
Abstract
Background
Preterm neonates are usually not part of a traditional drug development programme, however they are frequently administered medicines. Developing modelling and simulation tools, such as physiologically based pharmacokinetic (PBPK) models that incorporate developmental physiology and maturation of drug metabolism, can be used to predict drug exposure in this group of patients, and may help to optimize drug dose adjustment.
Objective
The aim of this study was to assess and verify the predictability of a preterm PBPK model using compounds that undergo diverse renal and/or hepatic clearance based on the knowledge of their disposition in adults.
Methods
A PBPK model was developed in the Simcyp Simulator V17 to predict the pharmacokinetics (PK) of drugs in preterm neonates. Drug parameters for alfentanil, midazolam, caffeine, ibuprofen, gentamicin and vancomycin were collated from the literature. Predicted PK parameters and profiles were compared against the observed data.
Results
The preterm PBPK model predicted the PK changes of the six compounds using ontogeny functions for cytochrome P450 (CYP) 1A2, CYP2C9 and CYP3A4 after oral and intravenous administrations. For gentamicin and vancomycin, the maturation of renal function was able to predict the exposure of these two compounds after intravenous administration. All PK parameter predictions were within a twofold error criteria.
Conclusion
While the developed preterm model for the prediction of PK behaviour in preterm patients is not intended to replace clinical studies, it can potentially help with deciding on first-time dosing in this population and study design in the absence of clinical data.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Knowledge regarding the safe and effective use of medicines in neonates has increased substantially over recent years, as evidenced by the increasing number of published studies [1, 2]. However, despite this, understanding of developmental pharmacokinetics (PK) and pharmacodynamics (including toxicity) in preterm neonates is still lacking. Consequently, many drugs used in the neonatal population. Particularly those used in neonatal intensive care units, are unlicensed or off-label. Drug regulators have released guidance on the investigation of medicinal products in the paediatric population [3, 4], however to date this has resulted in few label changes in preterm neonates.
The lack of knowledge of clinical pharmacology in this age group has led to either use of suboptimal doses or drug toxicity [5]. Renal damage in preterm neonates has been linked to the administration of some drugs, such as vancomycin, acyclovir, methotrexate and non-steroidal anti-inflammatory drugs (NSAIDs) [6]. Early postnatal exposure to opioids was found to be associated with a higher risk of adverse outcomes in extremely preterm infants [7]. Due to immaturity of their blood–brain barrier, preterm patients are also more susceptible to kernicterus (entry of bilirubin into the brain, causing yellow discoloration of brain tissue, seizures and death). Lopinavir/ritonavir oral solution toxicity in preterm subjects was reported in different case studies due to the high content of both ethanol and propylene glycol in this drug. Accumulation of these compounds led to toxic levels since ethanol inhibits the already limited metabolism of propylene glycol in preterm neonates [8, 9]. The presence of ‘sensitive windows’ in preterm organ development, where chances of toxicity are increased due to direct or indirect (breast milk) drug administration, means that care is needed in prescribing drugs to both mother and child [10, 11].
The higher volume of extracellular fluid [12] in preterm per kilogram of body weight, in comparison with older children, shows an increased distribution volume of hydrophilic drugs [13]. Conversely, the absence or lower expression of metabolizing enzymes in neonates and a lower glomerular filtration rate (GFR) contributes to a higher exposure [14]. These physiological changes can significantly affect the drug kinetics and an appropriate dose adjustment is often needed, either dose reduction, less frequent dosing, or a combination of both. There is no widely accepted approach for appropriate optimization of the dose in preterm neonates; the widely used approach of scaling the dose based solely on body surface area or body weight often leads to inappropriate dose administration. Modelling and simulation is required to address preterm safety, dosing and pharmaceutical quality for neonatal studies that evaluate medicinal products in this population [15]. In addition, inclusion of available knowledge on physiological changes that can affect drug PK is an important step during drug development [16].
Physiologically based PK (PBPK) models marry the complex interplay of physiological parameters (demographics, organ sizes and blood flows, etc.) with drug-related properties (lipid solubility, enzyme and transporter kinetics, etc.) and thus represent a mechanistic approach to quantitatively predict the PK of specific drugs in different populations [17]. The PBPK approach has been adopted within pharmaceutical companies [18] and drug regulatory agencies [19, 20]. Neonatal preterm and full-term paediatric PBPK models additionally account for the rapid development of organs and the ontogeny of specific enzymes and transporters that determine the age-related PK [21,22,23].
Progress has been made in the application of PBPK models to describe drug PK in full-term neonates [22, 24]. This approach has been shown to give more accurate dose prediction compared with an empirical approach when there are no clinical studies to guide the requirements [25]. PBPK models are useful not only in dosage adjustment but also in developing drugs to treat preterm-specific conditions (e.g. ductus arteriosus, bronchopulmonary dysplasia, necrotizing enterocolitis and retinopathy of prematurity). Information on the magnitude of anatomical, physiological and biochemical changes during the preterm period required to build a preterm PBPK model have already been reported [26].
The aim of this work was to assess and verify a preterm PBPK model using five different drugs commonly used therapeutically in premature neonates who undergo diverse renal and/or hepatic clearance. The substrates selected were caffeine (a cytochrome P450 [CYP] 1A2 substrate), midazolam and alfentanil (CYP3A4 substrates), gentamicin and vancomycin (mainly renally cleared) and ibuprofen (CYP2C9 substrate), as case examples, and to compare predicted PK with clinical observations.
2 Materials and Methods
2.1 Development of the Preterm Population
Preterm population parameters required for building preterm PBPK models have been previously collated, analysed and integrated [26]. The preterm model was incorporated into the Simcyp Simulator V17 Release 1, which was used for all predictions in the current study. The preterm model is integrated with the distribution models currently available within the Simulator, whereby the user can predict or assign volume of distribution for the compound. Predicted volume of distribution can be used with either the minimal or full PBPK model, where the drug partition to different body tissues is predicted according to Berezhkovskiy [27] or Rodgers and Rowland [28]. In the latter, one can account for drug ionization and binding to the cellular components.
The minimal PBPK model (mPBPK) is a ‘lumped’ PBPK model that has only four compartments, predicting only the systemic, portal vein and liver concentrations. The fourth compartment is a non-physiological single adjusting compartment, which permits adjustment to the drug concentration profile in the Systemic compartment, where the latter represents a lumping of all tissues, excluding the liver and portal vein. The full PBPK distribution model makes use of a number of time-based differential equations in order to simulate the concentrations in various organ compartments—the blood (plasma), adipose, bone, brain, gut, heart, kidney, liver, lung, muscle, pancreas, skin and spleen. Interindividual variability is introduced through tissue volume predictions using age, sex, weight and height as covariates through a Monte Carlo sampling that takes into account correlations between these covariates.
2.2 Development of the Compound Files
The compound files that are not part of the Simcyp Simulator compound library (alfentanil, gentamicin and vancomycin) were first developed and verified in adults using the healthy volunteer population, and then further verified in the paediatric population. Prediction of the drug kinetics in preterm populations was performed via matched clinical settings/designs to the original studies. In all simulations, the number of subjects in the corresponding clinical study has been used within each trial.
