ketamine administration was reported to rapidly (within 30 min of administration) increase total BDNF protein levels [43, 301] and to enhance BDNF levels in synaptoneurosomal fractions 24 hours following administration [45] in the hippocampus of rodents. Similarly, (2R,6R)-HNK administration increases hippocampal synaptoneurosomal BDNF levels at 24 h post-injection in mice [45]. There is also considerable evidence showing increased BDNF levels following electroconvulsive shock in the hippocampus of rodents [237, 281, 302,303,304,305,306,307,308,309]. Finally, sub-chronic (5 days) administration of a 5-HT2C antagonist [239], as well as partial activation of the GABAA receptor (via inhibition of GLO1) [179], increase BDNF protein levels in the mPFC and/or hippocampus of mice. In addition, administration of ketamine, GluN2-NMDAR antagonists, mGluR2/3 antagonists, and electroconvulsive shock reversed a chronic stress-induced reduction in BDNF levels in the PFC [95, 198] and hippocampus [198, 310,311,312] of rodents, suggesting that BDNF induction could be considered as a marker of rapid antidepressant efficacy.
Short-term (6–48 h) sleep deprivation has been shown to both increase [303, 313] or decrease [314, 315] hippocampal BDNF levels in stress-naïve rats, and has been shown to restore decreased BDNF levels in the hippocampus following chronic stress [316]. Moreover, although scopolamine administration has been shown to exert its antidepressant actions via a mechanism requiring activity-dependent increased BDNF release [298], there are controversial results showing decreased BDNF levels following scopolamine administration [317,318,319,320,321,322].
Ketamine and other putative rapid antidepressant drugs also increase the phosphorylation (activation) of hippocampal and/or mPFC TrkB [43, 198, 298], suggesting a BDNF-TrkB-dependent mechanism of rapid antidepressant action. Notably, there is also evidence that isoflurane, an anesthetic drug, may possess relatively rapid antidepressant actions in treatment-resistant patients [323,324,325] and antidepressant behavioral responses in the learned helplessness and the novelty-suppressed feeding tests in rodents [326], plausibly via a TrkB-dependent mechanism [326]. Further studies are required to assess for additional mechanisms whereby rapid and/or sustained antidepressant actions of isoflurane exposure may converge with other rapid-acting antidepressants.
5.2.1 Eukaryotic Elongation Factor 2 (eEF2)
Increased BDNF signaling following ketamine administration has been proposed to depend on decreases in the spontaneous activation of postsynaptic NMDARs [43]. Under physiological conditions, NMDAR-dependent activation of eukaryotic elongation factor 2 kinase (eEF2K), which is involved in protein synthesis and synaptic plasticity [327], causes an inactivation (phosphorylation) of its substrate protein, eEF2 (Thr 56), leading to the blockade of the elongation phase of protein synthesis and thus inhibition of protein translation [328, 329]. Ketamine is proposed to block NMDAR-mediated spontaneous activation of eEF2K, thereby causing a de-phosphorylation of eEF2 and a consequent de-suppression of protein synthesis and enhancement of BDNF translation [43]. This hypothesis is supported by the finding that administration of eEF2K inhibitors induces antidepressant behavioral responses in mice using the 30-min forced-swim test [43].
Administration of (2R,6R)-HNK [45] also decreases phospho-eEF2 (Thr 56) levels in the hippocampus of mice 1 and 24 h after administration, suggesting that this pathway could be triggered independently from NMDAR inhibition. Sub-chronic administration of 5-HT2C antagonists or GLYX-13 also decreases phospho-eEF2 (Thr 56) levels in the mPFC [239] and reduces the chronic stress-induced enhancement of phospho-eEF2 (Thr 56) in the hippocampus of mice [330], respectively, further challenging NMDAR inhibition dependency for these downstream changes. Indeed, there are multiple mechanisms other than NMDAR inhibition that could be responsible for a de-phosphorylation of eEF2 [331,332,333,334]. Finally, 8 h of sleep deprivation causes a robust increase in phospho-eEF2 (Thr 56) levels in the PFC and hippocampus of rats [335]. Decreased phospho-eEF2 levels alone may not be sufficient for exerting rapid-acting antidepressant actions. Opal et al. demonstrated that sub-chronic (5-day) administration of citalopram did not exert antidepressant actions in mice, even though it significantly reduced phospho-eEF2 (Thr 56) levels in the mPFC [239].
5.2.2 Mechanistic Target of Rapamycin (mTOR)
Enhanced BDNF release and activation of TrkB trigger downstream pathways via an activation of the phosphatidylinositol 3-kinase, which in turn translocates Akt (protein kinase B) to the plasma membrane [336]. Tropomyosin receptor kinase B activation can also induce activation of the downstream mitogen-activated protein kinase/Erk signaling pathway. Both pathways promote protein synthesis through activation of the mechanistic target of rapamycin complex 1 (mTORC1) [337]. Among the mTOR-regulated proteins are several that regulate neurogenesis and dendrite spine growth via phosphorylation of the synaptic p70S6 kinase and suppression of 4E-binding proteins [338,339,340].
Mechanistic target of rapamycin complex 1 signaling has been implicated in rapid antidepressant actions. In particular, administration of ketamine [42, 52, 247, 341,342,343,344], mGluR2/3 antagonists [196], GluN2B-NMDAR antagonists [42], GLYX-13 [128, 330], scopolamine [85], 7-chlorokynurenic acid [97], and 5-HT2C antagonists [239] induces a fast-onset increase in levels of phospho-mTOR (Ser 2448), phospho-p70S6 kinase (Thr 389), and phospho-4E-binding protein 1 (Thr 37/46) in the hippocampus and/or mPFC of rodents. Enhanced mTORC1 signaling following administration of ketamine is transient [42], indicating that acute activation of mTORC1 and thus protein translation may transiently induce synaptic plasticity responsible for the prolonged effects of ketamine. Mechanistic target of rapamycin complex 1 activation was shown to be necessary for the behavioral antidepressant responses of ketamine, scopolamine, GLYX-13, and mGluR2/3 antagonists. Specifically, pre-treatment with the selective mTORC1 inhibitor rapamycin blocks ketamine-induced synaptic molecular changes [42], as well as the antidepressant actions of ketamine, scopolamine, Ro 25-6981, GLYX-13, and mGluR2/3 inhibition in rodents [42, 85, 128, 193, 345]. Importantly, AMPAR inhibition prior to ketamine administration not only blocks its antidepressant actions, but also blocks ketamine-induced actions on mTORC1 signaling [42]. There is evidence that long-term SSRI administration does not induce mTORC1 activation [42] (but see [239]), suggesting that mTORC1 is a point of convergence that is uniquely activated by rapid-acting antidepressants.
In addition to the direct activation through the BDNF/TrkB pathway, it is hypothesized that mTORC1 activation could also occur via alternate pathways. One alternate is upstream phosphorylation-dependent deactivation of glycogen synthase kinase-3 (GSK-3). Glycogen synthase kinase-3, which has two isoforms, α and β, with similar but not identical functions, has been extensively linked with the antidepressant actions of lithium [346, 347] and has been implicated in the rapid antidepressant actions of ketamine [51]. Ketamine administration increases the levels of phosphorylated GSK-3β (Ser 9) in the PFC and/or hippocampus in rodents [39, 51, 348]. Mice carrying a knock-in mutation of both α and β isoforms of GSK-3 that prevents their kinase activity do not show antidepressant behavioral responses to ketamine [51], and lack ketamine-induced upregulation of cell surface GluA1 in the hippocampus [349]. Moreover, when lithium (a non-selective GSK-3 inhibitor) is co-administered with ketamine at sub-effective doses, it induces an activation of the mTORC1 signaling pathway, phosphorylation of GSK-3, synaptogenesis, and greater antidepressant effects [39], indicating that a convergent mechanism between mTORC1 signaling and GSK-3 might be involved in the rapid antidepressant actions of ketamine. Similar to ketamine, a single electroconvulsive shock enhances phosphorylation of GSK-3β (Ser 9) in the PFC and/or hippocampus of rodents [350,351,352].
6 Conclusion
Elucidation of the neurobiological underpinnings of the rapid and persistent antidepressant actions of ketamine has been a major recent research focus in psychopharmacology, with the expectation that knowledge gained from such studies will lead to the development of novel pharmacotherapies for the effective rapid treatment of depression [353]. Here, we discussed clinical and pre-clinical findings demonstrating rapid-onset antidepressant actions of ketamine and other promising candidate drugs. We reviewed convergent mechanisms of actions underlying the induction of rapid antidepressant efficacy including NMDAR modulation, synaptic plasticity strengthening, and synaptogenesis, as well as the common downstream effector pathways such as AMPAR activation, enhanced BDNF-TrkB signaling, de-phosphorylation of eEF2, and activation of mTORC1. Pre-clinical and/or clinical findings suggest that other compounds, including scopolamine, (2R,6R)-HNK, GLYX-13, 4-chlorokynurenine, GluN2B-NMDAR antagonists, mGluR2/3 antagonists, and GABAA receptor negative allosteric modulators also possess fast-onset antidepressant efficacy (see Tables 1, 2).
It is important to better understand the convergent mechanisms underlying rapid antidepressant efficacy in preclinical models to maximize their antidepressant potency and ameliorate any undesirable side effects these drugs may currently display. Although NMDAR inhibition was long assumed to underlie the antidepressant actions of ketamine, recent evidence indicates that additional downstream mechanisms are likely to be involved. Indeed, it was recently shown that the (2S,6S;2R,6R)-HNK metabolite of ketamine is essential for its antidepressant actions and that (2R,6R)-HNK possesses robust antidepressant efficacy with low potency at the NMDAR [45, 107,108,109, 354].
A well-acknowledged point of convergence between distinct mechanistic hypotheses is the required activation of AMPARs for the emergence of rapid antidepressant actions. AMPA-preferring glutamate receptor activation promotes activation of downstream signaling pathways, including BDNF/TrkB signaling and activation of mTORC1, thereby promoting protein synthesis, neosynaptogenesis, and restoration of synaptic function in reward and mood-related circuits, where its impairment contributes to the symptoms of depression [355, 356]. The ultimate result of these processes is a sustained potentiation of excitatory synapses in cortico-mesolimbic brain circuits involved in the maintenance of mood and appropriate reactivity to stress [124].
