
Abstract
Background
Tacrolimus has a narrow therapeutic range and requires dose adjustment, usually based on the trough blood concentration but preferably on the area under the concentration–time curve over 12 h post-dose (AUC0–12h). The single-arm, multicentre, clinical study IMPAKT aimed: (i) to develop, in de novo kidney transplant recipients, pharmacokinetic models and maximum a-posteriori Bayesian estimators for a generic, immediate-release, oral formulation of tacrolimus to estimate tacrolimus AUC0–12h at different post-transplant periods using a limited sampling strategy, and considering the CYP3A5*3 polymorphism as a covariate and (ii) to compare the performance of these Bayesian estimators to those previously developed for the original formulation.
Methods
Thirty patients were enrolled and 29 provided nine blood samples over 9 h at day 7 and months 1 and 3 post-transplant. Tacrolimus blood profiles measured with liquid chromatography-tandem mass spectrometry were modelled using one-compartment, double gamma absorption, linear elimination models developed in-house. Different limited sampling strategies of three time-points within 4 h post-dose were tested for the maximum a-posteriori Bayesian estimator of tacrolimus AUC0–12h. The models and estimators were validated internally and their performance compared to that of models previously developed for the original formulation.
Results
The concentration–time curves, AUC0–12h/dose and trough blood concentration/dose exhibited wide inter-individual variability. The covariate-free pharmacokinetic models developed for the three post-transplant periods closely fitted the individual profiles. Maximum a-posteriori Bayesian estimators based on three different limited sampling strategies and no covariate yielded accurate AUC0–12h estimates, including for the five cytochrome P450 3A5 expressers and for the four patients without corticosteroids. The 0–1 h–3 h strategy finally chosen had very low bias (− 4.0 to − 2.5%) and imprecision (root mean square error 5.5–9.2%). The maximum a-posteriori Bayesian estimators previously developed for the reference product fitted the generic profiles with similar performance.
Conclusions
We developed original pharmacokinetic models and accurate maximum a-posteriori Bayesian estimators to estimate patient exposure and adjust the dose of generic tacrolimus, and confirmed that the robust tools previously developed for the original formulation can be applied to this generic.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
This paper reports the first population pharmacokinetic models and Bayesian estimators developed for Adoport®, a generic oral formulation of tacrolimus |
A one-compartment, double gamma absorption, linear elimination model closely fitted the individual profiles |
Bayesian estimators combined with the 0–1 h–3 h limited sampling strategy yielded accurate and precise estimation of the area under the concentration–time curve over 12 h post-dose |
Pharmacokinetic models and Bayesian estimators previously developed for the original formulation fitted Adoport® profiles and estimated the inter-dose area under the concentration–time curve as well as the ad hoc tools These tools are now available to the kidney transplant community through the ISBA expert system: https://pharmaco.chu-limoges.fr |
1 Introduction
The bioequivalence of generic drugs with the reference product has to be demonstrated in healthy volunteers on the basis of the area under the concentration–time curve (AUC) and maximal concentration of the concentration–time profile, generally after a single drug dose. This standard scheme cannot guarantee that the generic formulation will yield the same pre-dose concentration at steady state, i.e. after repeated dosing, even less so in patients. This may be of concern for narrow therapeutic index drugs, in particular those requiring therapeutic drug monitoring, which is often based on the drug pre-dose blood concentration. The fact that some of these drugs can be dose adjusted based on their inter-dose AUC can be reassuring in this respect, as the AUC is one of the bioequivalence criteria, but it still raises the question of the applicability of the tools developed for AUC estimation of the reference product to generic formulations.
Tacrolimus is a calcineurin inhibitor widely used for the prevention of allograft rejection in solid organ transplantation, as well as in other indications across the world [1]. It is characterised by a narrow therapeutic index and large inter-individual pharmacokinetic (PK) variability. Therapeutic drug monitoring is recommended for tacrolimus (and is even compulsory in many countries) to minimise the risk of acute rejection and the occurrence of adverse effects (mainly nephrotoxicity and, to a lesser extent, neurotoxicity) and it is generally based on trough whole blood tacrolimus concentrations (C0). However, retrospective studies showed that C0 was not [2,3,4], or was only slightly [5], associated with acute graft rejection in kidney or liver transplant recipients. In contrast, a few papers reported that tacrolimus AUC was significantly associated with acute rejection [6, 7]. The recent consensus report of a consortium of European experts on tacrolimus therapeutic drug monitoring recommended using the inter-dose AUC over 12 h post-dose (AUC0–12h), as it is expected to be the best marker of tacrolimus exposure [1]. Actually, tacrolimus monitoring based on the full AUC0–12h is difficult to set up routinely, owing to clinical constraints and the necessity of multiple samples. Our and other groups previously reported PK models and Bayesian estimators to calculate tacrolimus inter-dose AUC using a limited sampling strategy (LSS), i.e. a few blood samples collected in the first hours post-dose, for three different tacrolimus formulations in different transplant settings [8,9,10,11,12,13,14,15]. We have proposed them to the transplant community though a free website since 2007 and reported the results obtained in hundreds of patients [16, 17].