2.3 Performance Verification of Preterm Physiologically Based Pharmacokinetics (PBPK)
The preterm PBPK model predictions were compared with different sets of clinical observations available in the literature for alfentanil, midazolam, caffeine, ibuprofen, gentamicin and vancomycin. Selection of these probes was based on the availability of the relevant data, i.e. PK parameters and concentration–time profiles, from preterm patients and the information regarding their gestational age (GA), postnatal age (PNA) and/or postmenstrual age and administered doses. The drug kinetics in preterm populations were predicted using matched trial settings/designs to the original studies.
2.3.1 Alfentanil
Alfentanil undergoes extensive hepatic metabolism mainly through CYP3A, and approximately 0.4% of alfentanil is excreted unchanged in the urine [29, 30]. In the alfentanil model, in vivo intrinsic clearance (CLint) values were reversely translated by back calculation of CLint from clinically reported intravenous adult clearance data [31]. In vivo CLint values were separated into CYP3A4 and non-CYP3A4 [included as additional human liver microsomes option (HLM)] components. The fraction of alfentanil metabolised by CYP3A4 (fmCYP3A4) = 0.93 was calculated from in vitro chemical inhibition study data [32], and is in line with the fm value derived from the in vivo drug–drug interaction (DDI) study [33]. A summary of the in vitro and PK parameters for alfentanil are shown in Table 1.
The alfentanil model was verified using clinical data in adults. The CYP3A4 ontogeny function is described using a linear relationship with postmenstrual age in preterm neonates based on the in vitro expression and activity data in fetal and neonate liver microsomes [39,40,41]. To predict alfentanil PK in preterm neonates, the Simcyp Simulator trial design was set to match the clinical studies [42, 43] as described below.
Trial design A Single intravenous infusion of 0.025 mg/kg over 5 min; 10 trials of six preterm subjects, 50% female, 28–34 gestational weeks, PNA was uniformly distributed between 0 and 3 days.
Trial design B Continuous intravenous infusion, loading dose of 0.008 mg/kg over 10 min, followed by 0.03 mg/kg for 6 h, then 0.015 mg/kg for 6 h, and finally 0.075 mg/kg for 15 h; 10 trials of 13 subjects, 38.5% female, 35–40 gestational weeks, PNA was uniformly distributed between 0 and 28 days.
2.3.2 Midazolam
Midazolam is cleared mainly by CYP3A4- and CYP3A5-mediated metabolism and a minor contribution from uridine diphosphate glucuronosyltransferase (UGT) 1A4 (< 5%); < 1% is excreted unchanged in urine.
The default midazolam compound file was selected from the Simcyp compound library. The first-order model with an absorption rate constant (ka) of 3 h−1 [44] was used to describe the absorption of midazolam after oral administration. The distribution was described using the mPBPK model, with a user input volume of distribution at steady state (Vss) of 0.88 L/kg [45]. The hepatic and intestinal ontogeny function for CYP3A4 were as described for alfentanil. The CYP3A5 expression levels in the fetal liver samples showed large variability, partially due to the CYP3A5 genetic polymorphisms [46]. In the present model, due to the overlapping substrate specificities and the predominant frequency of CYP3A5 poor metabolizers in the Caucasian population, the preterm CYP3A5 level was assumed to be either at adult levels or to follow the same ontogeny as CYP3A4 in the liver and intestine. Both scenarios have been explored.
The UGT1A4 ontogeny function was defined at the level of the full-term neonate at birth, i.e. 5% of the adult abundances.
To predict midazolam PK in preterm individuals, the Simulator trial design was set to match the clinical studies after intravenous [47] and oral [48] administration, as described below.
Trial design A Single intravenous infusion of 0.1 mg/kg over 30 min; 10 trials of 24 preterm neonates, 50% female, 28–34 weeks of GA, and PNA was uniformly distributed between 3 and 11 days.
Trial design B Single oral dose of 0.1 mg/kg; 20 trials of 15 preterm neonates, 50% female, 28–31 weeks of GA, and PNA was uniformly distributed between 3 and 13 days.
2.3.3 Caffeine
Caffeine is mainly a CYP1A2 substrate. PK studies of caffeine in neonates showed that its disposition varies with PNA and can differ markedly between premature and term neonates [24].
To predict the PK in preterm individuals, the default caffeine compound file was selected from the Simcyp compound library. The model consisted of first-order oral absorption with a ka of 1.48 h−1 [49], and an mPBPK distribution model with a reported Vss of 0.85 L/kg [49]. The default in vitro elimination data using enzyme kinetics, mainly CYP1A2 (fm CYP1A2 ~ 98%), was used to predict caffeine clearance. The trial design was set to match the clinical studies, and the following trial designs were replicated.
Trial design A A single intravenous dose of 15 mg/kg over a period of 5–10 min was administered to 13 preterm patients with a PNA of 3 days (range 1–42) and born at an average of 31 weeks of GA (range 25–34) and with a PNA of 1–8 days [50]. The original study reported a profile for a female preterm aged 3 days, who was born at 32 weeks of GA. A trial simulation of 10 virtual trials with 10 subjects in each trial matching the age of this female preterm was used. The infusion duration was set to 5 min in the simulation.
Trial design B 10 mg/kg caffeine on day 1, followed by 2.5 mg/kg/day for 6 days, administered either orally or by intravenous infusion over 15 min to preterm neonates during their first 1–25 days after birth at 28 weeks of GA [49]. A trial simulation setting of 10 trials of 10 virtual preterm neonates, all male subjects, 28 weeks of GA, and run for 7 days. PNA was uniformly distributed between 1 and 25 days.
Trial design C 40 mg/kg caffeine on day 1, followed by 10 mg/kg/day for 6 days, administered either orally or by intravenous infusion over 15 min [49]. The trial simulation settings were similar to trial design B, except the loading and maintenance doses were updated.
Given the length of trials B and C, the redefining subject over time features were selected for trial designs B and C to allow the growth of individual physiology alongside the simulation progress. An ontogeny profile for CYP1A2 was derived for a preterm population of 28–40 weeks’ postmenstrual age based on a previously reported equation [51]. The developed model was used to compare PK parameters for the two dosage regimens stated above.
The same designs were used to predict caffeine in preterm, but using the full PBPK model after verification of the method in the adult population using the Rodgers and Rowland method [28], with a Kp scalar of 1.7 in the adult model; we kept this setting for preterm prediction.
2.3.4 Ibuprofen
Ibuprofen undergoes elimination in adults mainly via the CYP2C9 enzyme, with limited involvement of CYP2C8, UGT2B7 and UGT1A9 [52, 53]. An ibuprofen compound file has been generated (Table 2). The compound model performance was compared against the adult and paediatric clinical PK data.
The PK of ibuprofen were predicted in a preterm population following intravenous and oral administration as described below.
Intravenous To mimic the clinical studies as closely as possible, a total of 130 virtual subjects (50% female), grouped into 10 trials with a mean PNA of 3 days and gestational weeks of 28.6 were included in the simulation. Ibuprofen was administered on days 1, 2, and 3 by a 15-min intravenous infusion of 10, 5 and 5 mg/kg, respectively [57, 58]. The redefining subject over time feature was selected to allow the growth of individuals alongside the simulation progress. The current model does not account for the presence of patent ductus arteriosus (PDA) and the mutual impact of ibuprofen exposure on the time-dependent PDA closure.