Rapid-acting antidepressants hold a promising future for the effective treatment of depression. Although ketamine is increasingly being used as a treatment [357], there is not a single rapid-acting antidepressant medication that is approved for the treatment of major depression to date. (S)-ketamine and GLYX-13 are currently in phase III clinical trials for the treatment of depression. However, a significant amount of research remains to be performed to delineate the exact mechanisms responsible for the emergence of rapid antidepressant efficacy and to define the most efficacious dose regimens for achieving the desired clinical effects, with fewer side effects. Moreover, future clinical studies should aim to include measures that will indicate whether drugs are active at the proposed target in vivo (i.e., target engagement), to make definitive conclusions regarding the mechanism of action, and to properly interpret the relevance of negative findings. The identification of additional putative rapid-acting antidepressants in pre-clinical tests that lack ketamine-like side effects, including the metabolite of ketamine (2R,6R)-HNK, which does not possess NMDAR inhibition-mediated side effects in rodents, opens new paths for the treatment of depression. An important aspect for consideration and an area of future research is the tolerability and efficacy of these treatments following long-term administration. It is important to identify rapid-acting antidepressant medications that can be routinely administered to patients with depression and provide rapid and sustained relief of their mood symptoms without detrimental effects associated with long-term use. Additionally, work is needed to determine approaches that may extend the therapeutic effects of rapid-acting drugs such as ketamine and avoid either short- or long-term relapse.
References
Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289(23):3095–105.
Insel TR, Wang PS. The STAR*D trial: revealing the need for better treatments. Psychiatr Serv. 2009;60(11):1466–7.
Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Pychiatry. 2006;163(11):1905–17.
Gelenberg AJ, Chesen CL. How fast are antidepressants? J Clin Psychiatry. 2000;61(10):712–21.
Insel TR, Scolnick EM. Cure therapeutics and strategic prevention: raising the bar for mental health research. Mol Psychiatry. 2006;11(1):11–7.
Short B, Fong J, Galvez V, Shelker W, Loo CK. Side-effects associated with ketamine use in depression: a systematic review. Lancet Psychiatry. 2018;5(1):65–78.
Segman RH, Shapira B, Gorfine M, Lerer B. Onset and time course of antidepressant action: psychopharmacological implications of a controlled trial of electroconvulsive therapy. Psychopharmacology (Berl). 1995;119(4):440–8.
Husain MM, Rush AJ, Fink M, Knapp R, Petrides G, Rummans T, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65(4):485–91.
Kellner CH, Knapp RG, Petrides G, Rummans TA, Husain MM, Rasmussen K, et al. Continuation electroconvulsive therapy vs pharmacotherapy for relapse prevention in major depression: a multisite study from the Consortium for Research in Electroconvulsive Therapy (CORE). Arch Gen Psychiatry. 2006;63(12):1337–44.
Dierckx B, Heijnen WT, van den Broek WW, Birkenhager TK. Efficacy of electroconvulsive therapy in bipolar versus unipolar major depression: a meta-analysis. Bipolar Disord. 2012;14(2):146–50.
Post RM, Uhde TW, Rubinow DR, Huggins T. Differential time course of antidepressant effects after sleep deprivation, ECT, and carbamazepine: clinical and theoretical implications. Psychiatry Res. 1987;22(1):11–9.
Nutt DJ, Gleiter CH, Glue P. Neuropharmacological aspects of ECT: in search of the primary mechanism of action. Convuls Ther. 1989;5(3):250–60.
Nobler MS, Sackeim HA, Moeller JR, Prudic J, Petkova E, Waternaux C. Quantifying the speed of symptomatic improvement with electroconvulsive therapy: comparison of alternative statistical methods. Convuls Ther. 1997;13(4):208–21.
Houck W, Abonour R, Vance G, Einhorn LH. Secondary leukemias in refractory germ cell tumor patients undergoing autologous stem-cell transplantation using high-dose etoposide. J Clin Oncol. 2004;22(11):2155–8.
Wu JC, Bunney WE. The biological basis of an antidepressant response to sleep deprivation and relapse: review and hypothesis. Am J Psychiatry. 1990;147(1):14–21.
Wiegand M, Riemann D, Schreiber W, Lauer CJ, Berger M. Effect of morning and afternoon naps on mood after total sleep deprivation in patients with major depression. Biol Psychiatry. 1993;33(6):467–76.
Zarate CA Jr, Mathews DC, Furey ML. Human biomarkers of rapid antidepressant effects. Biol Psychiatry. 2013;73(12):1142–55.
Sinner B, Graf BM. Ketamine. Handb Exp Pharmacol. 2008;182:313–33.
Mion G. History of anaesthesia: the ketamine story: past, present and future. Eur J Anaesthesiol. 2017;34(9):571–5.
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351–4.
Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63(8):856–64.
Price RB, Iosifescu DV, Murrough JW, Chang LC, Al Jurdi RK, Iqbal SZ, et al. Effects of ketamine on explicit and implicit suicidal cognition: a randomized controlled trial in treatment-resistant depression. Depress Anxiety. 2014;31(4):335–43.
DiazGranados N, Ibrahim LA, Brutsche NE, Ameli R, Henter ID, Luckenbaugh DA, et al. Rapid resolution of suicidal ideation after a single infusion of an N-methyl-d-aspartate antagonist in patients with treatment-resistant major depressive disorder. J Clin Psychiatry. 2010;71(12):1605–11.
Lapidus KA, Levitch CF, Perez AM, Brallier JW, Parides MK, Soleimani L, et al. A randomized controlled trial of intranasal ketamine in major depressive disorder. Biol Psychiatry. 2014;76(12):970–6.
Newport DJ, Carpenter LL, McDonald WM, Potash JB, Tohen M, Nemeroff CB. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950–66.
McGirr A, Berlim MT, Bond DJ, Fleck MP, Yatham LN, Lam RW. A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychol Med. 2015;45(4):693–704.
Romeo B, Choucha W, Fossati P, Rotge JY. Meta-analysis of short- and mid-term efficacy of ketamine in unipolar and bipolar depression. Psychiatry Res. 2015;230(2):682–8.
Kishimoto T, Chawla JM, Hagi K, Zarate CA, Kane JM, Bauer M, et al. Single-dose infusion ketamine and non-ketamine N-methyl-d-aspartate receptor antagonists for unipolar and bipolar depression: a meta-analysis of efficacy, safety and time trajectories. Psychol Med. 2016;46(7):1459–72.
Wilkinson ST, Ballard ED, Bloch MH, Mathew SJ, Murrough JW, Feder A, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2017. https://doi.org/10.1176/appi.ajp.2017.17040472. (epub ahead of print).
Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3):199–214.
Sanacora G, Frye MA, McDonald W, Mathew SJ, Turner MS, Schatzberg AF, et al. A consensus statement on the use of ketamine in the treatment of mood disorders. JAMA Psychiatry. 2017;74(4):399–405.
Ramaker MJ, Dulawa SC. Identifying fast-onset antidepressants using rodent models. Mol Psychiatry. 2017;22(5):656–65.
Mion G, Villevieille T. Ketamine pharmacology: an update (pharmacodynamics and molecular aspects, recent findings). CNS Neurosci Ther. 2013;19(6):370–80.
Kohrs R, Durieux ME. Ketamine: teaching an old drug new tricks. Anesth Analg. 1998;87(5):1186–93.
Singh JB, Fedgchin M, Daly E, Xi L, Melman C, De Bruecker G, et al. Intravenous esketamine in adult treatment-resistant depression: a double-blind, double-randomization, placebo-controlled study. Biol Psychiatry. 2016;80(6):424–31.
Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134–42.
Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, Khalife S, et al. A randomized add-on trial of an N-methyl-d-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. 2010;67(8):793–802.
Zarate CA Jr, Brutsche NE, Ibrahim L, Franco-Chaves J, Diazgranados N, Cravchik A, et al. Replication of ketamine’s antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol Psychiatry. 2012;71(11):939–46.
Liu R-J, Fuchikami M, Dwyer JM, Lepack AE, Duman RS, Aghajanian GK. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology. 2013;38(11):2268–77.
Lally N, Nugent AC, Luckenbaugh DA, Ameli R, Roiser JP, Zarate CA. Anti-anhedonic effect of ketamine and its neural correlates in treatment-resistant bipolar depression. Transl Psychiatry. 2014;4(10):e469.
Ballard ED, Ionescu DF, Vande Voort JL, Niciu MJ, Richards EM, Luckenbaugh DA, et al. Improvement in suicidal ideation after ketamine infusion: relationship to reductions in depression and anxiety. J Psychiatr Res. 2014;58:161–6.
Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–64.
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475(7354):91–5.
Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev. 2012;64(2):238–58.
Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533(7604):481–6.
Zanos P, Bhat S, Terrillion CE, Smith RJ, Tonelli LH, Gould TD. Sex-dependent modulation of age-related cognitive decline by the L-type calcium channel gene Cacna1c (Cav 1.2). Eur J Neurosci. 2015;42(8):2499–507.
Maeng S, Zarate CA Jr, Du J, Schloesser RJ, McCammon J, Chen G, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63(4):349–52.
Belujon P, Grace AA. Restoring mood balance in depression: ketamine reverses deficit in dopamine-dependent synaptic plasticity. Biol Psychiatry. 2014;76(12):927–36.
Koike H, Iijima M, Chaki S. Involvement of the mammalian target of rapamycin signaling in the antidepressant-like effect of group II metabotropic glutamate receptor antagonists. Neuropharmacology. 2011;61(8):1419–23.
Zhang H, Xue W, Wu R, Gong T, Tao W, Zhou X, et al. Rapid antidepressant activity of ethanol extract of Gardenia jasminoides Ellis is associated with upregulation of BDNF expression in the hippocampus. Evid Based Complement Alternat Med. 2015;2015:761238.
Beurel E, Song L, Jope RS. Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry. 2011;16(11):1068–70.
Carrier N, Kabbaj M. Sex differences in the antidepressant-like effects of ketamine. Neuropharmacology. 2013;70:27–34.
Gideons ES, Kavalali ET, Monteggia LM. Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc Natl Acad Sci USA. 2014;111(23):8649–54.
Iijima M, Fukumoto K, Chaki S. Acute and sustained effects of a metabotropic glutamate 5 receptor antagonist in the novelty-suppressed feeding test. Behav Brain Res. 2012;235(2):287–92.
Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, et al. Glutamate N-methyl-d-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69(8):754–61.
Louderback KM, Wills TA, Muglia LJ, Winder DG. Knockdown of BNST GluN2B-containing NMDA receptors mimics the actions of ketamine on novelty-induced hypophagia. Transl Psychiatry. 2013;3(3):e331.
Burgdorf J, Zhang XL, Nicholson KL, Balster RL, Leander JD, Stanton PK, et al. GLYX-13, a NMDA receptor glycine-site functional partial agonist, induces antidepressant-like effects without ketamine-like side effects. Neuropsychopharmacology. 2013;38(5):729–42.
Yang B, Zhang JC, Han M, Yao W, Yang C, Ren Q, et al. Comparison of R-ketamine and rapastinel antidepressant effects in the social defeat stress model of depression. Psychopharmacology (Berl). 2016;233(19–20):3647–57.
Donahue RJ, Muschamp JW, Russo SJ, Nestler EJ, Carlezon WA Jr. Effects of striatal DeltaFosB overexpression and ketamine on social defeat stress-induced anhedonia in mice. Biol Psychiatry. 2014;76(7):550–8.