One of the most important sources of inter-individual variability in tacrolimus pharmacokinetics is a single nucleotide polymorphism within intron 3 of cytochrome P450 (CYP) 3A5, a metabolic enzyme expressed in the intestinal mucosa and the liver highly involved in the oral tacrolimus first-pass effect. Patients who carry at least one CYP3A5*1 allele (CYP3A5 expressers, about 12% of the white European population [18]) show a 1.5- to two-fold higher tacrolimus oral clearance than CYP3A5*3/*3 carriers (non-expressers) [1]. Corticosteroids that induce CYP450s and P-glycoprotein might also affect tacrolimus pharmacokinetics [19].
Adoport® (Sandoz, Levallois-Perret, France) is a generic, twice-daily, immediate-release formulation of tacrolimus, whose pharmaceutical equivalence and bioequivalence with the reference product Prograf® (Astellas Pharma, Levallois-Perret, France) was demonstrated in healthy volunteers as well as in a kidney transplant population [20]. However, it has never been demonstrated that the PK tools (including Bayesian estimators) developed and used in a clinical setting for an original formulation can also be used for its generics. For instance, the LSSs used with maximum a posteriori Bayesian estimation (MAP-BE) generally include sampling points on both sides of the peak, i.e. before and after the time to maximum concentration (Tmax), while bioequivalence studies cannot guarantee that Tmax is similar between formulations (no statistical tests are required by the regulatory agencies for Tmax). This question is of particular importance for patient security and our liability in the context of the ISBA expert system (as well as similar web services), where on the one hand, one cannot guarantee that all patients are actually on the original formulation and, on the other hand, one cannot continue to refuse patients on generic formulations.
The objectives of the present clinical study were: (i) to develop, in de novo kidney transplant recipients on this generic formulation, population PK models and Bayesian estimators enabling the assessment of tacrolimus AUC0–12h at different post-transplant periods, on the basis of a limited number of samples collected during the first few hours post-dose, and considering the CYP3A5*3 polymorphism and corticosteroid cotreatment as covariates, if possible and useful, and (ii) to compare the performance of these MAP-BEs and LSSs to those we previously developed for the original formulation.
2 Material and Methods
2.1 Study Design and Patient Population
The “Pharmacokinetic Study of Adoport® (Tacrolimus) in Patients With de Novo Kidney Transplantation” (IMPAKT) study was sponsored by Limoges University Hospital, funded by Sandoz France, authorised by the Ethics Committee CPP16-058 on 8 December, 2016 and registered on ClinicalTrials.gov (Identifier: NCT03076151, registered on 6 March, 2017). We enrolled 30 kidney transplant recipients on immunosuppression regimes including Adoport®, from five French kidney transplant centres (university hospitals of Amiens, Clermont-Ferrand Limoges, Poitiers and Tours). Patients had to be aged > 18 years, male or female, recipients of a first kidney graft, transplanted for less than 7 days at the time of enrolment, affiliated to a health insurance system and they had to sign the study informed consent form. The exclusion criteria were: all contraindications to tacrolimus according to the summary of product characteristics; patients with pre-transplant, donor-specific, anti-human leukocyte antigen antibodies; recipients of any allograft other than the kidney; pregnant (positive β-HCG test) or lactating women; women without any method of contraception, except for those with no childbearing potential (according to the guidelines of the Clinical Trial Facilitation Group, related to contraception and pregnancy tests in clinical trials) [21]; patients participating in any other interventional clinical study; patients under judicial protection; and patients unable to understand the purposes and risks of the study, who could not provide written informed consent or who were unwilling to comply with the study protocol.
Each patient had to participate in three PK investigations, at 7 ± 3 days (D7), 1 month ± 1 week (M1) and 3 months ± 1 week (M3) post-transplantation. During each of these study visits, nine blood samples were collected in EDTA tubes, just before (C0) and then 20 and 40 minutes, 1, 2, 3, 4, 6 and 9 h after the morning dose of generic tacrolimus administration.