Oral Similarly, 10 trials of 13 (50% female) and 20 (40% female) preterm neonates of gestational weeks 28.6 and 30.5, respectively, with a PNA of 0–3 days, have been included in the simulation, and 10 mg/kg of ibuprofen was administered orally as a single dose in both studies [59, 60]. The first-order absorption model for ibuprofen was parametrized using the mechanistic permeability approach [61], where the adjusted adult value for effective permeability was used. The model then predicts the ka value for each preterm individual using the individual intestinal physiology, i.e. radius and length of the gut segment as described earlier [26]. An ontogeny profile for CYP2C9 was used for the preterm population of 28–40 weeks’ postmenstrual age as previously reported [51].
2.3.5 Gentamicin
Gentamicin is predominately eliminated by renal excretion through glomerular filtration; less than 10% is bound to plasma protein. The rapidly changing GFR is expected to affect the drug disposition in the preterm population. The PBPK model of gentamicin was built and verified using clinical data from adults, children and full-term neonates. A summary of the in vitro and PK parameters for the gentamicin model are shown in Table 3.
To predict the PK of gentamicin in preterm patients, the trial design was set to match the clinical studies [65, 66].
Trial design A Single intravenous infusion of 4 mg/kg over 5 min; 10 trials of 20 preterm subjects, 50% female, 28–40 gestational weeks, PNA was uniformly distributed between 0 and 28 days.
Trial design B Multiple intravenous boluses of 2.5 mg/kg every 12 h for 5 days; 10 trials of 18 subjects, 33% female, 35–40 gestational weeks, PNA was uniformly distributed between 0 and 4 days.
Trial design C Multiple intravenous boluses of 4 mg/kg every 24 h for 5 days; 10 trials of 18 subjects, 33% female, 35–40 gestational weeks, PNA was uniformly distributed between 0 and 4 days.
For the multiple-dose trials, the redefining subject over time feature was selected.
2.3.6 Vancomycin
Vancomycin also undergoes renal elimination through glomerular filtration. The systemic exposure is expected to be modified by the maturation of GFR in the preterm population. The PBPK model of vancomycin was built and verified using clinical data in adults, children and full-term subjects. A summary of the in vitro and PK parameters for the vancomycin model are shown in Table 4.
To predict the PK of vancomycin in preterm patients, the trial design was set to match the clinical studies [70, 71] as described below.
Trial design A Single intravenous infusion of 10 mg/kg over 30 min, 50% female, 10 trials of seven preterm subjects, 32 gestational weeks, PNA of 3.3 days.
Trial design B Single intravenous infusion of 15 mg/kg over 30 min, 50% female, 10 trials of seven preterm subjects, 34 gestational weeks, PNA of 4.7 days.
Trial design C Multiple intravenous infusions of 12.6 mg/kg over 30 min every 8 h for 3 days, 50% female, 10 trials of 10 preterm subjects, 28–34 gestational weeks, PNA was uniformly distributed between 7 and 28 days.
The redefining subject over time feature was selected for the multiple-dose designs.
Maturation of vancomycin clearance with age was also replicated and compared with reported observations from clinical studies [72, 73].
2.4 Data Analysis
Performance verification of the new compounds in adult and older paediatric subjects was carried out to ensure that the compound models adequately describe the observed data before applying the physiological changes in the preterm population. The adequacy of the compound files was justified by predicting adult and paediatric data, where available, based on the 90th predictive percentiles. The same criterion was applied to check the performance of the preterm model, i.e. observed concentrations based on the 90th percentile predictive interval. Where PK parameters such as area under the curve (AUC), maximum concentration (Cmax) and systemic clearance were available for the compound, this information was compared with the predicted parameters using the twofold boundary criterion. Where the administered doses were reported as mean and standard deviation (SD), the mean value was used for the simulation, and, if the dose range was reported, the median value was used for performing the simulation. If a study reported a narrow range of GA and another range for PNA, the PNA range was used, with a median value of GA. If the original study grouped the preterm subjects according to the different GA and PNA ranges, then subpopulations of separate GA and different PNA ranges were used within the simulation.
Published preterm studies reported their results for PK parameters, such as clearance, in different units, i.e. mL/min/kg or mL/min, and at different GA or PNA, without individual data on covariates. We tried to report the prediction in the same way as it is difficult to standardize the results across these different compounds.
3 Results
3.1 Development of the Compound Files
Performance verification for the compound files that are not part of the Simcyp Simulator compound library (alfentanil, gentamicin and vancomycin), are given in the electronic supplementary material (ESM). They have been verified in adults using the healthy volunteer population, and then further verified in the paediatric population.
3.2 Performance Verification of Preterm PBPK
3.2.1 Alfentanil
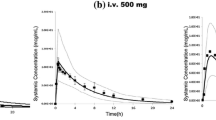
The PBPK prediction of observed alfentanil plasma concentration–time profiles in preterm neonates after single and multiple dosing are given in Fig. 1a, b. Observed values are within the 5th and 95th predicted percentiles. In neonates (from 28 to 40 gestation weeks), the predicted clearance values were in good agreement with the literature data (Fig. 1c). Only three observed clearance data fell out of the predicted 5th–95th percentiles for the simulated virtual preterm populations. Table 5 shows a comparison between the predicted and reported PK parameters for alfentanil in preterm populations.

Prediction of alfentanil pharmacokinetics in neonates. a, b Predicted alfentanil concentration–time profiles in preterm neonates corresponding to trial designs A and B in the Methods section, respectively. The solid line is the predicted mean of the simulated population and the dashed lines represent the 5th and 95th percentiles of the total virtual population. Open circles are the observed data for the mean (a) and individual (b) alfentanil concentration in plasma over time. (c) Predicted versus observed (yellow triangles, individual data [43]; red circle, mean value [42]; blue diamonds, mean value [74]; square, mean value [75]) clearance (mL/min/kg) values of alfentanil in preterm neonates between 28 and 40 weeks’ GA. The error bars represent the standard deviation of the observed data. The predicted means are given as filled black circles, with the associated predicted standard deviation. The dashed lines represent the minimum and maximum, and the shaded area represents the 5th–95th percentiles, with the solid line representing the median for the predicted values. GA gestational age
3.2.2 Midazolam
The PBPK model prediction of observed midazolam mean plasma concentration–time data in premature neonates is given in Fig. 2a, b (black lines), assuming CYP3A5 has the same ontogeny as CYP3A4. Assuming CYP3A5 levels were the same as in the adult populations led to a larger variability of the simulated PK, but had no significant impact on the mean PK profile compared with the same ontogeny as CYP3A4 (Fig. 2a, b, blue lines). The predicted changes in clearance of midazolam in the preterm population over a wide range of body weight was also in agreement with the observed clearance values (Fig. 2c). Table 5 shows a comparison between the predicted and reported PK parameters for midazolam in preterm populations.