Zhang JC, Yao W, Dong C, Yang C, Ren Q, Ma M, et al. Comparison of ketamine, 7,8-dihydroxyflavone, and ANA-12 antidepressant effects in the social defeat stress model of depression. Psychopharmacology (Berl). 2015;232(23):4325–35.
Brachman RA, McGowan JC, Perusini JN, Lim SC, Pham TH, Faye C, et al. Ketamine as a prophylactic against stress-induced depressive-like behavior. Biol Psychiatry. 2016;79(9):776–86.
Gould TD, Georgiou P, Brenner LA, Brundin L, Can A, Courtet P, et al. Animal models to improve our understanding and treatment of suicidal behavior. Transl Psychiatry. 2017;7(4):e1092.
Daly EJ, Singh JB, Fedgchin M, Cooper K, Lim P, Shelton RC, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: A randomized clinical trial. JAMA Psychiatry. 2018;75(2):139–48.
Yang C, Shirayama Y, Zhang JC, Ren Q, Yao W, Ma M, et al. R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl Psychiatry. 2015;5:e632.
Fukumoto K, Toki H, Iijima M, Hashihayata T, J-i Yamaguchi, Hashimoto K, et al. Antidepressant potential of (R)-ketamine in rodent models: comparison with (S)-ketamine. J Pharmacol Exp Ther. 2017;361(1):9–16.
Zanos P, Gould TD. Intracellular signaling pathways involved in (S)- and (R)-ketamine antidepressant actions. Biol Psychiatry. 2018;83(1):2–4
Morgan CJ, Mofeez A, Brandner B, Bromley L, Curran HV. Ketamine impairs response inhibition and is positively reinforcing in healthy volunteers: a dose-response study. Psychopharmacology (Berl). 2004;172(3):298–308.
Malhotra AK, Pinals DA, Weingartner H, Sirocco K, Missar CD, Pickar D, et al. NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology. 1996;14(5):301–7.
Curran HV, Monaghan L. In and out of the K-hole: a comparison of the acute and residual effects of ketamine in frequent and infrequent ketamine users. Addiction. 2001;96(5):749–60.
Wolff K, Winstock AR. Ketamine : from medicine to misuse. CNS Drugs. 2006;20(3):199–218.
Burke RE. The relative selectivity of anticholinergic drugs for the M1 and M2 muscarinic receptor subtypes. Mov Disord. 1986;1(2):135–44.
Browne RG. Effects of antidepressants and anticholinergics in a mouse “behavioral despair” test. Eur J Pharmacol. 1979;58(3):331–4.
Betin C, DeFeudis FV, Blavet N, Clostre F. Further characterization of the behavioral despair test in mice: positive effects of convulsants. Physiol Behav. 1982;28(2):307–11.
Kasper S, Moises HW, Beckmann H. The anticholinergic biperiden in depressive disorders. Pharmacopsychiatria. 1981;14(6):195–8.
Gillin JC, Sutton L, Ruiz C, Darko D, Golshan S, Risch SC, et al. The effects of scopolamine on sleep and mood in depressed patients with a history of alcoholism and a normal comparison group. Biol Psychiatry. 1991;30(2):157–69.
Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry. 2006;63(10):1121–9.
Drevets WC, Furey ML. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010;67(5):432–8.
Furey ML, Khanna A, Hoffman EM, Drevets WC. Scopolamine produces larger antidepressant and antianxiety effects in women than in men. Neuropsychopharmacology. 2010;35(12):2479–88.
Howland RH. The antidepressant effects of anticholinergic drugs. J Psychosoc Nurs Ment Health Serv. 2009;47(6):17–20.
Gillin JC, Lauriello J, Kelsoe JR, Rapaport M, Golshan S, Kenny WM, et al. No antidepressant effect of biperiden compared with placebo in depression: a double-blind 6-week clinical trial. Psychiatry Res. 1995;58(2):99–105.
Navarria A, Wohleb ES, Voleti B, Ota KT, Dutheil S, Lepack AE, et al. Rapid antidepressant actions of scopolamine: Role of medial prefrontal cortex and M1-subtype muscarinic acetylcholine receptors. Neurobiol Dis. 2015;82:254–61.
Wohleb ES, Wu M, Gerhard DM, Taylor SR, Picciotto MR, Alreja M, et al. GABA interneurons mediate the rapid antidepressant-like effects of scopolamine. J Clin Invest. 2016;126(7):2482–94.
Geoffroy M, Scheel-Kruger J, Christensen AV. Effect of imipramine in the “learned helplessness” model of depression in rats is not mimicked by combinations of specific reuptake inhibitors and scopolamine. Psychopharmacology (Berl). 1990;101(3):371–5.
Anisman H, Remington G, Sklar LS. Effect of inescapable shock on subsequent escape performance: catecholaminergic and cholinergic mediation of response initiation and maintenance. Psychopharmacology (Berl). 1979;61(2):107–24.
Voleti B, Navarria A, Liu RJ, Banasr M, Li N, Terwilliger R, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74(10):742–9.
Witkin JM, Overshiner C, Li X, Catlow JT, Wishart GN, Schober DA, et al. M1 and m2 muscarinic receptor subtypes regulate antidepressant-like effects of the rapidly acting antidepressant scopolamine. J Pharmacol Exp Ther. 2014;351(2):448–56.
Martinowich K, Jimenez DV, Zarate CA Jr, Manji HK. Rapid antidepressant effects: moving right along. Mol Psychiatry. 2013;18(8):856–63.
Vyklicky V, Korinek M, Smejkalova T, Balik A, Krausova B, Kaniakova M, et al. Structure, function, and pharmacology of NMDA receptor channels. Physiol Res. 2014;63(Suppl. 1):S191–203.
Trullas R, Skolnick P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur J Pharmacol. 1990;185(1):1–10.
Paul IA, Nowak G, Layer RT, Popik P, Skolnick P. Adaptation of the N-methyl-d-aspartate receptor complex following chronic antidepressant treatments. J Pharmacol Exp Ther. 1994;269(1):95–102.
Nowak G, Li Y, Paul IA. Adaptation of cortical but not hippocampal NMDA receptors after chronic citalopram treatment. Eur J Pharmacol. 1996;295(1):75–85.
Murrough JW, Abdallah CG, Mathew SJ. Targeting glutamate signalling in depression: progress and prospects. Nat Rev Drug Discov. 2017;16(7):472–86.
Jimenez-Sanchez L, Campa L, Auberson YP, Adell A. The role of GluN2A and GluN2B subunits on the effects of NMDA receptor antagonists in modeling schizophrenia and treating refractory depression. Neuropsychopharmacology. 2014;39(11):2673–80.
Kiselycznyk C, Jury NJ, Halladay LR, Nakazawa K, Mishina M, Sprengel R, et al. NMDA receptor subunits and associated signaling molecules mediating antidepressant-related effects of NMDA-GluN2B antagonism. Behav Brain Res. 2015;287:89–95.
Li SX, Han Y, Xu LZ, Yuan K, Zhang RX, Sun CY, et al. Uncoupling DAPK1 from NMDA receptor GluN2B subunit exerts rapid antidepressant-like effects. Mol Psychiatry. 2017. https://doi.org/10.1038/mp.2017.85. (epub ahead of print).
Yang Y, Cui Y, Sang K, Dong Y, Ni Z, Ma S, et al. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature. 2018;554:317
Zhu W-L, Wang S-J, Liu M-M, Shi H-S, Zhang R-X, Liu J-F, et al. Glycine site N-methyl-d-aspartate receptor antagonist 7-CTKA produces rapid antidepressant-like effects in male rats. J Psychiatry Neurosci. 2013;38(5):306–16.
Liu B-B, Luo L, Liu X-L, Geng D, Liu Q, Yi L-T. 7-Chlorokynurenic acid (7-CTKA) produces rapid antidepressant-like effects: through regulating hippocampal microRNA expressions involved in TrkB-ERK/Akt signaling pathways in mice exposed to chronic unpredictable mild stress. Psychopharmacology. 2015;232(3):541–50.
Zhang JC, Li SX, Hashimoto K. R (−)-ketamine shows greater potency and longer lasting antidepressant effects than S (+)-ketamine. Pharmacol Biochem Behav. 2014;116:137–41.
Yang C, Qu Y, Fujita Y, Ren Q, Ma M, Dong C, et al. Possible role of the gut microbiota-brain axis in the antidepressant effects of (R)-ketamine in a social defeat stress model. Transl Psychiatry. 2017;7(12):1294.
Sarkar A, Kabbaj M. Sex differences in effects of ketamine on behavior, spine density, and synaptic proteins in socially isolated rats. Biol Psychiatry. 2016;80(6):448–56.
Can A, Zanos P, Moaddel R, Kang HJ, Dossou KSS, Wainer IW, et al. Effects of ketamine and ketamine metabolites on evoked striatal dopamine release, dopamine receptors, and monoamine transporters. J Pharmacol Exp Ther. 2016;359(1):159–70.
Zarate CA Jr, Brutsche N, Laje G, Luckenbaugh DA, Venkata SL, Ramamoorthy A, et al. Relationship of ketamine’s plasma metabolites with response, diagnosis, and side effects in major depression. Biol Psychiatry. 2012;72(4):331–8.
Pham TH, Defaix C, Xu X, Deng S-X, Fabresse N, Alvarez J-C, et al. Common neurotransmission recruited in recruited in (R,S)-ketamine and (2R,6R)-hydroxynorketamine-induced sustained antidepressant-like effects. Biol Psychiatry. 2017. S0006-3223(17)32130–3. https://doi.org/10.1016/j.biopsych.2017.10.020. (epub ahead of print).
Yang C, Qu Y, Abe M, Nozawa D, Chaki S, Hashimoto K. (R)-Ketamine shows greater potency and longer lasting antidepressant effects than its metabolite (2R,6R)-hydroxynorketamine. Biol Psychiatry. 2017;82(5):e43–4.
Shirayama Y, Hashimoto K. Lack of antidepressant effects of (2R,6R)-hydroxynorketamine in a rat learned helplessness model: comparison with (R)-ketamine. Int J Neuropsychopharmacol. 2018;21(1):84–8.
Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, et al. Reply to: effects of a ketamine metabolite on synaptic NMDAR function. Nature. 2017;546(7659):E4–5.
Suzuki K, Nosyreva E, Hunt KW, Kavalali ET, Monteggia LM. Effects of a ketamine metabolite on synaptic NMDAR function. Nature. 2017;546(7659):E1–3.
Morris PJ, Moaddel R, Zanos P, Moore CE, Gould T, Zarate CA, et al. Synthesis and N-methyl-d-aspartate (NMDA) receptor activity of ketamine metabolites. Org Lett. 2017;19(17):4572–5.
Zarate CA Jr, Singh JB, Quiroz JA, De Jesus G, Denicoff KK, Luckenbaugh DA, et al. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry. 2006;163(1):153–5.