2.2 Tacrolimus Determination and Pharmacokinetic Modelling
Tacrolimus blood determination was centralised at the Laboratory of Pharmacology-Toxicology of Limoges University Hospital (CHU Limoges) and performed using a fully validated, liquid chromatography–tandem mass spectrometry technique, as previously described [11]. Briefly, after precipitation of 100 µL of whole blood with 25 g/L of zinc sulphate in water/methanol (70:30, v/v), tacrolimus was extracted and separated using two-dimension high-performance liquid chromatography and detected using tandem mass spectrometry in the positive-ion, selected reaction monitoring mode. [13C,D2]-Tacrolimus was employed as the internal standard. The lower limit of quantification was 1 ng/mL, the method was linear between 1 and 100 µg/L (r > 0.99) and the intra- and inter-day coefficients of variation (CV%) were < 18%.
Pharmacokinetic modelling was performed using a parametric approach in the iterative two-stage individual modelling software (developed locally by one of us, JD). We used a single-compartment model with linear elimination, and biphasic absorption, as previously described for other formulations of tacrolimus in different transplant settings [8, 10, 14, 15].
In this model, the absorption rate at time t is described as the sum of two gamma distributions:
With
The estimated population parameters were: Ĉ0, the model-estimated trough concentration after administration of a theoretical dose D0 = 1000 mg (the real trough concentration can be calculated by dividing this value by 1000 and multiplying by the patient dose); MAT1 and MAT2, the mean absorption times of the two absorption phases (defined as \(\frac{{a_{i} }}{{b_{i} }}\)); SDAT1 and SDAT2, the standard deviations of absorption times (defined as \(\frac{{\sqrt {a_{i} } }}{{b_{i} }}\)); r, the fraction of the dose absorbed by the faster method; FAIV = F × D0/Vd, where F is the bioavailability factor, AIV is the intravenous coefficient, Vd is the volume of distribution, and b is the elimination constant. The error model used was 0.0001 + 0.05 × C (C = drug concentration). Different sets of parameters were estimated for the different post-transplantation periods (D7, M1 and M3). The robustness of the model was checked using the traditional predicted vs observed and individually predicted vs observed concentrations, and the weighted residuals vs time. The stability of the results was investigated using iterative random splitting of the database (circular permutation) to obtain at each round one group for model development (75% of the concentration vs time profiles) and another group for model validation (the remaining 25% profiles). Additionally, the final models were evaluated using visual predictive checks after 1000 Monte-Carlo simulations and extraction of their 90% prediction intervals, using the vpc R package (https://CRAN.R-project.org/package=vpc).
The optimal combinations of sampling times for Bayesian estimation were obtained by comparing the accuracy and precision of tacrolimus AUC0–12h estimates obtained with several combinations of three sampling times within the first few hours post-dose, as compared to the reference tacrolimus AUC0–12h obtained using all available time-points. In addition, we evaluated the ability of Bayesian estimators previously developed for the reference product [16] to predict tacrolimus AUCs measured in patients on the generic formulation in the present study.
2.3 Cytochrome P450 3A5 Genotyping
Genotyping of CYP3A5*3 was also centralised in the Department of Pharmacology-Toxicology at Limoges University Hospital. DNA was isolated from EDTA-treated blood and genotyped for CYP3A5 rs776746 (CYP3A5*1/*3 allele) using a validated TaqMan allelic discrimination assay on a Rotor Gene Q® (Qiagen, Courtaboeuf, France), as previously described [22].
2.4 Statistical Analysis
The performance of the population PK models was evaluated by linear regression of measured vs estimated concentrations in the validation set (successive subgroups of 25% patients not involved in model development). The performance of the models in the subgroups of CYP3A5 expressers and patients without corticosteroids was checked by visually comparing them on the diagnostic plots with the predominant group of non-expressers on corticosteroids.
We used a two-way analysis of variance (patients*LSS) followed by the Dunnett’s multiple comparison test to compare the different LSS for MAP-BE of tacrolimus AUC0–12h with the reference AUC0–12h obtained using all time-points. For those providing estimates not statistically different from the references, we calculated the relative mean prediction error, or bias, and the relative root mean square error (RMSE). For the LSS finally selected, we computed the correlation and difference (Bland–Altman) plots with the reference, the minimum and maximum differences, the bias, RMSE and the number of differences out of the ± 20% acceptable range. The performance of the Bayesian estimators in patients carrying the minor CYP3A5*1 allele or receiving no corticosteroids was assessed visually on the correlation and Bland–Altman plots, but no statistical test was performed because of their small numbers in our population.