Prediction of midazolam kinetics in the preterm population. a, b Predicted midazolam concentration–time profiles in preterm neonates corresponding to trial designs A and B in the Methods section, respectively. Different ontogeny functions of CYP3A5 were applied in the simulation (black lines, same ontogeny as CYP3A4; blue lines, same level as in adults). The solid line is the predicted mean of the simulated population and the dashed lines represent the 5th and 95th percentiles of the total virtual population. Open circles are the observed data for the mean midazolam concentration in plasma over time. The error bars represent the standard deviation of the observed data. c Predicted (grey circle) versus observed individual clearance values (black square [47] and blue triangle [76], extracted from [77]; red diamond [78]) of midazolam clearance in neonates from 28 to 40 weeks’ GA and body weight of 1–4 kg. Broken lines represent the 5th and 95th predictive intervals. CYP cytochrome P450, GA gestational age
3.2.3 Caffeine
Simulated profiles for caffeine after single-dose intravenous administration are shown in Fig. 3a, after a loading dose followed by multiple intravenous doses are shown in Fig. 3b, and after a loading dose followed by multiple oral doses are shown in Fig. 3c. All simulations are in agreement with the observed data. The predicted Ctrough concentrations at day 7 were in agreement with the observed data (Fig. 3d). All these results demonstrated that the model replicated the disposition kinetics adequately. Table 5 shows a comparison between the predicted and reported PK parameters for caffeine in preterm populations. Results after running the full PBPK distribution model are given in the ESM.

Plasma concentration profiles of caffeine in preterm neonates following a a 15 mg/kg single intravenous infusion over 5 min, b an intravenous infusion of a loading dose of 10 mg/kg over 15 min followed by a 2.5 mg/kg/day maintenance dose, and c an oral loading dose of 40 mg/kg followed by a 10 mg/kg/day maintenance dose. Solid lines are the predicted mean and dashed lines are the 5th and 95th percentiles. Observed mean data were from Gorodischer and Karplus [50] for a single intravenous dose (red filled circles), and individual observed data for multiple intravenous doses (orange filled circles) and oral doses (orange filled squares) were from Charles et al. [49]. Plot d is the predicted (grey open circle) versus observed (orange open circle) individual plasma caffeine concentration [79] Ctrough at day 7 following a multiple intravenous dose, against body weight
3.2.4 Ibuprofen
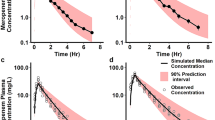
Simulated ibuprofen concentration profiles for multiple-dose intravenous administration (Fig. 4a) and single-dose oral administration (Fig. 4b, c) were comparable with the clinical observations reported in preterm populations. For intravenous ibuprofen [42], the ratios of predicted versus observed mean clearance, AUC from time zero to 24 h (AUC24) and Cmax, for the first dose before closure of the PDA, were 1.9, 1.2 and 1.4, while the ratios for the third dose after PDA closure were 1.5, 1.1 and 1.0, respectively. Predicted and observed individual half-life and peak plasma concentrations of oral ibuprofen in preterm neonates were plotted against their body weight (Fig. 4d). The results suggest that the model can also recover the absorption parameters reasonably well. Table 5 shows a comparison between the predicted and reported PK parameters for ibuprofen in preterm populations.

Prediction of ibuprofen pharmacokinetics in preterm neonates. a After intravenous infusion of 10, 5 and 5 mg/kg over 15 min on days 1, 2 and 3, respectively, to subjects with a mean GA of 28.6 weeks and PNA of 3 days, with observed data (circles with bars are mean ± SD [58], and triangles are individual values [57]); b after a single oral dose of 10 mg/kg to preterm patients aged 0–3 days, who were born at a mean of 28.6 weeks’ GA [59]; and c after a single oral dose of 10 mg/kg to preterm patients aged 0–3 days who were born at a mean of 30.5 weeks’ GA [60]. Circles are observed concentrations, solid lines are predicted concentrations, and dashed lines represent the 5th and 95th percentiles of the total virtual population. d Ibuprofen predicted versus observed individual oral half-life and Cmax against body weight [60]. Grey and red open circles represent predicted and observed half-life, respectively; yellow and black open triangles represent the predicted and observed Cmax, respectively. GA gestational age, PNA postnatal age, SD standard deviation, Cmax maximum concentration
3.2.5 Gentamicin
The PBPK model prediction of gentamicin plasma concentration–time profiles after single (Fig. 5a) and multiple doses (Fig. 5b, c) show good agreement with the observed data. In neonates (from 28 to 40 gestation weeks), the predicted weight-normalized clearances were in good agreement with the reported data in the literature (Fig. 5d). Approximately 80% of observed clearance data fits within the 5th and 95th percentile of the predictions in virtual preterm populations. Table 5 shows a comparison between the predicted and reported PK parameters for gentamicin in preterm populations.

Gentamicin concentration–time profiles. a–c Predicted gentamicin concentration–time profiles in preterm neonates correspond to trial designs A, B and C in the Methods section, respectively. The solid line is the predicted mean of the simulated population and the dashed lines represent the 5th and 95th percentiles of the total virtual population. Open circles are the observed individual (a) or mean (b, c) data, and the error bars are the standard deviation of the observed data. d Predicted median weight normalized clearance (black line) versus the observed individual value in 28–40 weeks’ GA in preterm subjects (yellow triangle [80], purple square [81] and blue cross [82]. The dashed lines represent the minimum and maximum, and the shaded area represents the 5th–95th percentile for the predicted values
3.2.6 Vancomycin
The PBPK model predictions of vancomycin plasma concentration profiles in the preterm population were in good agreement with the observed data after both single (Fig. 6a, b) and multiple doses (Fig. 6c). In neonates (from 28 to 40 gestation weeks’), the predicted weight-normalized clearance (Fig. 6d) was in good agreement with the literature data. Approximately 80% of observed clearance data fit within the 5th and 95th percentiles of the predictions in the virtual preterm populations. Table 5 shows a comparison between the predicted and reported PK parameters for vancomycin in preterm populations.

Prediction of vancomycin pharmacokinetics. a–c Predicted vancomycin concentration–time profiles in preterm neonates corresponding to trial designs A, B and C in the Methods section, respectively. The solid line is the predicted mean of the simulated population and the dashed lines represent the 5th and 95th percentiles of the total virtual population. Open circles are the observed data. d Predicted median weight-normalized clearance (black line) versus the observed individual value in preterm neonates (green diamonds [72], red circles (Serum Creatinine > 0.7 mg/dL) and blue triangles (Serum Creatinine < 0.7 mg/dL) [73]). The dashed lines represent the minimum and maximum, and the shaded area represents the 5th–95th percentile for the predicted values
The predicted PK parameters (AUC, Cmax and clearance) were compared with the observed data and are summarized in Table 5. The predicted mean/median PK parameters were all within twofold of the clinically reported data. Thirty of the 34 predictions were within the 0.67- to 1.5-fold range, and 21 of the 34 predictions were within the 0.8- to 1.25-fold range. In addition, the simulated range and coefficient of variation of the PK parameters covered the range of clinically reported data.