Lenze EJ, Skidmore ER, Begley AE, Newcomer JW, Butters MA, Whyte EM. Memantine for late-life depression and apathy after a disabling medical event: a 12-week, double-blind placebo-controlled pilot study. Int J Geriatr Psychiatry. 2012;27(9):974–80.
Ferguson JM, Shingleton RN. An open-label, flexible-dose study of memantine in major depressive disorder. Clin Neuropharmacol. 2007;30(3):136–44.
Moaddel R, Abdrakhmanova G, Kozak J, Jozwiak K, Toll L, Jimenez L, et al. Sub-anesthetic concentrations of (R,S)-ketamine metabolites inhibit acetylcholine-evoked currents in alpha7 nicotinic acetylcholine receptors. Eur J Pharmacol. 2013;698(1–3):228–34.
Sanacora G, Smith MA, Pathak S, Su HL, Boeijinga PH, McCarthy DJ, et al. Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol Psychiatry. 2014;19(9):978–85.
Sanacora G, Johnson MR, Khan A, Atkinson SD, Riesenberg RR, Schronen JP, et al. Adjunctive lanicemine (AZD6765) in patients with major depressive disorder and history of inadequate response to antidepressants: a randomized, placebo-controlled study. Neuropsychopharmacology. 2017;42(4):844–53.
Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-d-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28(6):631–7.
Hashimoto K. Comments on “An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-d-aspartate antagonist, CP-101,606 in patients with treatment-refractory major depressive disorder”. J Clin Psychopharmacol. 2009;29(4):411–2 (author reply 2).
Hashimoto K, London ED. Further characterization of [3H]ifenprodil binding to sigma receptors in rat brain. Eur J Pharmacol. 1993;236(1):159–63.
Hashimoto K, Ishiwata K. Sigma receptor ligands: possible application as therapeutic drugs and as radiopharmaceuticals. Curr Pharm Des. 2006;12(30):3857–76.
Stahl SM. The sigma enigma: can sigma receptors provide a novel target for disorders of mood and cognition? J Clin Psychiatry. 2008;69(11):1673–4.
Hashimoto K. Sigma-1 receptors and selective serotonin reuptake inhibitors: clinical implications of their relationship. Cent Nerv Syst Agents Med Chem. 2009;9(3):197–204.
Ibrahim L, Diaz Granados N, Jolkovsky L, Brutsche N, Luckenbaugh DA, Herring WJ, et al. A randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J Clin Psychopharmacol. 2012;32(4):551–7.
Sanacora G. What are we learning from early-phase clinical trials with glutamate targeting medications for the treatment of major depressive disorder. JAMA Psychiatry. 2016;73(7):651–2.
Thompson SM, Kallarackal AJ, Kvarta MD, Van Dyke AM, LeGates TA, Cai X. An excitatory synapse hypothesis of depression. Trends Neurosci. 2015;38(5):279–94.
Zanos P, Gould TD. Mechanisms of ketamine action as an antidepressant. Mol Psychiatry. (in press).
Moskal JR, Kuo AG, Weiss C, Wood PL, Hanson AO, Kelso S, et al. GLYX-13: a monoclonal antibody-derived peptide that acts as an N-methyl-d-aspartate receptor modulator. Neuropharmacology. 2005;49(7):1077–87.
Preskorn S, Macaluso M, Mehra DO, Zammit G, Moskal JR, Burch RM, et al. Randomized proof of concept trial of GLYX-13, an N-methyl-d-aspartate receptor glycine site partial agonist, in major depressive disorder nonresponsive to a previous antidepressant agent. J Psychiatr Pract. 2015;21(2):140–9.
Liu RJ, Duman C, Kato T, Hare B, Lopresto D, Bang E, et al. GLYX-13 produces rapid antidepressant responses with key synaptic and behavioral effects distinct from ketamine. Neuropsychopharmacology. 2017;42(6):1231–42.
Chen L, Muhlhauser M, Yang CR. Glycine tranporter-1 blockade potentiates NMDA-mediated responses in rat prefrontal cortical neurons in vitro and in vivo. J Neurophysiol. 2003;89(2):691–703.
Huang CC, Wei IH, Huang CL, Chen KT, Tsai MH, Tsai P, et al. Inhibition of glycine transporter-I as a novel mechanism for the treatment of depression. Biol Psychiatry. 2013;74(10):734–41.
Heresco-Levy U, Gelfin G, Bloch B, Levin R, Edelman S, Javitt DC, et al. A randomized add-on trial of high-dose d-cycloserine for treatment-resistant depression. Int J Neuropsychopharmacol. 2013;16(3):501–6.
Wilhelm S, Buhlmann U, Tolin DF, Meunier SA, Pearlson GD, Reese HE, et al. Augmentation of behavior therapy with d-cycloserine for obsessive-compulsive disorder. Am J Psychiatry. 2008;165(3):335–41 (quiz 409).
Poleszak E, Wlaź P, Szewczyk B, Wlaź A, Kasperek R, Wróbel A, et al. A complex interaction between glycine/NMDA receptors and serotonergic/noradrenergic antidepressants in the forced swim test in mice. J Neural Transm. 2011;118(11):1535–46.
van Berckel BN, Lipsch C, Timp S, Gispen-de Wied C, Wynne H, van Ree JM, et al. Behavioral and neuroendocrine effects of the partial NMDA agonist d-cycloserine in healthy subjects. Neuropsychopharmacology. 1997;16(5):317–24.
Trullas R, Folio T, Young A, Miller R, Boje K, Skolnick P. 1-aminocyclopropanecarboxylates exhibit antidepressant and anxiolytic actions in animal models. Eur J Pharmacol. 1991;203(3):379–85.
Moskal JR, Burch R, Burgdorf JS, Kroes RA, Stanton PK, Disterhoft JF, et al. GLYX-13, an NMDA receptor glycine site functional partial agonist enhances cognition and produces antidepressant effects without the psychotomimetic side effects of NMDA receptor antagonists. Expert Opin Investig Drugs. 2014;23(2):243–54.
Rajagopal L, Burgdorf JS, Moskal JR, Meltzer HY. GLYX-13 (rapastinel) ameliorates subchronic phencyclidine- and ketamine-induced declarative memory deficits in mice. Behav Brain Res. 2016;15(299):105–10.
Shu Y, Hasenstaub A, McCormick DA. Turning on and off recurrent balanced cortical activity. Nature. 2003;423(6937):288–93.
Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5(10):793–807.
Williams SM, Goldman-Rakic PS, Leranth C. The synaptology of parvalbumin-immunoreactive neurons in the primate prefrontal cortex. J Comp Neurol. 1992;320(3):353–69.
Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378(6552):75–8.
Miles R, Toth K, Gulyas AI, Hajos N, Freund TF. Differences between somatic and dendritic inhibition in the hippocampus. Neuron. 1996;16(4):815–23.
Cardin JA, Carlen M, Meletis K, Knoblich U, Zhang F, Deisseroth K, et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459(7247):663–7.
Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459(7247):698–702.
de Lima AD, Morrison JH. Ultrastructural analysis of somatostatin-immunoreactive neurons and synapses in the temporal and occipital cortex of the macaque monkey. J Comp Neurol. 1989;283(2):212–27.
Kawaguchi Y, Kubota Y. Physiological and morphological identification of somatostatin- or vasoactive intestinal polypeptide-containing cells among GABAergic cell subtypes in rat frontal cortex. J Neurosci. 1996;16(8):2701–15.
Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17(8):2921–7.
Lorrain DS, Baccei CS, Bristow LJ, Anderson JJ, Varney MA. Effects of ketamine and N-methyl-d-aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience. 2003;117(3):697–706.
Chowdhury GM, Zhang J, Thomas M, Banasr M, Ma X, Pittman B, et al. Transiently increased glutamate cycling in rat PFC is associated with rapid onset of antidepressant-like effects. Mol Psychiatry. 2017;22(1):120–6.
Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27(43):11496–500.
Ling DS, Benardo LS. Recruitment of GABAA inhibition in rat neocortex is limited and not NMDA dependent. J Neurophysiol. 1995;74(6):2329–35.
Grunze HC, Rainnie DG, Hasselmo ME, Barkai E, Hearn EF, McCarley RW, et al. NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci. 1996;16(6):2034–43.
Hiyoshi T, Kambe D, Karasawa J, Chaki S. Involvement of glutamatergic and GABAergic transmission in MK-801-increased gamma band oscillation power in rat cortical electroencephalograms. Neuroscience. 2014;7(280):262–74.
Jones NC, Anderson P, Rind G, Sullivan C, van den Buuse M, O’Brien TJ. Effects of aberrant gamma frequency oscillations on prepulse inhibition. Int J Neuropsychopharmacol. 2014;17(10):1671–81.
Kocsis B. Differential role of NR2A and NR2B subunits in N-methyl-d-aspartate receptor antagonist-induced aberrant cortical gamma oscillations. Biol Psychiatry. 2012;71(11):987–95.
Kocsis B. State-dependent increase of cortical gamma activity during REM sleep after selective blockade of NR2B subunit containing NMDA receptors. Sleep. 2012;35(7):1011–6.
Sapkota K, Mao Z, Synowicki P, Lieber D, Liu M, Ikezu T, et al. GluN2D N-methyl-d-aspartate receptor subunit contribution to the stimulation of brain activity and gamma oscillations by ketamine: implications for schizophrenia. J Pharmacol Exp Ther. 2016;356(3):702–11.
Disney AA, Reynolds JH. Expression of m1-type muscarinic acetylcholine receptors by parvalbumin-immunoreactive neurons in the primary visual cortex: a comparative study of rat, guinea pig, ferret, macaque, and human. J Comp Neurol. 2014;522(5):986–1003.
McCormick DA, Prince DA. Two types of muscarinic response to acetylcholine in mammalian cortical neurons. Proc Nat Acad Sci USA. 1985;82(18):6344–8.
Amar M, Lucas-Meunier E, Baux G, Fossier P. Blockade of different muscarinic receptor subtypes changes the equilibrium between excitation and inhibition in rat visual cortex. Neuroscience. 2010;169(4):1610–20.
Towers SK, Gloveli T, Traub RD, Driver JE, Engel D, Fradley R, et al. Alpha 5 subunit-containing GABAA receptors affect the dynamic range of mouse hippocampal kainate-induced gamma frequency oscillations in vitro. J Physiol. 2004;559(Pt 3):721–8.
Fischell J, Van Dyke AM, Kvarta MD, LeGates TA, Thompson SM. Rapid antidepressant action and restoration of excitatory synaptic strength after chronic stress by negative modulators of alpha5-containing GABAA receptors. Neuropsychopharmacology. 2015;40(11):2499–509.
Carreno FR, Collins GT, Frazer A, Lodge DJ. Selective pharmacological augmentation of hippocampal activity produces a sustained antidepressant-like response without abuse-related or psychotomimetic effects. Int J Neuropsychopharmacol. 2017;20(6):504–9. https://doi.org/10.1093/ijnp/pyx003.