3 Results
One patient was withdrawn from the study owing to numerous severe adverse events (AEs), before any PK profile was collected. The other 29 patients completed the study and their characteristics are described in Table 1. Twenty-three patients received tacrolimus in combination with mycophenolate mofetil and 26 with corticosteroids at one study visit at least, whereas only two received tacrolimus as a monotherapy over the whole study period. The average tacrolimus daily dose decreased from 9.4 mg on day 7 to 6.2 mg at month 3 (with up to eight-fold variability across the study population at M1 and M3), while C0 and AUC0–12h tended to increase at M1 and decrease at M3. Plasma creatinine, haematocrit and haemoglobin improved over time (Table 1). The AEs reported by the investigators are reported in Table 1 of the Electronic Supplementary Material (ESM). All patients except for one had at least one AE, with a total of 260 AEs reported, 24 of which were considered as serious AEs. However, none of these serious AEs was deemed attributable to tacrolimus by the investigators and the Clinical Trial Pharmacovigilance Unit of Limoges University Hospital. Three acute graft rejection episodes were reported in three patients, two of which were confirmed by a biopsy (both categorised as acute T-cell-mediated rejection, grade 1b). These acute rejection episodes occurred 2 weeks, 3 weeks and 3 months post-transplantation.
A total of 87 PK profiles of tacrolimus were analysed. The concentration–time curves of Adoport® exhibited large inter-individual variability at each time period, and so did C0 (CV% = 38%, 35% and 26% at D7, M1 and M3, respectively) and AUC0–12h (CV% = 32%, 31% and 26%, respectively). The variability was even larger for C0/dose (CV% = 47%, 76% and 43%, respectively) and AUC0–12h/dose (CV% = 41%, 65% and 40%, respectively). In contrast, the AUC/C0 ratio showed much lower inter-individual variability (CV% = 21%, 18% and 18%, respectively) and intra-individual variability over the first 3 months post-transplant (CV% = 14%).
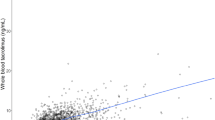
Reasonable correlation was found between morning C0 and the reference AUC0–12h at all post-transplant periods (r2 = 0.6857 at D7, 0.7912 at M1 and 0.6441 at M3) (Fig. 1). Accordingly, the AUC0–12h/C0 ratio varied mildly with time and between patients: mean (inter-individual CV%) = 18.58 h (21.31%) at D7, 18.17 h (18.36%) at M1 and 18.28 h (17.89%) at M3. The mean AUC0–12h/C0 ratio was not different in CYP3A5 expressers and/or patients not on steroids, as compared with the other patients (20.5 h vs 18.8 h, respectively) and the datapoints of these patients were not further apart in the AUC0–12h vs C0 regression lines than the others (Fig. 1). One of the five CYP3A5 expressers experienced T-cell-mediated graft rejection on day 21 post-transplant, i.e. 2 weeks after the D7 visit where the tacrolimus AUC was 108 h·µg/L and 1 week before the M1 visit where the AUC was 111 h·µg/L (as compared to mean values of 166 and 186 h·µg/L in the study population, respectively). However, the other two rejection episodes were reported in non-expressers, one at week 2 just after a very high AUC value (247 h·µg/L) and the other at M3 with AUC = 137 h·µg/L (for a population average of 141 h·µg/L).

Linear regression and correlation between tacrolimus C0 and AUC0-12h at the different post-transplant periods. The 4 patients carrying the CYP3A5*1 allele (i.e. CYP3A5 expressers) and who received corticosteroids are represented with blue diamonds, the only CYP3A5 expresser without steroids with black diamonds and the CYP3A5 non-expressers (or presumably so, for one) without steroids with red diamonds
The three PK models developed (one for each of the three post-transplant periods) adequately described the data, as shown by the goodness-of-fit plots (Fig. 2). The model-predicted concentrations were clearly overestimated in the five CYP3A5 expressers, whether on corticosteroids (n = 4) or not (n = 1), but the individually predicted concentrations were as tightly fitted by the model as those of the dominant group of non-expressers on corticosteroids (n = 23) and the weighted residuals were very similar. The three other patients not on steroids were CYP3A5 non-expressers (or presumably so for one of them who was not genotyped) and their model-predicted concentrations were not significantly different from, and the individually predicted concentrations and Weighted residuals (WRES) as good as, those of the other non-expressers, confirming that corticosteroids had no significant influence on tacrolimus pharmacokinetics in this study. The visual predictive checks showed that the simulated data matched the observed concentration–time profiles for the three post-transplant periods (Fig. 3).