4 Discussion
A preterm PBPK model was successfully developed within the Simcyp Simulator to predict PK in the preterm population and to account for interindividual variability. The model performance was verified for different compounds, namely alfentanil, midazolam, caffeine, ibuprofen, gentamicin and vancomycin, administered to preterm subjects at different PNAs and born at different gestational weeks. PBPK models combine demographic, genetic, physiological and pathological information in adults with in vitro data on human drug metabolism and transport to predict population PK and the extent of metabolic DDIs. In the past, such models were used to predict drug kinetics and the associated variability in adult populations, as well as in the paediatric population born full-term (> 36 gestational weeks). By incorporating information on preterm developmental physiology and the early ontogeny of specific CYPs [24], the models have now been extended to preterm babies.
An understanding of drug kinetics in preterm populations is becoming the focus of different research groups, especially in the area of antimicrobial therapy, anaesthesia, and treatment of ductus arteriosus and neonatal abstinence syndrome [21, 83,84,85,86]. Hence, a PBPK model for preterm subjects was built [26] and the model performance was verified in this work using different compounds covering different PK features. An advantage of preterm PBPK models is that they have the ability to predict the tissue concentration, and hence the pharmacodynamic response (or toxicity) can be linked to the local operative concentration in the tissue.
While the predicted PK profiles and the derived PK parameters for these different compounds were in good agreement with the observed data, there are limitations encountered during the verification process. For example, the reported doses were provided either as a range or average, with variability, and the studied populations were under different disease conditions and co-medications. Simulations were performed without accounting for these conditions, for example the alfentanil study involved administering multiple doses to respiratory distressed preterm subjects, and no account was taken of the effects of respiratory distress on drug disposition. Sometimes the clinical data were limited and, in the alfentanil case above, concentration–time data were only available for the last dose.
No data from multiple doses of midazolam could be found. The predicted Cmax after a single oral administration seems over the observed value, however the observed data for Cmax varied from 21 to 448 ng/mL [48]. For multiple-dose caffeine studies, the maximum simulated PNA was 30 days, while the reported value was 45 days and the reported gestational weeks ranged from 24 to 29 weeks (mean 27.6; simulated GA was 28 weeks). In addition, the original data were not stratified based on these ages, but in terms of the dosage regimen. Use of the mPBPK distribution model with user-input values lumps the organs together, apart from liver, and hence does not account for any age-dependency in tissue-to-plasma partition ratio (Kps). However, a scaling element is implicit within this model as it is parameterized in litres per kilogram. In contrast, the full PBPK distribution model that considers the change in organ volume and composition does account for Kps age-dependency. For the ibuprofen multiple-dose case, high variability in clearance was observed in the clinical studies, which was successfully replicated in the simulated scenarios. While comorbidity and comedications for these patients are potential contributing factors, the role of CYP2C9 genotype polymorphism may also contribute to the observed variability in clearance among these individuals, as suggested in the literature [58, 60]. The CYP2C protein content was not detectable in human liver samples from fetuses during the third trimester and was barely detectable in newborns aged less than 24 h. Nonetheless, it increases steadily in the first week of life [87] and may start to separate clearance of these two populations. Dose 3 observed better prediction of PK parameters than dose 1, which could be due to the fact that about 63–66% of patients have their ductus arteriosus closed after ibuprofen treatment. Mean ibuprofen apparent volume of distribution was found to be significantly lower, and clearance was higher, after the third dose compared with the first dose during ductus closure [58]. It is highly probable that infants with haemodynamically significant PDA may have altered drug disposition. The presence of haemodynamically significant PDA may affect drug disposition [88] by causing reduced blood flow to drug-metabolizing organs (i.e. liver), as well as introducing fluid overload, as data are available on the influence of physiological changes induced by PDA on the PK of gentamicin [89, 90], ibuprofen [55, 57], mefenamic acid [91] and indomethacin [86]. Others suggested no impact of PDA on indomethacin kinetics [92]. It should be noted that the high interindividual PK variability that can be observed in the preterm population can mask the impact of PDA on the drug kinetics [80].
During the course of treatment of PDA with ibuprofen or indomethacin, these perfusion changes can result in a feedback effect on the clearance and volume of distribution of these compounds. If the preterm patient received comedication therapy such as antibiotics, then the PK of the comedicated drug will be affected by these haemodynamic changes during the course of PDA closure. Other feedback changes in the system cannot be ruled out. Mechanisms such as the inhibition of cyclooxygenase cascade due to NSAIDs (i.e. ibuprofen) results in a reduction of renal prostaglandins and renal perfusion, i.e. GFR, and, finally, reduction of renal clearance of drugs eliminated by the kidney, such as gentamicin and amikacin [93]. It should be pointed out that the renal function model incorporated in the current preterm PBPK model accounts for GFR maturation, but not the active secretion.
Where the Vss is predicted using the full minimal or full PBPK distribution model, the tissue-to-plasma partition coefficient (Kp) is calculated based on the tissue composition, which is age-dependent in this population (see Part I), therefore tissue Kp values change as a function of age. The Kp scalar was applied to the calculated tissue Kps in order to recover the adult clinical reported Vss and ensure the compound recovers the adult concentration profile. Compounds that have a Kp scalar applied for the adult model retain the same value for preterm prediction. The fraction of unbound drugs was predicted in the preterm population using the concentration of the binding proteins (albumin or acid glycoprotein [AGP]) in preterm according to their maturation functions [26].
To date, two other preterm PBPK models have been published (Claassen et al. [94] and Fisher et al. [95]), but both presented limited application. The paper by Claassen et al. shows prediction of acetaminophen and amikacin, while the paper by Fisher et al. shows prediction of piperacillin and tazobactam. In this study, we have presented the prediction of another six compounds in the preterm population.
One advantage of the PBPK approach is that sensitivity analysis can be applied on system parameters to investigate their impact on the PK parameters and profiles. For example, sensitivity analysis can be performed on haemodynamic changes caused by PDA, and also to investigate the effect of cyclooxygenase on renal function. Moving forward, it will be highly beneficial to connect PBPK models to pharmacodynamics and disease progression models to be able to propagate and investigate the impact of physiological changes.
5 Conclusions
The developed preterm PBPK model was verified using six compounds with diverse PK properties, administered to preterm subjects at different PNAs and born at different gestational weeks. The model successfully described observed average and, where available, individual concentration–time profiles of these compounds, as well as the derived PK parameters. For relatively long trials, the time-varying physiology features were used to more realistically simulate interaction between individual physiological parameters and compound characteristics. Application of an in vitro–in vivo extrapolation approach, in conjunction with PBPK modelling under a mechanistic systems biology approach, can help to predict drug exposure and inform the design of clinical studies, as well as allow safety and efficacy risk assessment in the preterm patient population.
References
Allegaert K. Better medicines for neonates: Improving medicine development, testing, and prescribing. Early Hum Dev. 2017;114:22–5.
Girardi A, Galletti S, Raschi E, Koci A, Poluzzi E, Faldella G, et al. Pattern of drug use among preterm neonates: results from an Italian neonatal intensive care unit. Ital J Pediatr. 2017;43:37.