Zanos P, Nelson ME, Highland JN, Krimmel SR, Georgiou P, Gould TD, et al. A negative allosteric modulator for alpha5 subunit-containing GABA receptors exerts a rapid and persistent antidepressant-like action without the side effects of the NMDA receptor antagonist ketamine in mice. eNeuro. 2017;4(1). https://doi.org/10.1523/eneuro.0285-16.2017.
Xu NZ, Ernst M, Treven M, Cerne R, Wakulchik M, Li X, et al. Negative allosteric modulation of alpha 5-containing GABAA receptors engenders antidepressant-like effects and selectively prevents age-associated hyperactivity in tau-depositing mice. Psychopharmacology. 2018. https://doi.org/10.1007/s00213-018-4832-9
Atack JR, Maubach KA, Wafford KA, O’Connor D, Rodrigues AD, Evans DC, et al. In vitro and in vivo properties of 3-tert-butyl-7-(5-methylisoxazol-3-yl)-2-(1-methyl-1H-1,2,4-triazol-5-ylmethoxy)- pyrazolo[1,5-d]-[1,2,4]triazine (MRK-016), a GABAA receptor alpha5 subtype-selective inverse agonist. J Pharmacol Exp Ther. 2009;331(2):470–84.
Malherbe P, Sigel E, Baur R, Persohn E, Richards JG, Mohler H. Functional expression and sites of gene transcription of a novel alpha subunit of the GABAA receptor in rat brain. FEBS Lett. 1990;260(2):261–5.
Lingford-Hughes A, Hume SP, Feeney A, Hirani E, Osman S, Cunningham VJ, et al. Imaging the GABA-benzodiazepine receptor subtype containing the alpha5-subunit in vivo with [11C]Ro15 4513 positron emission tomography. J Cereb Blood Flow Metab. 2002;22(7):878–89.
Ren Z, Pribiag H, Jefferson SJ, Shorey M, Fuchs T, Stellwagen D, et al. Bidirectional homeostatic regulation of a depression-related brain state by gamma-aminobutyric acidergic deficits and ketamine treatment. Biol Psychiatry. 2016;80(6):457–68.
Shen Q, Lal R, Luellen BA, Earnheart JC, Andrews AM, Luscher B. γ-Aminobutyric acid-type A receptor deficits cause hypothalamic-pituitary-adrenal axis hyperactivity and antidepressant drug sensitivity reminiscent of melancholic forms of depression. Biol Psychiatry. 2010;68(6):512–20.
Smith KS, Rudolph U. Anxiety and depression: mouse genetics and pharmacological approaches to the role of GABA(A) receptor subtypes. Neuropharmacology. 2012;62(1):54–62.
Fuchs T, Jefferson SJ, Hooper A, Yee P-HP, Maguire J, Luscher B. Disinhibition of somatostatin-positive GABAergic interneurons results in an anxiolytic and antidepressant-like brain state. Mol Psychiatry. 2017;22(6):920–30.
Möhler H. The GABA system in anxiety and depression and its therapeutic potential. Neuropharmacology. 2012;62(1):42–53.
Kalueff AV, Nutt DJ. Role of GABA in anxiety and depression. Depress Anxiety. 2007;24(7):495–517.
Klumpers UM, Veltman DJ, Drent ML, Boellaard R, Comans EF, Meynen G, et al. Reduced parahippocampal and lateral temporal GABAA-[11C]flumazenil binding in major depression: preliminary results. Eur J Nucl Med Mol Imaging. 2010;37(3):565–74.
Sanacora G, Mason GF, Rothman DL, Krystal JH. Increased occipital cortex GABA concentrations in depressed patients after therapy with selective serotonin reuptake inhibitors. Am J Psychiatry. 2002;159(4):663–5.
Fava M, Schaefer K, Huang H, Wilson A, Iosifescu DV, Mischoulon D, et al. A post hoc analysis of the effect of nightly administration of eszopiclone and a selective serotonin reuptake inhibitor in patients with insomnia and anxious depression. J Clin Psychiatry. 2011;72(4):473–9.
Distler MG, Plant LD, Sokoloff G, Hawk AJ, Aneas I, Wuenschell GE, et al. Glyoxalase 1 increases anxiety by reducing GABAA receptor agonist methylglyoxal. J Clin Invest. 2012;122(6):2306–15.
McMurray KMJ, Ramaker MJ, Barkley-Levenson AM, Sidhu PS, Elkin PK, Reddy MK, et al. Identification of a novel, fast-acting GABAergic antidepressant. Mol Psychiatry. 2017. https://doi.org/10.1038/mp.2017.14. (epub ahead of print).
Piantadosi SC, French BJ, Poe MM, Timic T, Markovic BD, Pabba M, et al. Sex-dependent anti-stress effect of an alpha5 subunit containing GABAA receptor positive allosteric modulator. Front Pharmacol. 2016;7:446.
Bespalov AY, van Gaalen MM, Sukhotina IA, Wicke K, Mezler M, Schoemaker H, et al. Behavioral characterization of the mGlu group II/III receptor antagonist, LY-341495, in animal models of anxiety and depression. Eur J Pharmacol. 2008;592(1):96–102.
Chaki S, Yoshikawa R, Hirota S, Shimazaki T, Maeda M, Kawashima N, et al. MGS0039: a potent and selective group II metabotropic glutamate receptor antagonist with antidepressant-like activity. Neuropharmacology. 2004;46(4):457–67.
Witkin JM, Monn JA, Schoepp DD, Li X, Overshiner C, Mitchell SN, et al. The rapidly acting antidepressant ketamine and the mGlu2/3 receptor antagonist LY341495 rapidly engage dopaminergic mood circuits. J Pharmacol Exp Ther. 2016;358(1):71–82.
Yoshimizu T, Shimazaki T, Ito A, Chaki S. An mGluR2/3 antagonist, MGS0039, exerts antidepressant and anxiolytic effects in behavioral models in rats. Psychopharmacology. 2006;186(4):587.
Ohishi H, Ogawa-Meguro R, Shigemoto R, Kaneko T, Nakanishi S, Mizuno N. Immunohistochemical localization of metabotropic glutamate receptors, mGluR2 and mGluR3, in rat cerebellar cortex. Neuron. 1994;13(1):55–66.
Petralia RS, Wang YX, Niedzielski AS, Wenthold RJ. The metabotropic glutamate receptors, MGLUR2 and MGLUR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience. 1996;71(4):949–76.
Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, et al. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17(19):7503–22.
Chen Y-L, Huang C-C, Hsu K-S. Time-dependent reversal of long-term potentiation by low-frequency stimulation at the hippocampal mossy fiber–CA3 synapses. J Neurosci. 2001;21(11):3705–14.
Tzounopoulos T, Janz R, Südhof TC, Nicoll RA, Malenka RC. A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron. 1998;21(4):837–45.
Ohishi H, Shigemoto R, Nakanishi S, Mizuno N. Distribution of the mRNA for a metabotropic glutamate receptor (mGluR3) in the rat brain: an in situ hybridization study. J Comp Neurol. 1993;335(2):252–66.
Ciccarelli R, Sureda FX, Casabona G, Di Iorio P, Caruso A, Spinella F, et al. Opposite influence of the metabotropic glutamate receptor subtypes mGlu3 and -5 on astrocyte proliferation in culture. Glia. 1997;21(4):390–8.
Aronica E, Gorter JA, Ijlst-Keizers H, Rozemuller AJ, Yankaya B, Leenstra S, et al. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur J Neurosci. 2003;17(10):2106–18.
Dwyer JM, Lepack AE, Duman RS. mTOR activation is required for the antidepressant effects of mGluR(2)/(3) blockade. Int J Neuropsychopharmacol. 2012;15(4):429–34.
Koike H, Fukumoto K, Iijima M, Chaki S. Role of BDNF/TrkB signaling in antidepressant-like effects of a group II metabotropic glutamate receptor antagonist in animal models of depression. Behav Brain Res. 2013;1(238):48–52.
Fukumoto K, Iijima M, Chaki S. Serotonin-1A receptor stimulation mediates effects of a metabotropic glutamate 2/3 receptor antagonist, 2S-2-amino-2-(1S,2S-2-carboxycycloprop-1-yl)-3-(xanth-9-yl)propanoic acid (LY341495), and an N-methyl-d-aspartate receptor antagonist, ketamine, in the novelty-suppressed feeding test. Psychopharmacology (Berl). 2014;231(11):2291–8.
Dwyer JM, Lepack AE, Duman RS. mGluR2/3 blockade produces rapid and long-lasting reversal of anhedonia caused by chronic stress exposure. J Mol Psychiatry. 2013;1(1):15.
Ago Y, Yano K, Araki R, Hiramatsu N, Kita Y, Kawasaki T, et al. Metabotropic glutamate 2/3 receptor antagonists improve behavioral and prefrontal dopaminergic alterations in the chronic corticosterone-induced depression model in mice. Neuropharmacology. 2013;65:29–38.
Dong C, J-c Zhang, Yao W, Ren Q, Ma M, Yang C, et al. Rapid and sustained antidepressant action of the mGlu2/3 receptor antagonist MGS0039 in the social defeat stress model: comparison with ketamine. Int J Neuropsychopharmacol. 2017;20(3):228–36.
Umbricht D, Niggli M, Sanwald-Ducray P, Deptula D, Moore R, Grünbauer W, et al. Results of a double-blind placebo-controlled study of the antidepressant effects of the mGLU2 negative allosteric modulator RG1578. Eur Neuropsychopharmacol. 2015;25:S447.
Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci USA. 1992;89(10):4363–7.
Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232(2):331–56.
Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377(6545):115–8.
Alfarez DN, Joels M, Krugers HJ. Chronic unpredictable stress impairs long-term potentiation in rat hippocampal CA1 area and dentate gyrus in vitro. Eur J Neurosci. 2003;17(9):1928–34.
Joels M, Karst H, Alfarez D, Heine VM, Qin Y, van Riel E, et al. Effects of chronic stress on structure and cell function in rat hippocampus and hypothalamus. Stress. 2004;7(4):221–31.
Pavlides C, Nivon LG, McEwen BS. Effects of chronic stress on hippocampal long-term potentiation. Hippocampus. 2002;12(2):245–57.
Duman RS, Aghajanian GK, Sanacora G, Krystal JH. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med. 2016;22(3):238–49.
Wang M, Yang Y, Dong Z, Cao J, Xu L. NR2B-containing N-methyl-d-aspartate subtype glutamate receptors regulate the acute stress effect on hippocampal long-term potentiation/long-term depression in vivo. Neuroreport. 2006;17(12):1343–6.
Leuner B, Shors TJ. Stress, anxiety, and dendritic spines: what are the connections? Neuroscience. 2013;22(251):108–19.