Diagnostic plots of the population pharmacokinetic models. The 4 patients carrying the CYP3A5*1 allele (i.e. CYP3A5 expressers) and who received corticosteroids are represented with blue diamonds, the only CYP3A5 expresser without steroids with black diamonds and the CYP3A5 non-expressers (or presumably so, for one) without steroids with red diamonds

Visual predictive checks of the PK models developed for generic tacrolimus for the three different post-transplant periods (day 7, month 1 and month 3)
Bayesian AUC estimates obtained with six different three-point LSSs were compared with the reference AUC values using a two-way analysis of variance (patients*LSS), showing significant between-patient variability (p < 0.0001) but only a trend for differences among the LSSs (p = 0.0641). Dunnett’s multiple comparisons showed that the 0–1 h–2 h, 0–1 h–3 h and 0–2 h–4 h LSS yielded estimates not statistically different from the reference AUCs (Table 2). The Bland–Altman plots (Fig. 4) and the very similar mean prediction error (1.1%, 2.0% and 1.2%, respectively) and RMSE (9.2%, 7.8% and 8.6%, respectively) suggest that these strategies are equally acceptable, including for the patients carrying the CYP3A5*1 allele and/or without corticosteroids. Owing to its better Bland–Altman plot (Fig. 4) and lower RMSE, and for the sake of consistency with the LSS used routinely for the original formulation, we finally chose the 0–1 h–3 h LSS for the rest of this study. The AUC estimates obtained in the validation set obtained by circular permutation showed a bias between 0.2 and 3.6%, and imprecision RMSE between 6.0% and 8.6%, depending on the post-transplant period. Only one AUC value at M1, and two at M3, were estimated with inaccuracy >20% (Table 3). The best and worst AUC estimates at each post-transplant period are presented in Fig. 5. The PK models and Bayesian estimators previously developed for the reference product were able to fit closely the full concentration–time profiles obtained with the generic formulation Adoport® (Figs. 1 and 2 of the ESM), as well as to estimate accurately the AUC0–12h using the same LSS (Table 3).

Linear regression and Bland-Altman plots showing the correlation and the relative difference between reference AUCs and Bayesian AUCs obtained using different 3-point strategies (limited to the first 4 hours post-dose). The 4 patients carrying the CYP3A5*1 allele (i.e. CYP3A5 expressers) and who received corticosteroids are represented with blue diamonds, the only CYP3A5 expresser without steroids with black diamonds and the CYP3A5 non-expressers (or presumably so for one) without steroids with red diamonds

Best and worst AUC0–12 h estimates in the internal validation population at each post-transplant period using Bayesian estimation and the pre-dose, 1 h and 3 h limited sampling strategy. The profiles estimated using all sampling times are in thin lines, the 3-point Bayesian estimates in thick lines
4 Discussion
We conducted a PK study of Adoport®, a generic formulation of tacrolimus in adult kidney transplant recipients and developed PK models and Bayesian estimators, with the aim of estimating, at early (day 7) and more stable (months 1 and 3) post-transplant periods, tacrolimus AUC0–12h on the basis of a limited number of samples collected during the first few hours post-dose. The PK models obtained showed excellent fit with the observed profiles. In addition, the Bayesian estimators and the LSS previously developed in a similar population for Prograf®, the reference product, showed almost identical performance in this study. It is important to acknowledge that, in addition to being bioequivalent with Prograf®, the generic formulation tested in this study yields very similar Tmax values, as previously shown in a bioequivalence study in kidney transplant recipients [20], which may represent favourable conditions for the application of our initial Bayesian estimators. The question as to whether this can be extrapolated to all generic formulations of immediate-release tacrolimus, particular those with a shift in Tmax or more variable Tmax, should be further investigated. However, several LSSs yielded equivalent performance for AUC0–12h estimation in the present study, confirming that Bayesian estimation is flexible with respect to sampling times and suggesting that it can cope with situations where Tmax is not framed by the time-points (such as with the 0–2 h–4 h LSS for instance, where all time-points are after the peak for most patients). Altogether, the present results are very reassuring with regard to the thousands of individual dose adjustment requests posted on the ISBA website (https://pharmaco.chu-limoges.fr). Indeed, even if it was clearly stated that the tools available were only valid for the reference product and that the drug brand had to be completed on the request form, we could not be sure that all patients actually received the original formulation.
