
Abstract
Objective
The aims of this study were to (1) determine whether opportunistically collected data can be used to develop physiologically based pharmacokinetic (PBPK) models in pediatric patients; and (2) characterize age-related maturational changes in drug disposition for the renally eliminated and hepatically metabolized antibiotic trimethoprim (TMP)–sulfamethoxazole (SMX).
Methods
We developed separate population PBPK models for TMP and SMX in children after oral administration of the combined TMP–SMX product and used sparse and opportunistically collected plasma concentration samples to validate our pediatric model. We evaluated predictability of the pediatric PBPK model based on the number of observed pediatric data out of the 90% prediction interval. We performed dosing simulations to target organ and tissue (skin) concentrations greater than the methicillin-resistant Staphylococcus aureus (MRSA) minimum inhibitory concentration (TMP 2 mg/L; SMX 9.5 mg/L) for at least 50% of the dosing interval.
Results
We found 67–87% and 71–91% of the observed data for TMP and SMX, respectively, were captured within the 90% prediction interval across five age groups, suggesting adequate fit of our model. Our model-rederived optimal dosing of TMP at the target tissue was in the range of recommended dosing for TMP–SMX in children in all age groups by current guidelines for the treatment of MRSA.
Conclusion
We successfully developed a pediatric PBPK model of the combination antibiotic TMP–SMX using sparse and opportunistic pediatric pharmacokinetic samples. This novel and efficient approach has the potential to expand the use of PBPK modeling in pediatric drug development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Opportunistic pharmacokinetic data were successfully used to evaluate the predictive accuracy of a trimethoprim (TMP)–sulfamethoxazole (SMX) pediatric physiologically based pharmacokinetic (PBPK) model. |
The model predicted TMP and SMX exposures reasonably well and recommended doses within the range of current Infectious Diseases Society of America (IDSA) guidance for methicillin-resistant Staphylococcus aureus (MRSA) treatment. |
Given the greater access to opportunistic pediatric pharmacokinetic data, this method holds great promise to increase the development of PBPK models in children. |
1 Introduction
Trimethoprim (TMP)–sulfamethoxazole (SMX) is a combination of two synthetic antibiotics that inhibit protein and nucleic acid synthesis [1]. Both TMP and SMX are Biopharmaceutics Classification System (BCS) class 2 compounds (low solubility and high permeability) [2, 3]. In adults, after oral administration, TMP and SMX are rapidly absorbed, with a mean time to peak plasma concentration (Cmax) of 1–4 h [4,5,6,7,8,9]. In adults, the apparent clearance of TMP is 0.084–0.168 L/kg/h and volume of distribution is 1.4–1.8 L/kg, and the apparent clearance of SMX is 0.0126–0.015 L/kg/h and volume of distribution is 0.25–0.27 L/kg [6, 7, 10].
TMP–SMX is labeled for adults to treat Gram-negative organisms and certain Gram-positive aerobic organisms, and it is commonly used off-label in children with methicillin-resistant Staphylococcus aureus (MRSA) infections [11]. In in vitro time–kill studies, TMP–SMX demonstrated time-dependent bactericidal activity against MRSA at drug concentrations four times the mean inhibitory concentration (MIC) [12,13,14]. However, in vivo pediatric pharmacokinetic data are limited to small studies of < 20 children each [15, 16]. This paucity of pharmacokinetic data exposes children to dosing risks, including inadequate treatment or toxicity; however, filling this gap remains challenging. Prospective pharmacokinetic studies characterizing drug disposition across the pediatric development continuum remain hampered by informed consent barriers, rigidity of traditional trial design, and cost, among other factors [17,18,19].