Kallarackal AJ, Kvarta MD, Cammarata E, Jaberi L, Cai X, Bailey AM, et al. Chronic stress induces a selective decrease in AMPA receptor-mediated synaptic excitation at hippocampal temporoammonic-CA1 synapses. J Neurosci. 2013;33(40):15669–74.
Bliss TVP, Cooke SF. Long-term potentiation and long-term depression: a clinical perspective. Clinics. 2011;66(Suppl. 1):3–17.
Cai X, Kallarackal AJ, Kvarta MD, Goluskin S, Gaylor K, Bailey AM, et al. Local potentiation of excitatory synapses by serotonin and its alteration in rodent models of depression. Nat Neurosci. 2013;16(4):464–72.
Izumi Y, Zorumski CF. Metaplastic effects of subanesthetic ketamine on CA1 hippocampal function. Neuropharmacology. 2014;86:273–81.
Yao N, Skiteva O, Zhang X, Svenningsson P, Chergui K. Ketamine and its metabolite (2R,6R)-hydroxynorketamine induce lasting alterations in glutamatergic synaptic plasticity in the mesolimbic circuit. Mol Psychiatry. 2017. https://doi.org/10.1038/mp.2017.239. (epub ahead of print).
Moaddel R, Sanghvi M, Dossou KS, Ramamoorthy A, Green C, Bupp J, et al. The distribution and clearance of (2S,6S)-hydroxynorketamine, an active ketamine metabolite, in Wistar rats. Pharmacol Res Perspect. 2015;3(4):e00157.
Zhang XL, Sullivan JA, Moskal JR, Stanton PK. A NMDA receptor glycine site partial agonist, GLYX-13, simultaneously enhances LTP and reduces LTD at Schaffer collateral-CA1 synapses in hippocampus. Neuropharmacology. 2008;55(7):1238–50.
Burgdorf J, Zhang XL, Weiss C, Matthews E, Disterhoft JF, Stanton PK, et al. The N-methyl-d-aspartate receptor modulator GLYX-13 enhances learning and memory, in young adult and learning impaired aging rats. Neurobiol Aging. 2011;32(4):698–706.
Burgdorf J, Kroes RA, X-l Zhang, Gross AL, Schmidt M, Weiss C, et al. Rapastinel (GLYX-13) has therapeutic potential for the treatment of post-traumatic stress disorder: characterization of a NMDA receptor-mediated metaplasticity process in the medial prefrontal cortex of rats. Behav Brain Res. 2015;294:177–85.
Burgdorf J, X-l Zhang, Weiss C, Gross A, Boikess SR, Kroes RA, et al. The long-lasting antidepressant effects of rapastinel (GLYX-13) are associated with a metaplasticity process in the medial prefrontal cortex and hippocampus. Neuroscience. 2015;308:202–11.
Pöschel B, Wroblewska B, Heinemann U, Manahan-Vaughan D. The metabotropic glutamate receptor mGluR3 is critically required for hippocampal long-term depression and modulates long-term potentiation in the dentate gyrus of freely moving rats. Cereb Cortex. 2005;15(9):1414–23.
Hascup ER, Hascup KN, Stephens M, Pomerleau F, Huettl P, Gratton A, et al. Rapid microelectrode measurements and the origin and regulation of extracellular glutamate in rat prefrontal cortex. J Neurochem. 2010;115(6):1608–20.
Chana G, Landau S, Beasley C, Everall IP, Cotter D. Two-dimensional assessment of cytoarchitecture in the anterior cingulate cortex in major depressive disorder, bipolar disorder, and schizophrenia: evidence for decreased neuronal somal size and increased neuronal density. Biol Psychiatry. 2003;53(12):1086–98.
Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb Cortex. 2002;12(4):386–94.
Cotter D, Mackay D, Landau S, Kerwin R, Everall I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry. 2001;58(6):545–53.
Cotter D, Hudson L, Landau S. Evidence for orbitofrontal pathology in bipolar disorder and major depression, but not in schizophrenia. Bipolar Disord. 2005;7(4):358–69.
Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45(9):1085–98.
Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY, et al. Cellular changes in the postmortem hippocampus in major depression. Biol Psychiatry. 2004;56(9):640–50.
Magarinos AM, McEwen BS, Flugge G, Fuchs E. Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J Neurosci. 1996;16(10):3534–40.
Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531(1–2):225–31.
Watanabe Y, Gould E, McEwen BS. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 1992;588(2):341–5.
Cook SC, Wellman CL. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J Neurobiol. 2004;60(2):236–48.
Radley JJ, Sisti HM, Hao J, Rocher AB, McCall T, Hof PR, et al. Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience. 2004;125(1):1–6.
Wellman CL. Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J Neurobiol. 2001;49(3):245–53.
Goldwater DS, Pavlides C, Hunter RG, Bloss EB, Hof PR, McEwen BS, et al. Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience. 2009;164(2):798–808.
Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med. 2012;18(9):1413–7.
Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA, et al. The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry. 2009;14(8):764–73.
Cavalleri L, Merlo Pich E, Millan MJ, Chiamulera C, Kunath T, Spano PF, et al. Ketamine enhances structural plasticity in mouse mesencephalic and human iPSC-derived dopaminergic neurons via AMPAR-driven BDNF and mTOR signaling. Mol Psychiatry. 2017. https://doi.org/10.1038/mp.2017.241. (epub ahead of print).
Vaidya VA, Siuciak JA, Du F, Duman RS. Hippocampal mossy fiber sprouting induced by chronic electroconvulsive seizures. Neuroscience. 1999;89(1):157–66.
Acosta-Pena E, Camacho-Abrego I, Melgarejo-Gutierrez M, Flores G, Drucker-Colin R, Garcia-Garcia F. Sleep deprivation induces differential morphological changes in the hippocampus and prefrontal cortex in young and old rats. Synapse. 2015;69(1):15–25.
Opal MD, Klenotich SC, Morais M, Bessa J, Winkle J, Doukas D, et al. Serotonin 2C receptor antagonists induce fast-onset antidepressant effects. Mol Psychiatry. 2014;19(10):1106–14.
Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338(6103):68–72.
Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8(2):101–13.
Henley JM, Wilkinson KA. Synaptic AMPA receptor composition in development, plasticity and disease. Nat Rev Neurosci. 2016;17(6):337–50.
Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–43.
Koike H, Chaki S. Requirement of AMPA receptor stimulation for the sustained antidepressant activity of ketamine and LY341495 during the forced swim test in rats. Behav Brain Res. 2014;1(271):111–5.
Koike H, Iijima M, Chaki S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res. 2011;224(1):107–11.
Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B, et al. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology. 2013;38(9):1609–16.
Zhou W, Wang N, Yang C, Li XM, Zhou ZQ, Yang JJ. Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur Psychiatry. 2014;29(7):419–23.
Martin AE, Schober DA, Nikolayev A, Tolstikov VV, Anderson WH, Higgs RE, et al. Further evaluation of mechanisms associated with the antidepressant-like signature of scopolamine in mice. CNS Neurol Disord Drug Targets. 2017;16(4):492–500.
Karasawa J, Shimazaki T, Kawashima N, Chaki S. AMPA receptor stimulation mediates the antidepressant-like effect of a group II metabotropic glutamate receptor antagonist. Brain Res. 2005;1042(1):92–8.
Wolak M, Siwek A, Szewczyk B, Poleszak E, Pilc A, Popik P, et al. Involvement of NMDA and AMPA receptors in the antidepressant-like activity of antidepressant drugs in the forced swim test. Pharmacol Rep. 2013;65(4):991–7.
Whittington MA, Traub RD, Kopell N, Ermentrout B, Buhl EH. Inhibition-based rhythms: experimental and mathematical observations on network dynamics. Int J Psychophysiol. 2000;38(3):315–36.
Cunningham MO, Davies CH, Buhl EH, Kopell N, Whittington MA. Gamma oscillations induced by kainate receptor activation in the entorhinal cortex in vitro. J Neurosci. 2003;23(30):9761–9.
Muthukumaraswamy SD, Shaw AD, Jackson LE, Hall J, Moran R, Saxena N. Evidence that subanesthetic doses of ketamine cause sustained disruptions of NMDA and AMPA-mediated frontoparietal connectivity in humans. J Neurosci. 2015;35(33):11694–706.
Lazarewicz MT, Ehrlichman RS, Maxwell CR, Gandal MJ, Finkel LH, Siegel SJ. Ketamine modulates theta and gamma oscillations. J Cogn Neurosci. 2010;22(7):1452–64.
Ahnaou A, Ver Donck L, Drinkenburg WHIM. Blockade of the metabotropic glutamate (mGluR2) modulates arousal through vigilance states transitions: evidence from sleep–wake EEG in rodents. Behav Brain Res. 2014;270:56–67.
Buzsáki G, Wang X-J. Mechanisms of gamma oscillations. Annu Rev Neurosci. 2012;35:203–25.
Kumar S, Black SJ, Hultman R, Szabo ST, DeMaio KD, Du J, et al. Cortical control of affective networks. J Neurosci. 2013;33(3):1116–29.
Fuchikami M, Thomas A, Liu R, Wohleb ES, Land BB, DiLeone RJ, et al. Optogenetic stimulation of infralimbic PFC reproduces ketamine’s rapid and sustained antidepressant actions. Proc Nat Acad Sci. 2015;112(26):8106–11.
Nosyreva E, Szabla K, Autry AE, Ryazanov AG, Monteggia LM, Kavalali ET. Acute suppression of spontaneous neurotransmission drives synaptic potentiation. J Neurosci. 2013;33(16):6990–7002.
Bjorkholm C, Jardemark K, Schilstrom B, Svensson TH. Ketamine-like effects of a combination of olanzapine and fluoxetine on AMPA and NMDA receptor-mediated transmission in the medial prefrontal cortex of the rat. Eur Neuropsychopharmacol. 2015;25(10):1842–7.
El Iskandrani KS, Oosterhof CA, El Mansari M, Blier P. Impact of subanesthetic doses of ketamine on AMPA-mediated responses in rats: an in vivo electrophysiological study on monoaminergic and glutamatergic neurons. J Psychopharmacol. 2015;29(7):792–801.
Xie M, Li C, He C, Yang L, Tan G, Yan J, et al. Short-term sleep deprivation disrupts the molecular composition of ionotropic glutamate receptors in entorhinal cortex and impairs the rat spatial reference memory. Behav Brain Res. 2016;300:70–6.
Xie M, Yan J, He C, Yang L, Tan G, Li C, et al. Short-term sleep deprivation impairs spatial working memory and modulates expression levels of ionotropic glutamate receptor subunits in hippocampus. Behav Brain Res. 2015;286:64–70.
Zhang K, Xu T, Yuan Z, Wei Z, Yamaki VN, Huang M, et al. Essential roles of AMPA receptor GluA1 phosphorylation and presynaptic HCN channels in fast-acting antidepressant responses of ketamine. Sci Signal. 2016;9(458):ra123.