Similar to all the other formulations of tacrolimus, Adoport® oral administration in kidney transplant patients resulted in wide inter-individual variability of the concentration–time curves at each time period. Even in this rather small patient cohort, dose-standardised AUC0–12h varied five-fold on day 7, 11-fold at month 1 and eight-fold at month 3 post-transplantation, owing to the individual dose adjustments required to obtain exposure in a narrower range. Remarkably, the AUC/C0 ratio varied much less between patients (only two-fold at all time periods) and was very stable with time (intra-individual CV% = 14%). We have recently recommended using this metrics to determine individual C0 targets based on AUC and C0 determination at least twice, in the early and stable post-transplant periods [1, 17, 23]. The current findings are in favour of the robustness of the AUC/C0 ratio and also show that the individual relationship between C0 and AUC does not vary much over time.
Three rejection episodes were reported in the per-protocol population of 29 patients, one of whom was a CYP3A5 expresser and had rather low tacrolimus exposure, whereas the other two had normal or high AUCs. No clear conclusion can be made about the exposure–efficacy relationship, so much so that the study was not powered for this.
Owing to a 12% frequency in the White European population, about four carriers of the CYP3A5*1 allele were expected in this study, and we found five expressers out of 25 patients whose genotype was available. The AUC vs C0 regression, AUC/C0 ratio, PK models and Bayesian estimators developed were specifically evaluated in this subgroup, showing equivalent behaviour and performance as compared to the rest of the population. Similarly, only four patients did not receive steroids over the course of the study (except a bolus of methylprednisolone on day 21 for one of them), one of whom carried the CYP3A5*1 allele. The model-predicted concentrations were not different in this small subgroup from those of the dominant group of non-expressers on corticosteroids (n = 23) and their AUC vs C0 regression, AUC/C0 ratio, and Bayesian estimators showed equivalent behaviour and performance, suggesting that the influence of corticosteroids on tacrolimus was negligible in this study. This prevented us from trying to develop specific PK models and Bayesian estimators for this subpopulation, or even to use the CYP3A5 genotype or treatment with corticosteroids as covariates in the models or estimators. More importantly, this study confirms that robust PK models and Bayesian estimators can accurately estimate the inter-dose AUC of a drug with high inter-patient PK variability without considering covariates as strong as the CYP3A5 genotype (that doubles tacrolimus oral clearance in expressers). With our strategy of designing models and Bayesian estimators for each combination of transplanted organ, age group (paediatrics or adults), post-transplant period (early or stable) and analytical method [24], and with the three-point LSS employed, we never found additional covariates that could improve Bayesian AUC estimation or help to reduce the number of sampling points [8,9,10,11,12,13,14,15]. This confirms the reference demonstration made more than 40 years ago by Sheiner et al. for digoxin that “information from plasma concentrations is far more valuable for forecasting than that from observable patient features-sex, age, and the like; use of all the latter information does not improve accuracy and precision as much as only 1 plasma concentration” [25].
5 Conclusions
We developed population PK models and Bayesian estimators for Adoport®, one of the most prescribed generic formulation of tacrolimus, which tightly fitted the data without the need for covariates, not even the CYP3A5 genotype, and we confirmed that Bayesian estimators previously developed for the original formulation had very similar performance. These models and MAP-BE (now all available on the ISBA website) can also cope with different combinations of sampling times, suggesting that they are applicable to other generic formulations with a different distribution of Tmax values (as Tmax is not statistically compared in bioequivalence studies). We also consider that the model developed for the latest post-transplant period here (M3) is also suited for the steady-state situation, despite subtle changes that may occur in tacrolimus pharmacokinetics up to 1-year post-transplant.
References
Brunet M, van Gelder T, Åsberg A, Haufroid V, Hesselink DA, Langman L, et al. Therapeutic drug monitoring of tacrolimus-personalized therapy: second consensus report. Ther Drug Monit. 2019;41(3):261–307.
Bouamar R, Shuker N, Hesselink DA, Weimar W, Ekberg H, Kaplan B, et al. Tacrolimus predose concentrations do not predict the risk of acute rejection after renal transplantation: a pooled analysis from three randomized-controlled clinical trials. Am J Transplant. 2013;13(5):1253–61.