European Medicines Agency. Concept paper on the need for revision of the guideline on the investigation of medicinal products in the term and preterm neonate. 2018. https://www.ema.europa.eu/documents/scientific-guideline/concept-paper-need-revision-guideline-investigation-medicinal-products-term-preterm-neonate-revision_en.pdf. Accessed 21 Oct 2018.
US FDA. Guidance for Industry; General Clinical Pharmacology Considerations for Pediatric Studies for drugs and Biological Products. 2014. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-clinical-pharmacology-considerations-pediatric-studies-drugs-and-biological-products.
Leroux S, Zhao W, Betremieux P, Pladys P, Saliba E, Jacqz-Aigrain E, et al. Therapeutic guidelines for prescribing antibiotics in neonates should be evidence-based: a French national survey. Arch Dis Child. 2015;100:394–8.
Girardi A, Raschi E, Galletti S, Poluzzi E, Faldella G, Allegaert K, et al. Drug-induced renal damage in preterm neonates: state of the art and methods for early detection. Drug Saf. 2015;38:535–51.
Shah PS, Dunn M, Lee SK, Allen AC, Singhal N, Canadian Neonatal N. Early opioid infusion and neonatal outcomes in preterm neonates ≤ 28 weeks’ gestation. Am J Perinatol. 2011;28:361–6.
US FDA. Serious health problems seen in premature babies given Kaletra (lopinavir/ritonavir) oral solution. 2011. www.fda.gov/drugs/drugsafety/ucm246002.htm. Accessed 21 Oct 2018,
European Medicines Agency. Assessment report; Kaletra. EMA/CHMP/443456/2017. 2017. https://www.ema.europa.eu/documents/variation-report/kaletra-h-c-368-ii-0161-g-epar-assessment-report-variation_en.pdf. Accessed 21 Oct 2018.
Madadi P, Ross CJ, Hayden MR, Carleton BC, Gaedigk A, Leeder JS, et al. Pharmacogenetics of neonatal opioid toxicity following maternal use of codeine during breastfeeding: a case-control study. Clin Pharmacol Ther. 2009;85:31–5.
McPherson C, Haslam M, Pineda R, Rogers C, Neil JJ, Inder TE. Brain injury and development in preterm infants exposed to fentanyl. Ann Pharmacother. 2015;49:1291–7.
Friis-Hansen B. Body water compartments in children: changes during growth and related changes in body composition. Pediatrics. 1961;28:169–81.
Fuchs A, Guidi M, Giannoni E, Werner D, Buclin T, Widmer N, et al. Population pharmacokinetic study of gentamicin in a large cohort of premature and term neonates. Br J Clin Pharmacol. 2014;78:1090–101.
Gonzalez D, Delmore P, Bloom BT, Cotten CM, Poindexter BB, McGowan E, et al. Clindamycin pharmacokinetics and safety in preterm and term infants. Antimicrob Agents Chemother. 2016;60:2888–94.
Ward RM, Benjamin D, Barrett JS, Allegaert K, Portman R, Davis JM, et al. Safety, dosing, and pharmaceutical quality for studies that evaluate medicinal products (including biological products) in neonates. Pediatr Res. 2017;81:692–711.
Ku LC, Smith PB. Dosing in neonates: special considerations in physiology and trial design. Pediatr Res. 2015;77:2–9.
Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, Rostami-Hodjegan A. The Simcyp population-based ADME simulator. Expert Opin Drug Metab Toxicol. 2009;5:211–23.
Shebley M, Sandhu P, Emami Riedmaier A, Jamei M, Narayanan R, Patel A, et al. Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin Pharmacol Ther. 2018;104:88–110.
Luzon E, Blake K, Cole S, Nordmark A, Versantvoort C, Berglund EG. Physiologically based pharmacokinetic modeling in regulatory decision-making at the European Medicines Agency. Clin Pharmacol Ther. 2016;102:98–105.
Wagner C, Zhao P, Pan Y, Hsu V, Grillo J, Huang SM, et al. Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA public workshop on PBPK. CPT Pharmacometr Syst Pharmacol. 2015;4:226–30.
Brussee JM, Yu H, Krekels EHJ, de Roos B, Brill MJE, van den Anker JN, et al. First-pass CYP3A-mediated metabolism of midazolam in the gut wall and liver in preterm neonates. CPT Pharmacometr Syst Pharmacol. 2018;7:374–83.
Emoto C, Johnson TN, Neuhoff S, Hahn D, Vinks AA, Fukuda T. PBPK model of morphine incorporating developmental changes in hepatic OCT1 and UGT2B7 proteins to explain the variability in clearances in neonates and small infants. CPT Pharmacometr Syst Pharmacol. 2018;7:464–73.
Michelet R, Van Bocxlaer J, Allegaert K, Vermeulen A. The use of PBPK modeling across the pediatric age range using propofol as a case. J Pharmacokinet Pharmacodyn. 2018;45:765–85.
Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. 2006;45:931–56.
Templeton IE, Jones NS, Musib L. Pediatric dose selection and utility of PBPK in determining dose. AAPS J. 2018;20:31.
Abduljalil K, Pan X, Pansari A, Jamei M, Johnson TN. A Preterm physiologically-based pharmacokinetic model. Part I: physiological parameters and model building. Clin Pharmacokinet. 2019. https://doi.org/10.1007/s40262-019-00825-6.
Berezhkovskiy LM. Volume of distribution at steady state for a linear pharmacokinetic system with peripheral elimination. J Pharm Sci. 2004;93:1628–40.
Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95:1238–57.
Schuttler J, Stoeckel H. Clinical pharmacokinetics of alfentanyl (author’s transl) [in German]. Anaesthesist. 1982;31:10–4.
Meuldermans W, Van Peer A, Hendrickx J, Woestenborghs R, Lauwers W, Heykants J, et al. Alfentanil pharmacokinetics and metabolism in humans. Anesthesiology. 1988;69:527–34.
Kharasch ED, Walker A, Isoherranen N, Hoffer C, Sheffels P, Thummel K, et al. Influence of CYP3A5 genotype on the pharmacokinetics and pharmacodynamics of the cytochrome P4503A probes alfentanil and midazolam. Clin Pharmacol Ther. 2007;82:410–26.
Labroo R, Kharasch ED. Gas chromatographic-mass spectrometric analysis of alfentanil metabolites. Application to human liver microsomal alfentanil biotransformation. J Chromatogr B Biomed Appl. 1994;660:85–94.
Kharasch ED, Hoffer C, Whittington D, Walker A, Bedynek PS. Methadone pharmacokinetics are independent of cytochrome P4503A (CYP3A) activity and gastrointestinal drug transport: insights from methadone interactions with ritonavir/indinavir. Anesthesiology. 2009;110:660–72.
Mather LE. Clinical pharmacokinetics of fentanyl and its newer derivatives. Clin Pharmacokinet. 1983;8:422–46.
Meuldermans WE, Hurkmans RM, Heykants JJ. Plasma protein binding and distribution of fentanyl, sufentanil, alfentanil and lofentanil in blood. Arch Int Pharmacodyn Ther. 1982;257:4–19.