Lindholm JS, Autio H, Vesa L, Antila H, Lindemann L, Hoener MC, et al. The antidepressant-like effects of glutamatergic drugs ketamine and AMPA receptor potentiator LY 451646 are preserved in bdnf(+)/(−) heterozygous null mice. Neuropharmacology. 2012;62(1):391–7.
Schechter LE, Ring RH, Beyer CE, Hughes ZA, Khawaja X, Malberg JE, et al. Innovative approaches for the development of antidepressant drugs: current and future strategies. NeuroRx. 2005;2(4):590–611.
Li X, Tizzano JP, Griffey K, Clay M, Lindstrom T, Skolnick P. Antidepressant-like actions of an AMPA receptor potentiator (LY392098). Neuropharmacology. 2001;40(8):1028–33.
Akinfiresoye L, Tizabi Y. Antidepressant effects of AMPA and ketamine combination: role of hippocampal BDNF, synapsin, and mTOR. Psychopharmacology (Berl). 2013;230(2):291–8.
Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274(5290):1133–8.
Mamounas LA, Altar CA, Blue ME, Kaplan DR, Tessarollo L, Lyons WE. BDNF promotes the regenerative sprouting, but not survival, of injured serotonergic axons in the adult rat brain. J Neurosci. 2000;20(2):771–82.
Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2(1):24–32.
Pattwell SS, Bath KG, Perez-Castro R, Lee FS, Chao MV, Ninan I. The BDNF Val66Met polymorphism impairs synaptic transmission and plasticity in the infralimbic medial prefrontal cortex. J Neurosci. 2012;32(7):2410–21.
Bocchio-Chiavetto L, Bagnardi V, Zanardini R, Molteni R, Nielsen MG, Placentino A, et al. Serum and plasma BDNF levels in major depression: a replication study and meta-analyses. World J Biol Psychiatry. 2010;11(6):763–73.
Molendijk ML, Bus BA, Spinhoven P, Penninx BW, Kenis G, Prickaerts J, et al. Serum levels of brain-derived neurotrophic factor in major depressive disorder: state-trait issues, clinical features and pharmacological treatment. Mol Psychiatry. 2011;16(11):1088–95.
Karlovic D, Serretti A, Jevtovic S, Vrkic N, Seric V, Peles AM. Diagnostic accuracy of serum brain derived neurotrophic factor concentration in antidepressant naive patients with first major depression episode. J Psychiatr Res. 2013;47(2):162–7.
Yoshida T, Ishikawa M, Niitsu T, Nakazato M, Watanabe H, Shiraishi T, et al. Decreased serum levels of mature brain-derived neurotrophic factor (BDNF), but not its precursor proBDNF, in patients with major depressive disorder. PLoS One. 2012;7(8):e42676.
Castren E, Antila H. Neuronal plasticity and neurotrophic factors in drug responses. Mol Psychiatry. 2017;22(8):1085–95.
Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54(7):597–606.
Manji HK, Moore GJ, Rajkowska G, Chen G. Neuroplasticity and cellular resilience in mood disorders. Mol Psychiatry. 2000;5(6):578–93.
Castren E, Voikar V, Rantamaki T. Role of neurotrophic factors in depression. Curr Opin Pharmacol. 2007;7(1):18–21.
Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15(11):7539–47.
Shimizu E, Hashimoto K, Okamura N, Koike K, Komatsu N, Kumakiri C, et al. Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol Psychiatry. 2003;54(1):70–5.
Bocchio-Chiavetto L, Zanardini R, Bortolomasi M, Abate M, Segala M, Giacopuzzi M, et al. Electroconvulsive therapy (ECT) increases serum brain derived neurotrophic factor (BDNF) in drug resistant depressed patients. Eur Neuropsychopharmacol. 2006;16(8):620–4.
Adachi M, Barrot M, Autry AE, Theobald D, Monteggia LM. Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol Psychiatry. 2008;63(7):642–9.
Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S, et al. Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry. 2007;61(2):187–97.
Koponen E, Rantamaki T, Voikar V, Saarelainen T, MacDonald E, Castren E. Enhanced BDNF signaling is associated with an antidepressant-like behavioral response and changes in brain monoamines. Cell Mol Neurobiol. 2005;25(6):973–80.
Hoshaw BA, Malberg JE, Lucki I. Central administration of IGF-I and BDNF leads to long-lasting antidepressant-like effects. Brain Res. 2005;1037(1–2):204–8.
Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22(8):3251–61.
Schmidt HD, Duman RS. Peripheral BDNF produces antidepressant-like effects in cellular and behavioral models. Neuropsychopharmacology. 2010;35(12):2378–91.
Taliaz D, Loya A, Gersner R, Haramati S, Chen A, Zangen A. Resilience to chronic stress is mediated by hippocampal brain-derived neurotrophic factor. J Neurosci. 2011;31(12):4475–83.
Rantamaki T, Hendolin P, Kankaanpaa A, Mijatovic J, Piepponen P, Domenici E, et al. Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cgamma signaling pathways in mouse brain. Neuropsychopharmacology. 2007;32(10):2152–62.
Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald E, et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci. 2003;23(1):349–57.
Groves JO. Is it time to reassess the BDNF hypothesis of depression? Mol Psychiatry. 2007;12(12):1079–88.
Wook Koo J, Labonte B, Engmann O, Calipari ES, Juarez B, Lorsch Z, et al. Essential role of mesolimbic brain-derived neurotrophic factor in chronic social stress-induced depressive behaviors. Biol Psychiatry. 2016;80(6):469–78.
Lepack AE, Fuchikami M, Dwyer JM, Banasr M, Duman RS. BDNF release is required for the behavioral actions of ketamine. Int J Neuropsychopharmacol. 2014;18(1). https://doi.org/10.1093/ijnp/pyu033.
Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314(5796):140–3.
Liu RJ, Lee FS, Li XY, Bambico F, Duman RS, Aghajanian GK. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry. 2012;71(11):996–1005.
Ghosal S, Bang E, Yue W, Hare BD, Lepack AE, Girgenti MJ, et al. Activity-dependent BDNF release is required for the rapid antidepressant actions of scopolamine. Biol Psychiatry. 2018;83(1):29–37.
Kato T, Fogaca MV, Deyama S, Li X-Y, Fukumoto K, Duman RS. BDNF release and signaling are required for the antidepressant actions of GLYX-13. Mol Psychiatry. 2017. https://doi.org/10.1038/mp.2017.220. (epub ahead of print).
Laje G, Lally N, Mathews D, Brutsche N, Chemerinski A, Akula N, et al. Brain-derived neurotrophic factor Val66Met polymorphism and antidepressant efficacy of ketamine in depressed patients. Biol Psychiatry. 2012;72(11):e27–8.
Garcia LS, Comim CM, Valvassori SS, Reus GZ, Barbosa LM, Andreazza AC, et al. Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):140–4.
Newton SS, Collier EF, Hunsberger J, Adams D, Terwilliger R, Selvanayagam E, et al. Gene profile of electroconvulsive seizures: induction of neurotrophic and angiogenic factors. J Neurosci. 2003;23(34):10841–51.
Conti B, Maier R, Barr AM, Morale MC, Lu X, Sanna PP, et al. Region-specific transcriptional changes following the three antidepressant treatments electro convulsive therapy, sleep deprivation and fluoxetine. Mol Psychiatry. 2007;12(2):167–89.
Altar CA, Whitehead RE, Chen R, Wortwein G, Madsen TM. Effects of electroconvulsive seizures and antidepressant drugs on brain-derived neurotrophic factor protein in rat brain. Biol Psychiatry. 2003;54(7):703–9.
Sartorius A, Hellweg R, Litzke J, Vogt M, Dormann C, Vollmayr B, et al. Correlations and discrepancies between serum and brain tissue levels of neurotrophins after electroconvulsive treatment in rats. Pharmacopsychiatry. 2009;42(6):270–6.
Balu DT, Hoshaw BA, Malberg JE, Rosenzweig-Lipson S, Schechter LE, Lucki I. Differential regulation of central BDNF protein levels by antidepressant and non-antidepressant drug treatments. Brain Res. 2008;23(1211):37–43.
Li B, Suemaru K, Cui R, Araki H. Repeated electroconvulsive stimuli have long-lasting effects on hippocampal BDNF and decrease immobility time in the rat forced swim test. Life Sci. 2007;80(16):1539–43.
Angelucci F, Aloe L, Jimenez-Vasquez P, Mathe AA. Electroconvulsive stimuli alter the regional concentrations of nerve growth factor, brain-derived neurotrophic factor, and glial cell line-derived neurotrophic factor in adult rat brain. J ECT. 2002;18(3):138–43.
Chen AC, Shin KH, Duman RS, Sanacora G. ECS-Induced mossy fiber sprouting and BDNF expression are attenuated by ketamine pretreatment. J ECT. 2001;17(1):27–32.
Luo J, Min S, Wei K, Cao J, Wang B, Li P, et al. Behavioral and molecular responses to electroconvulsive shock differ between genetic and environmental rat models of depression. Psychiatry Res. 2015;226(2–3):451–60.
Gersner R, Toth E, Isserles M, Zangen A. Site-specific antidepressant effects of repeated subconvulsive electrical stimulation: potential role of brain-derived neurotrophic factor. Biol Psychiatry. 2010;67(2):125–32.
Vollmayr B, Faust H, Lewicka S, Henn FA. Brain-derived-neurotrophic-factor (BDNF) stress response in rats bred for learned helplessness. Mol Psychiatry. 2001;6(4):471–4.
Fujihara H, Sei H, Morita Y, Ueta Y, Morita K. Short-term sleep disturbance enhances brain-derived neurotrophic factor gene expression in rat hippocampus by acting as internal stressor. J Mol Neurosci. 2003;21(3):223–32.
Guo L, Guo Z, Luo X, Liang R, Yang S, Ren H, et al. Phosphodiesterase 10A inhibition attenuates sleep deprivation-induced deficits in long-term fear memory. Neurosci Lett. 2016;635:44–50.
Guzman-Marin R, Ying Z, Suntsova N, Methippara M, Bashir T, Szymusiak R, et al. Suppression of hippocampal plasticity-related gene expression by sleep deprivation in rats. J Physiol. 2006;575(Pt 3):807–19.
Jiang Y, Zhu J. Effects of sleep deprivation on behaviors and abnormal hippocampal BDNF/miR-10B expression in rats with chronic stress depression. Int J Clin Exp Pathol. 2015;8(1):586–93.
Konar A, Shah N, Singh R, Saxena N, Kaul SC, Wadhwa R, et al. Protective role of Ashwagandha leaf extract and its component withanone on scopolamine-induced changes in the brain and brain-derived cells. PLoS One. 2011;6(11):e27265.