Physiologically based pharmacokinetic (PBPK) models are mathematical tools that integrate drug- and systems-specific information to predict the effect of certain factors on drug exposure [11, 20]. Owing to its physiologically based structure, PBPK models allow extrapolation of pharmacokinetic estimates between developmentally unique age groups [21]. This approach has been used to predict dosing regimens for pediatric clinical trials by accounting for developmental changes that affect drug absorption, distribution, metabolism, and excretion (ADME) parameters [11]. A major barrier to widespread use of PBPK models in pediatric regulatory submissions is the availability of drug concentration data to validate model predictions. We previously presented an approach that leverages sparse and opportunistically collected pharmacokinetic data to develop and validate a pediatric PBPK model for the hepatically eliminated drug clindamycin [22]. This resulted in acceptable model performance, but questions about generalizability remain, particularly regarding drugs with different elimination pathways. Here, in order to investigate pediatric dosing of TMP–SMX, we applied the adult PBPK model to a new pediatric model, altering key parameters in PK-Sim® (version 5.5; Bayer Technology Services, Leverkusen, Germany), and then tested it against opportunistically collected patient plasma concentrations.
2 Methods
2.1 Model Development Workflow
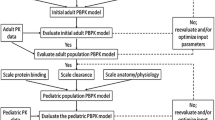
We followed US Food and Drug Administration (FDA) guidance of PBPK model development and workflow in children to build adult and pediatric PBPK models for TMP and SMX (Fig. 1) [23, 24]. We first developed an adult PBPK model and used concentration versus time data extracted from the literature to assess the quality of model predictions of both TMP and SMX. The final adult model served as a basis for developing the pediatric model. In the pediatric model, we maintained the physico-chemical and drug-specific ADME parameters for each drug and replaced the anthropomorphic and physiological information (such as age-dependent enzyme activity and plasma protein concentration) with pediatric values using established age-dependent algorithms in PK-Sim®. Given the sparse availability of opportunistic data in children, we assessed the model predictions of concentration versus time through simulations with virtual populations of children ranging from 2 months to 18 years of age. We excluded infants < 2 months old because TMP–SMX is contraindicated in this population due to the risk of hyperbilirubinemia and kernicterus [11]. We generated a prediction interval (5th to 95th percentile) of drug concentrations per timepoint for the population and quantified the number of observed concentrations outside the prediction interval.

Model-building workflow for pediatric physiologically based pharmacokinetic model
2.2 Clinical Data for Model Development
We identified relevant adult TMP and SMX concentration versus time data in the literature using a systematic search of PubMed with the terms ‘trimethoprim’, ‘sulfamethoxazole’, ‘pharmacokinetics’, and ‘pharmacokinetic’. We selected four adult studies with the most appropriate concentration versus time data based on administration route, study subjects, and analytical methods for measuring TMP and SMX concentrations. Pharmacokinetic parameters and patient demographics are summarized in the Electronic Supplementary Material (ESM) (Online Resource 1).
To develop the pediatric PBPK model, we obtained individual TMP and SMX plasma concentration versus time data after oral administration of TMP–SMX from an opportunistic pharmacokinetic study (POPS [Pharmacokinetics of Understudied Drugs Administered to Children Per Standard of Care], ClinicalTrials.gov identifier NCT01431326 [57]). We collected pharmacokinetic samples in children receiving TMP–SMX per standard of care with standard-of-care laboratory collections at prespecified sampling windows: 0.5–2 h, 2–8 h, and within 1 h prior to the next dose. We measured TMP and SMX concentrations separately using a validated bioanalytical assay [25]. We excluded concentration versus time data from pre-term infants; obese patients (body mass index > 95th percentile); those with abnormal laboratory values (serum creatinine > 1.2 mg/dL, albumin < 3 g/dL); patients who received TMP and SMX via nasojejunal, gastrostomy, or jejunostomy tubes; and those on extracorporeal membrane oxygenation support.
We used PK-Sim® for model development and simulation and STATA® (version 13.1, StataCorp LP, College Station, TX, USA) for output analysis.
2.3 Adult Model Development
Using the standard whole-body 15-organ PBPK model implemented in PK-Sim®, we predicted tissue/plasma partition coefficients (Kp) using the in silico tissue composition approach proposed by Rodgers and colleagues [26,27,28]. Organs were kinetically equivalent to well-stirred compartments. TMP was mainly eliminated renally: 61–85% was excreted unchanged in urine [6, 7, 10]. Conversely, only 10–12% of SMX was excreted unchanged in urine [7].