Beaumont K, Gardner I, Chapman K, Hall M, Rowland M. Toward an integrated human clearance prediction strategy that minimizes animal use. J Pharm Sci. 2011;100:4518–35.
Bower S, Hull CJ. Comparative pharmacokinetics of fentanyl and alfentanil. Br J Anaesth. 1982;54:871–7.
Meuldermans W, Woestenborghs R, Noorduin H, Camu F, van Steenberge A, Heykants J. Protein binding of the analgesics alfentanil and sufentanil in maternal and neonatal plasma. Eur J Clin Pharmacol. 1986;30:217–9.
Lacroix D, Sonnier M, Moncion A, Cheron G, Cresteil T. Expression of CYP3A in the human liver–evidence that the shift between CYP3A7 and CYP3A4 occurs immediately after birth. Eur J Biochem. 1997;247:625–34.
Stevens JC, Hines RN, Gu C, Koukouritaki SB, Manro JR, Tandler PJ, et al. Developmental expression of the major human hepatic CYP3A enzymes. J Pharmacol Exp Ther. 2003;307:573–82.
Hines RN. Ontogeny of human hepatic cytochromes P450. J Biochem Mol Toxicol. 2007;21:169–75.
Davis PJ, Killian A, Stiller RL, Cook DR, Guthrie RD, Scierka AM. Pharmacokinetics of alfentanil in newborn premature infants and older children. Dev Pharmacol Ther. 1989;13:21–7.
Wiest DB, Ohning BL, Garner SS. The disposition of alfentanil in neonates with respiratory distress. Pharmacotherapy. 1991;11:308–11.
Allonen H, Ziegler G, Klotz U. Midazolam kinetics. Clin Pharmacol Ther. 1981;30:653–61.
Kupferschmidt HH, Ha HR, Ziegler WH, Meier PJ, Krahenbuhl S. Interaction between grapefruit juice and midazolam in humans. Clin Pharmacol Ther. 1995;58:20–8.
Vyhlidal CA, Pearce RE, Gaedigk R, Calamia JC, Shuster DL, Thummel KE, et al. Variability in expression of CYP3A5 in human fetal liver. Drug Metab Dispos. 2015;43:1286–93.
de Wildt SN, Kearns GL, Hop WC, Murry DJ, Abdel-Rahman SM, van den Anker JN. Pharmacokinetics and metabolism of intravenous midazolam in preterm infants. Clin Pharmacol Ther. 2001;70:525–31.
de Wildt SN, Kearns GL, Hop WC, Murry DJ, Abdel-Rahman SM, van den Anker JN. Pharmacokinetics and metabolism of oral midazolam in preterm infants. Br J Clin Pharmacol. 2002;53:390–2.
Charles BG, Townsend SR, Steer PA, Flenady VJ, Gray PH, Shearman A. Caffeine citrate treatment for extremely premature infants with apnea: population pharmacokinetics, absolute bioavailability, and implications for therapeutic drug monitoring. Ther Drug Monit. 2008;30:709–16.
Gorodischer R, Karplus M. Pharmacokinetic aspects of caffeine in premature infants with apnoea. Eur J Clin Pharmacol. 1982;22:47–52.
Salem F, Johnson TN, Abduljalil K, Tucker GT, Rostami-Hodjegan A. A re-evaluation and validation of ontogeny functions for cytochrome P450 1A2 and 3A4 based on in vivo data. Clin Pharmacokinet. 2014;53:625–36.
Kuehl GE, Lampe JW, Potter JD, Bigler J. Glucuronidation of nonsteroidal anti-inflammatory drugs: identifying the enzymes responsible in human liver microsomes. Drug Metab Dispos. 2005;33:1027–35.
Chang SY, Li W, Traeger SC, Wang B, Cui D, Zhang H, et al. Confirmation that cytochrome P450 2C8 (CYP2C8) plays a minor role in (S)-(+)- and (R)-(−)-ibuprofen hydroxylation in vitro. Drug Metab Dispos. 2008;36:2513–22.
Beloica S, Cvijic S, Bogataj M, Parojcic J. In vitro-in vivo-in silico approach in biopharmaceutical characterization of ibuprofen IR and SR tablets. Eur J Pharm Sci. 2015;75:151–9.
Aranda JV, Varvarigou A, Beharry K, Bansal R, Bardin C, Modanlou H, et al. Pharmacokinetics and protein binding of intravenous ibuprofen in the premature newborn infant. Acta Paediatr. 1997;86:289–93.
Rainsford K. Ibuprofen: pharmacology, efficacy and safety. Inflammopharmacology. 2009;17:275–342.
Hirt D, Van Overmeire B, Treluyer JM, Langhendries JP, Marguglio A, Eisinger MJ, et al. An optimized ibuprofen dosing scheme for preterm neonates with patent ductus arteriosus, based on a population pharmacokinetic and pharmacodynamic study. Br J Clin Pharmacol. 2008;65:629–36.
Van Overmeire B, Touw D, Schepens PJ, Kearns GL, van den Anker JN. Ibuprofen pharmacokinetics in preterm infants with patent ductus arteriosus. Clin Pharmacol Ther. 2001;70:336–43.
Barzilay B, Youngster I, Batash D, Keidar R, Baram S, Goldman M, et al. Pharmacokinetics of oral ibuprofen for patent ductus arteriosus closure in preterm infants. Arch Dis Child Fetal Neonatal Ed. 2012;97:F116–9.
Sharma PK, Garg SK, Narang A. Pharmacokinetics of oral ibuprofen in premature infants. J Clin Pharmacol. 2003;43:968–73.
Yang J, Jamei M, Yeo KR, Tucker GT, Rostami-Hodjegan A. Prediction of intestinal first-pass drug metabolism. Curr Drug Metab. 2007;8:676–84.
Lee MG, Chen ML, Huang SM, Chiou WL. Pharmacokinetics of drugs in blood. I. Unusual distribution of gentamicin. Biopharm Drug Dispos. 1981;2:89–97.
Goodman LS, Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman’s the pharmacological basis of therapeutics. 10th ed. New York: McGraw-Hill; 2001.
Dickson CJ, Schwartzman MS, Bertino JS Jr. Factors affecting aminoglycoside disposition: effects of circadian rhythm and dietary protein intake on gentamicin pharmacokinetics. Clin Pharmacol Ther. 1986;39:325–8.
Nielsen EI, Sandstrom M, Honore PH, Ewald U, Friberg LE. Developmental pharmacokinetics of gentamicin in preterm and term neonates: population modelling of a prospective study. Clin Pharmacokinet. 2009;48:253–63.
Krishnan L, George SA. Gentamicin therapy in preterms: a comparison of two dosage regimens. Indian Pediatr. 1997;34:1075–80.
Shin WG, Lee MG, Lee MH, Kim ND. Pharmacokinetics of drugs in blood. VII: Unusual distribution and blood storage effect of vancomycin. Biopharm Drug Dispos. 1992;13:305–10.
Kees MG, Wicha SG, Seefeld A, Kees F, Kloft C. Unbound fraction of vancomycin in intensive care unit patients. J Clin Pharmacol. 2014;54:318–23.