Lee B, Sur B, Shim J, Hahm DH, Lee H. Acupuncture stimulation improves scopolamine-induced cognitive impairment via activation of cholinergic system and regulation of BDNF and CREB expressions in rats. BMC Complement Altern Med. 2014;17(14):338.
Chen W, Cheng X, Chen J, Yi X, Nie D, Sun X, et al. Lycium barbarum polysaccharides prevent memory and neurogenesis impairments in scopolamine-treated rats. PLoS One. 2014;9(2):e88076.
Kotani S, Yamauchi T, Teramoto T, Ogura H. Donepezil, an acetylcholinesterase inhibitor, enhances adult hippocampal neurogenesis. Chem Biol Interact. 2008;175(1–3):227–30.
Shi Z, Chen L, Li S, Chen S, Sun X, Sun L, et al. Chronic scopolamine-injection-induced cognitive deficit on reward-directed instrumental learning in rat is associated with CREB signaling activity in the cerebral cortex and dorsal hippocampus. Psychopharmacology (Berl). 2013;230(2):245–60.
Heo YM, Shin MS, Kim SH, Kim TW, Baek SB, Baek SS. Treadmill exercise ameliorates disturbance of spatial learning ability in scopolamine-induced amnesia rats. J Exerc Rehabil. 2014;10(3):155–61.
Weeks HR 3rd, Tadler SC, Smith KW, Iacob E, Saccoman M, White AT, et al. Antidepressant and neurocognitive effects of isoflurane anesthesia versus electroconvulsive therapy in refractory depression. PLoS One. 2013;8(7):e69809.
Langer G, Neumark J, Koinig G, Graf M, Schonbeck G. Rapid psychotherapeutic effects of anesthesia with isoflurane (ES narcotherapy) in treatment-refractory depressed patients. Neuropsychobiology. 1985;14(3):118–20.
Langer G, Karazman R, Neumark J, Saletu B, Schonbeck G, Grunberger J, et al. Isoflurane narcotherapy in depressive patients refractory to conventional antidepressant drug treatment. A double-blind comparison with electroconvulsive treatment. Neuropsychobiology. 1995;31(4):182–94.
Antila H, Ryazantseva M, Popova D, Sipila P, Guirado R, Kohtala S, et al. Isoflurane produces antidepressant effects and induces TrkB signaling in rodents. Sci Rep. 2017;7(1):7811.
Taha E, Gildish I, Gal-Ben-Ari S, Rosenblum K. The role of eEF2 pathway in learning and synaptic plasticity. Neurobiol Learn Mem. 2013;105:100–6.
Chotiner JK, Khorasani H, Nairn AC, O’Dell TJ, Watson JB. Adenylyl cyclase-dependent form of chemical long-term potentiation triggers translational regulation at the elongation step. Neuroscience. 2003;116(3):743–52.
Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, et al. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59(1):70–83.
Lu Y, Wang C, Xue Z, Li C, Zhang J, Zhao X, et al. PI3K/AKT/mTOR signaling-mediated neuropeptide VGF in the hippocampus of mice is involved in the rapid onset antidepressant-like effects of GLYX-13. Int J Neuropsychopharmacol. 2014;18(5). https://doi.org/10.1093/ijnp/pyu110.
Hizli AA, Chi Y, Swanger J, Carter JH, Liao Y, Welcker M, et al. Phosphorylation of eukaryotic elongation factor 2 (eEF2) by cyclin A-cyclin-dependent kinase 2 regulates its inhibition by eEF2 kinase. Mol Cell Biol. 2013;33(3):596–604.
Knebel A, Morrice N, Cohen P. A novel method to identify protein kinase substrates: eEF2 kinase is phosphorylated and inhibited by SAPK4/p38delta. EMBO J. 2001;20(16):4360–9.
Redpath NT, Foulstone EJ, Proud CG. Regulation of translation elongation factor-2 by insulin via a rapamycin-sensitive signalling pathway. EMBO J. 1996;15(9):2291–7.
Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001;20(16):4370–9.
Grønli J, Dagestad G, Milde AM, Murison R, Bramham CR. Post-transcriptional effects and interactions between chronic mild stress and acute sleep deprivation: regulation of translation factor and cytoplasmic polyadenylation element-binding protein phosphorylation. Behav Brain Res. 2012;235(2):251–62.
Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361(1473):1545–64.
Yoshii A, Constantine-Paton M. Post-synaptic BDNF-TrkB signaling in synapse maturation, plasticity and disease. Dev Neurobiol. 2010;70(5):304–22.
Duman RS, Li N, Liu RJ, Duric V, Aghajanian G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62(1):35–41.
Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–45.
Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33(2):67–75.
Paul RK, Singh NS, Khadeer M, Moaddel R, Sanghvi M, Green CE, et al. (R,S)-Ketamine metabolites (R,S)-norketamine and (2S,6S)-hydroxynorketamine increase the mammalian target of rapamycin (mTOR) function. Anesthesiology. 2014;121(1):149–59.
Miller OH, Yang L, Wang CC, Hargroder EA, Zhang Y, Delpire E, et al. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. Elife. 2014;23(3):e03581.
Yang C, Hu YM, Zhou ZQ, Zhang GF, Yang JJ. Acute administration of ketamine in rats increases hippocampal BDNF and mTOR levels during forced swimming test. Ups J Med Sci. 2013;118(1):3–8.
Zhang K, Yamaki VN, Wei Z, Zheng Y, Cai X. Differential regulation of GluA1 expression by ketamine and memantine. Behav Brain Res. 2017;1(316):152–9.
Holubova K, Kleteckova L, Skurlova M, Ricny J, Stuchlik A, Vales K. Rapamycin blocks the antidepressant effect of ketamine in task-dependent manner. Psychopharmacology (Berl). 2016;233(11):2077–97.
Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015;148:114–31.
Can A, Schulze TG, Gould TD. Molecular actions and clinical pharmacogenetics of lithium therapy. Pharmacol Biochem Behav. 2014;123:3–16.
Zhou W, Dong L, Wang N, Shi JY, Yang JJ, Zuo ZY, et al. Akt mediates GSK-3beta phosphorylation in the rat prefrontal cortex during the process of ketamine exerting rapid antidepressant actions. Neuroimmunomodulation. 2014;21(4):183–8.
Beurel E, Grieco SF, Amadei C, Downey K, Jope RS. Ketamine-induced inhibition of glycogen synthase kinase-3 contributes to the augmentation of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor signaling. Bipolar Disord. 2016;18(6):473–80.
Basar K, Eren-Kocak E, Ozdemir H, Ertugrul A. Effects of acute and chronic electroconvulsive shocks on glycogen synthase kinase 3β level and phosphorylation in mice. J ECT. 2013;29(4):265–70.
Roh MS, Kang UG, Shin SY, Lee YH, Jung HY, Juhnn YS, et al. Biphasic changes in the Ser-9 phosphorylation of glycogen synthase kinase-3beta after electroconvulsive shock in the rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(1):1–5.
Kang UG, Roh MS, Jung JR, Shin SY, Lee YH, Park JB, et al. Activation of protein kinase B (Akt) signaling after electroconvulsive shock in the rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(1):41–4.
Daly EJ, Singh JB, Fedgchin M, Cooper K, Lim P, Shelton RC, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: A randomized clinical trial. JAMA Psychiatry. 2018; 75(2):139–48
Zanos P, Moaddel R, Morris PJ, Wainer IW, Albuquerque EX, Thompson SM, et al. Reply to: Antidepressant actions of ketamine versus hydroxynorketamine. Biol Psychiatry. 2017;81(8):e69–71.
Hare BD, Ghosal S, Duman RS. Rapid acting antidepressants in chronic stress models: molecular and cellular mechanisms. Chron Stress (Thousand Oaks). 2017;1. https://doi.org/10.1177/2470547017697317. (epub 2017 Apr 10).
Wohleb ES, Gerhard D, Thomas A, Duman RS. Molecular and cellular mechanisms of rapid-acting antidepressants ketamine and scopolamine. Curr Neuropharmacol. 2017;15(1):11–20.
Wilkinson ST, Toprak M, Turner MS, Levine SP, Katz RB, Sanacora G. A survey of the clinical, off-label use of ketamine as a treatment for psychiatric disorders. Am J Psychiatry. 2017;174(7):695–6.
Sos P, Klirova M, Novak T, Kohutova B, Horacek J, Palenicek T. Relationship of ketamine’s antidepressant and psychotomimetic effects in unipolar depression. Neuro Endocrinol Lett. 2013;34(4):287–93.
Hu YD, Xiang YT, Fang JX, Zu S, Sha S, Shi H, et al. Single i.v. ketamine augmentation of newly initiated escitalopram for major depression: results from a randomized, placebo-controlled 4-week study. Psychol Med. 2016;46(3):623–35.
Singh JB, Fedgchin M, Daly EJ, De Boer P, Cooper K, Lim P, et al. A double-blind, randomized, placebo-controlled, dose-frequency study of intravenous ketamine in patients with treatment-resistant depression. Am J Psychiatry. 2016;173(8):816–26.
Lieberman JA, Papadakis K, Csernansky J, Litman R, Volavka J, Jia XD, et al. A randomized, placebo-controlled study of memantine as adjunctive treatment in patients with schizophrenia. Neuropsychopharmacology. 2009;34(5):1322–9.
Smith EG, Deligiannidis KM, Ulbricht CM, Landolin CS, Patel JK, Rothschild AJ. Antidepressant augmentation using the NMDA-antagonist memantine: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2013;74(10):966–73.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by a National Institutes of Health Grant (MH107615) and a Harrington Discovery Institute Scholar-Innovator Grant to Todd D. Gould, and a National Institutes of Health Grant (MH086828) to Scott M. Thompson.
Conflict of interest
Todd D. Gould has received consulting fees from Janssen Pharmaceuticals, and research funding from Janssen Pharmaceuticals and Roche Pharmaceuticals during the preceding 3 years. Ronald S. Duman has received consulting fees from Janssen Pharmaceuticals and Taisho, and research funding from Janssen Pharmaceuticals and Taisho, Allergan, Naurex, Navitor, and Relmada during the preceding 3 years. Carlos A. Zarate Jr. is listed as a co-inventor on a patent for the use of ketamine in major depression and suicidal ideation. Panos Zanos, Carlos A. Zarate Jr., and Todd D. Gould are listed as co-authors in patent applications related to the pharmacology and use of (2S,6S)- and (2R,6R)-hydroxynorketamine in the treatment of depression, anxiety, anhedonia, suicidal ideation, and post-traumatic stress disorders. Scott M. Thompson is listed as a co-inventor on a patent application for the use of negative allosteric modulators of GABAA receptors containing alpha 5 subunits as fast-acting antidepressants.
Rights and permissions
About this article

Cite this article
Zanos, P., Thompson, S.M., Duman, R.S. et al. Convergent Mechanisms Underlying Rapid Antidepressant Action. CNS Drugs 32, 197–227 (2018). https://doi.org/10.1007/s40263-018-0492-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-018-0492-x