Rodríguez-Perálvarez M, Germani G, Darius T, Lerut J, Tsochatzis E, Burroughs AK. Tacrolimus trough levels, rejection and renal impairment in liver transplantation: a systematic review and meta-analysis. Am J Transplant. 2012;12(10):2797–814.
Daher Abdi Z, Prémaud A, Essig M, Alain S, Munteanu E, Garnier F, et al. Exposure to mycophenolic acid better predicts immunosuppressive efficacy than exposure to calcineurin inhibitors in renal transplant patients. Clin Pharmacol Ther. 2014;96(4):508–15.
Israni AK, Riad SM, Leduc R, Oetting WS, Guan W, Schladt D, DeKAF Genomics Investigators, et al. Tacrolimus trough levels after month 3 as a predictor of acute rejection following kidney transplantation: a lesson learned from DeKAF Genomics. Transpl Int. 2013;26(10):982–9.
Undre NA, van Hooff J, Christiaans M, Vanrenterghem Y, Donck J, Heeman U, et al. Low systemic exposure to tacrolimus correlates with acute rejection. Transplant Proc. 1999;31(1–2):296–8.
Kuypers DR, Claes K, Evenepoel P, Maes B, Vanrenterghem Y. Clinical efficacy and toxicity profile of tacrolimus and mycophenolic acid in relation to combined long-term pharmacokinetics in de novo renal allograft recipients. Clin Pharmacol Ther. 2004;75(5):434–47.
Saint-Marcoux F, Knoop C, Debord J, et al. Pharmacokinetic study of tacrolimus in cystic fibrosis and non-cystic fibrosis lung transplant patients and design of Bayesian estimators using limited sampling strategies. Clin Pharmacokinet. 2005;44:1317–28.
Benkali K, Prémaud A, Picard N, Rérolle JP, Toupance O, Hoizey G, et al. Tacrolimus population pharmacokinetic-pharmacogenetic analysis and Bayesian estimation in renal transplant recipients. Clin Pharmacokinet. 2009;48(12):805–16.
Saint-Marcoux F, Debord J, Undre N, Rousseau A, Marquet P. Pharmacokinetic modeling and development of Bayesian estimators in kidney transplant patients receiving the tacrolimus once-daily formulation. Ther Drug Monit. 2010;32(2):129–35.
Benkali K, Rostaing L, Premaud A, Woillard JB, Saint-Marcoux F, Urien S, et al. Population pharmacokinetics and Bayesian estimation of tacrolimus exposure in renal transplant recipients on a new once-daily formulation. Clin Pharmacokinet. 2010;49(10):683–92.
Woillard JB, de Winter BC, Kamar N, Marquet P, Rostaing L, Rousseau A. Population pharmacokinetic model and Bayesian estimator for two tacrolimus formulations: twice daily Prograf and once daily Advagraf. Br J Clin Pharmacol. 2011;71(3):391–402.
Monchaud C, de Winter BC, Knoop C, Estenne M, Reynaud-Gaubert M, Pison C, et al. Population pharmacokinetic modelling and design of a Bayesian estimator for therapeutic drug monitoring of tacrolimus in lung transplantation. Clin Pharmacokinet. 2012;51(3):175–86.
Woillard JB, Debord J, Monchaud C, Saint-Marcoux F, Marquet P. Population pharmacokinetics and Bayesian estimators for refined dose adjustment of a new tacrolimus formulation in kidney and liver transplant patients. Clin Pharmacokinet. 2017;56(12):1491–8.
Riff C, Debord J, Monchaud C, Marquet P, Woillard JB. Population pharmacokinetic model and Bayesian estimator for 2 tacrolimus formulations in adult liver transplant patients. Br J Clin Pharmacol. 2019;85(8):1740–50.
Saint-Marcoux F, Woillard JB, Jurado C, Marquet P. Lessons from routine dose adjustment of tacrolimus in renal transplant patients based on global exposure. Ther Drug Monit. 2013;35(3):322–7.
Marquet P, Bedu A, Monchaud C, Saint-Marcoux F, Rérolle JP, Etienne I, et al. Pharmacokinetic therapeutic drug monitoring of Advagraf in more than 500 adult renal transplant patients, using an expert system online. Ther Drug Monit. 2018;40(3):285–91.
HapMap project. https://www.ncbi.nlm.nih.gov/probe/docs/projhapmap/. Accessed 7 May 2020.