Matzke GR, McGory RW, Halstenson CE, Keane WF. Pharmacokinetics of vancomycin in patients with various degrees of renal function. Antimicrob Agents Chemother. 1984;25:433–7.
Schaad UB, McCracken GH Jr, Nelson JD. Clinical pharmacology and efficacy of vancomycin in pediatric patients. J Pediatr. 1980;96:119–26.
Reed MD, Kliegman RM, Weiner JS, Huang M, Yamashita TS, Blumer JL. The clinical pharmacology of vancomycin in seriously ill preterm infants. Pediatr Res. 1987;22:360–3.
Asbury WH, Darsey EH, Rose WB, Murphy JE, Buffington DE, Capers CC. Vancomycin pharmacokinetics in neonates and infants: a retrospective evaluation. Ann Pharmacother. 1993;27:490–6.
Capparelli EV, Lane JR, Romanowski GL, McFeely EJ, Murray W, Sousa P, et al. The influences of renal function and maturation on vancomycin elimination in newborns and infants. J Clin Pharmacol. 2001;41:927–34.
Killian A, Davis PJ, Stiller RL, Cicco R, Cook DR, Guthrie RD. Influence of gestational age on pharmacokinetics of alfentanil in neonates. Dev Pharmacol Ther. 1990;15:82–5.
Marlow N, Weindling AM, Van Peer A, Heykants J. Alfentanil pharmacokinetics in preterm infants. Arch Dis Child. 1990;65:349–51.
Jacqz-Aigrain E, Daoud P, Burtin P, Desplanques L, Beaufils F. Placebo-controlled trial of midazolam sedation in mechanically ventilated newborn babies. Lancet. 1994;344:646–50.
Ince I, de Wildt SN, Wang C, Peeters MY, Burggraaf J, Jacqz-Aigrain E, et al. A novel maturation function for clearance of the cytochrome P450 3A substrate midazolam from preterm neonates to adults. Clin Pharmacokinet. 2013;52:555–65.
Mansoor N, Ahmad T, Alam Khan R, Sharib SM, Mahmood I. Prediction of clearance and dose of midazolam in preterm and term neonates: a comparative study between allometric scaling and physiologically based pharmacokinetic modeling. Am J Ther. 2016;26:e32–7.
Leon AE, Michienzi K, Ma CX, Hutchison AA. Serum caffeine concentrations in preterm neonates. Am J Perinatol. 2007;24:39–47.
Touw DJ, Proost JH, Stevens R, Lafeber HN, van Weissenbruch MM. Gentamicin pharmacokinetics in preterm infants with a patent and a closed ductus arteriosus. Pharm World Sci. 2001;23:200–4.
Rocha MJ, Almeida AM, Afonso E, Martins V, Santos J, Leitao F, et al. The kinetic profile of gentamicin in premature neonates. J Pharm Pharmacol. 2000;52:1091–7.
Edginton AN, Schmitt W, Voith B, Willmann S. A mechanistic approach for the scaling of clearance in children. Clin Pharmacokinet. 2006;45:683–704.
Anand KJ, Anderson BJ, Holford NH, Hall RW, Young T, Shephard B, et al. Morphine pharmacokinetics and pharmacodynamics in preterm and term neonates: secondary results from the NEOPAIN trial. Br J Anaesth. 2008;101:680–9.
Momper JD, Capparelli EV, Wade KC, Kantak A, Dhanireddy R, Cummings JJ, et al. Population pharmacokinetics of fluconazole in premature infants with birth weights less than 750 grams. Antimicrob Agents Chemother. 2016;60:5539–45.
Moore JN, Gastonguay MR, Ng CM, Adeniyi-Jones SC, Moody DE, Fang WB, et al. The pharmacokinetics and pharmacodynamics of buprenorphine in neonatal abstinence syndrome. Clin Pharmacol Ther. 2018;103:1029–37.
Gal P, Ransom JL, Weaver RL, Schall S, Wyble LE, Carlos RQ, et al. Indomethacin pharmacokinetics in neonates: the value of volume of distribution as a marker of permanent patent ductus arteriosus closure. Ther Drug Monit. 1991;13:42–5.
Treluyer JM, Gueret G, Cheron G, Sonnier M, Cresteil T. Developmental expression of CYP2C and CYP2C-dependent activities in the human liver: in vivo/in vitro correlation and inducibility. Pharmacogenetics. 1997;7:441–52.
Gal P, Gilman JT. Drug disposition in neonates with patent ductus arteriosus. Ann Pharmacother. 1993;27:1383–8.
Williams BS, Ransom JL, Gal P, Carlos RQ, Smith M, Schall SA. Gentamicin pharmacokinetics in neonates with patent ductus arteriosus. Crit Care Med. 1997;25:273–5.
Watterberg KL, Kelly HW, Johnson JD, Aldrich M, Angelus P. Effect of patent ductus arteriosus on gentamicin pharmacokinetics in very low birth weight (less than 1,500 g) babies. Dev Pharmacol Ther. 1987;10:107–17.
Ito K, Niida Y, Sato J, Owada E, Ito K, Umetsu M. Pharmacokinetics of mefenamic acid in preterm infants with patent ductus arteriosus. Acta Paediatr Jpn. 1994;36:387–91.
Al Za’abi M, Donovan T, Tudehope D, Woodgate P, Collie LA, Charles B. Orogastric and intravenous indomethacin administration to very premature neonates with patent ductus arteriosus: population pharmacokinetics, absolute bioavailability, and treatment outcome. Ther Drug Monit. 2007;29:807–14.
De Cock RF, Allegaert K, Schreuder MF, Sherwin CM, de Hoog M, van den Anker JN, et al. Maturation of the glomerular filtration rate in neonates, as reflected by amikacin clearance. Clin Pharmacokinet. 2012;51:105–17.
Claassen K, Thelen K, Coboeken K, Gaub T, Lippert J, Allegaert K, Willmann, et al. Development of a physiologically-based pharmacokinetic model for preterm neonates: evaluation with in vivo data. Curr Pharm Des. 2015;21(39):5688–98.
Fisher JW, Wu H, Cohen-Wolkowiez M, Watt K, Wang J, Burckart GJ, et al. Predicting the pharmacokinetics of piperacillin and tazobactam in preterm and term neonates using physiologically based pharmacokinetic modeling. Comput Toxicol. 2019;12:100104.
Acknowledgements
The authors thank Eleanor Savill and Rosalie Bower for their assistance with collecting the references and preparing the manuscript, and Ruth Clayton for her helpful comments and proofreading. The authors also thank the reviewers of this manuscript for their helpful suggestions and comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the preparation of this study.
Conflict of interest
Khaled Abduljalil, Xian Pan, Amita Pansari, Trevor N. Johnson and Masoud Jamei are full-time employees of Certara UK Limited, Simcyp Division. The activities of Certara are supported by a consortium of pharmaceutical companies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Abduljalil, K., Pan, X., Pansari, A. et al. Preterm Physiologically Based Pharmacokinetic Model. Part II: Applications of the Model to Predict Drug Pharmacokinetics in the Preterm Population. Clin Pharmacokinet 59, 501–518 (2020). https://doi.org/10.1007/s40262-019-00827-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00827-4