Lam S, Partovi N, Ting LS, Ensom MH. Corticosteroid interactions with cyclosporine, tacrolimus, mycophenolate, and sirolimus: fact or fiction? Ann Pharmacother. 2008;42(7):1037–47.
Alloway RR, Sadaka B, Trofe-Clark J, Wiland A, Bloom RD. A randomized pharmacokinetic study of generic tacrolimus versus reference tacrolimus in kidney transplant recipients. Am J Transplant. 2012;12(10):2825–31.
Heads of Medicines Agencies, Clinical Trial Facilitation Group (CTFG). Recommendations related to contraception and pregnancy testing in clinical trials. https://www.hma.eu/fileadmin/dateien/Human_Medicines/01-About_HMA/Working_Groups/CTFG/2014_09_HMA_CTFG_Contraception.pdf. Accessed 7 May 2020.
Le Meur Y, Djebli N, Szelag JC, et al. CYP3A5*3 influences sirolimus oral clearance in de novo and stable renal transplant recipients. Clin Pharmacol Ther. 2006;80(1):51–60.
Marquet P, Albano L, Woillard JB, Rostaing L, Kamar N, Sakarovitch C, et al. Comparative clinical trial of the variability factors of the exposure indices used for the drug monitoring of two tacrolimus formulations in kidney transplant recipients. Pharmacol Res. 2018;129:84–94.
Saint-Marcoux F, Debord J, Parant F, et al. Development and evaluation of a simulation procedure to take into account various assays for the Bayesian dose adjustment of tacrolimus. Ther Drug Monit. 2011;33(2):171–7.
Sheiner LB, Beal S, Rosenberg B, Marathe VV. Forecasting individual pharmacokinetics. Clin Pharmacol Ther. 1979;26(3):294–305.
Acknowledgements
We thank Sandoz France and Limoges University Hospital for funding this study and providing the study drug; Hélène Roussel for writing the study protocol and overseeing the investigation sites; Alexandre Garnier for excellent study data management; Chloé Barny for handling patient samples; Naïma Tafzi and the laboratory technicians of the Department of Pharmacology, Toxicology and Pharmacovigilance for tacrolimus determination; Hélène Arnion for pharmacogenetic analyses; and Karen Poole for revising the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Sandoz France and Limoges University Hospital.
Conflicts of interest/Competing Interests
Pierre Marquet received consulting fees and travel grants from Sandoz, Chiesi, Pfizer, and BMS and research grants from Sandoz and Chiesi. Caroline Monchaud received an honorarium from Chiesi to participate in a board of experts. Matthias Buchler received a travel grant to attend an international congress from Sandoz. Jean-Baptiste Woillard received speaker fees from Chiesi and Gilead and travel grants to attend symposia or congresses from Chiesi and Gilead, VIIV, Astellas and Abbvie. Alexandre Destere, Jean-Philippe Rérolle, Hakim Mazouz, Isabelle Etienne, Antoine Thierry, Nicolas Picard and Jean Debord have no conflicts of interest that are directly relevant to the content of this article.
Ethics approval
This study was approved by the National Research Ethics Committee CPP16-058 on 8 December, 2016 and was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments.
Consent to participate
Informed consent was obtained from all participants included in the study.
Consent for publication
Not applicable.
Availability of data and material
Raw data are available on request from the authors.
Code Availability
Not applicable.
Author Contributions
PM designed and was the principal investigator of the study, participated in result analysis and interpretation, and is the main author of the manuscript. AD participated in data analysis and participated in manuscript writing. CM participated in the study design, supervised the clinical research team, data collection and cleaning, and participated in manuscript writing. J-PR enrolled patients in the study and critically reviewed the manuscript. MB enrolled patients in the study and critically reviewed the manuscript. HM enrolled patients in the study and critically reviewed the manuscript. IE enrolled patients in the study and critically reviewed the manuscript. AT enrolled patients in the study and critically reviewed the manuscript. NP supervised pharmacogenetic testing and critically reviewed the manuscript. J-BW participated in the pharmacokinetic modelling and manuscript writing. JD performed most of the pharmacokinetic modelling and Bayesian estimation and participated in manuscript writing.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Marquet, P., Destère, A., Monchaud, C. et al. Clinical Pharmacokinetics and Bayesian Estimators for the Individual Dose Adjustment of a Generic Formulation of Tacrolimus in Adult Kidney Transplant Recipients. Clin Pharmacokinet 60, 611–622 (2021). https://doi.org/10.1007/s40262-020-00959-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-020-00959-y