The cytochrome P450 (CYP) enzymes (CYP2C9 and CYP3A4) are the liver enzymes contributing to TMP metabolism [29]. SMX is primarily eliminated by metabolism via CYP2C9 and N-acetyltransferase 2 (NAT2) [30,31,32]. We did not find information in the literature about the contribution of each enzyme to metabolism of TMP and SMX. Therefore, we assumed equal contribution (CYP3A4 and CYP2C9 for TMP; CYP2C9 and NAT2 for SMX). We incorporated literature values of drug physico-chemical parameters such as LogP, the negative logarithm of the acid dissociation constant (pKa), solubility, and ADME data for TMP and SMX separately in our initial adult model. We obtained other ADME data from DrugBank [33]. We used a Weibull function to describe the oral formulation dissolution process.
For both TMP and SMX, we approximated renal clearance using two methods: (1) total plasma clearance and percentage of dose recovered in urine as unchanged; and (2) glomerular filtration rate (GFR) and unbound fraction of drug in plasma. Method 1 reflects true renal clearance, whereas method 2 reflects renal clearance assuming the net effect of reabsorption and tubular secretion is zero. We then compared estimated clearance values to determine whether the model needed an active secretion or reabsorption process. SMX had a higher renal clearance estimated from method 2, so we assumed that renal clearance of SMX was the net effect of glomerular filtration and reabsorption. TMP had a higher renal clearance estimated from method 1, so we assumed that renal clearance of TMP consisted of GFR and tubular secretion.
We first simulated plasma concentration–time profiles after intravenous administration of TMP-SMX in an adult with age and body weight corresponding to the average values reported in adult pharmacokinetic studies by Dudley et al. [34] (ESM Online Resource 1). We then compared the observed concentration–time data from this study with the simulated data using the initial adult PBPK model. We evaluated model predictions using a visual check of any substantial discrepancy between the PBPK model and the observed mean intravenous concentration–time curve. Next, we optimized model parameters for TMP and SMX, including intrinsic clearance, renal filtration fraction, and tubular secretion, using the concentration–time data from this study [33]. Using the intermediate adult PBPK model with optimized parameter values predicting TMP and SMX concentrations after intravenous administration and default parameter values for absorption-related processes in PK-Sim®, we simulated concentration-time data in an adult with average age and body weight after oral administration of TMP–SMX from the selected adult pharmacokinetic studies [6, 7, 35]. We optimized dissolution time, dissolution shape, lag time, intestinal permeability, and solubility by comparing simulated versus observed concentration–time data from multiple oral dose studies and using findings from an in vitro dissolution study of TMP–SMX (dissolution time: 10–15 min), solubility of TMP (400 mg/L) and SMX (610 mg/L) in water, and an in vitro permeability study of TMP (0.001897 cm/min) and SMX (0.00079 cm/min) [33, 36, 37].
To account for inter-individual pharmacokinetic variability of critical physiological parameters in adults, we developed an adult population PBPK model for each drug by incorporating virtual populations while maintaining the physico-chemical and ADME parameters from the finalized adult PBPK model. We created virtual populations of adults (n = 100) using the demographic (sex, age, weight) distribution reported in the selected papers for TMP and SMX adult concentration–time data after oral administration of TMP–SMX [6, 7, 35]. We included inter-individual pharmacokinetic variability parameters (expressed as the percentage coefficient of variation [CV%]) in enzymes and transporters (CYP3A4 [81%], CYP2C9 [54%], and transporter for tubular secretion for TMP [25%]; CYP2C9 [54%] and NAT2 [20%] for SMX) extracted from the literature [38,39,40]. We evaluated the adult population PBPK model by comparing the distribution of observed plasma concentration data versus the simulated data from the virtual population. We considered a model adequate if the mean ± standard deviation (SD) values of the simulated data were consistent with the observed data.
2.4 Pediatric Model Development
2.4.1 Anatomical and Physiological Parameterization
We used pre-established age-dependent algorithms in PK-Sim® to generate anatomical and physiological parameters—including body weight, height, organ weights, blood flows, cardiac output, gastric emptying time, total body water, and lipid and protein concentrations—for children from 2 months to 18 years of age [41, 42]. We used default values of these parameters in our pediatric PBPK model.
2.4.2 Scaling Absorption
We assumed parameter values for dissolution shape, dissolution time, lag time, and intestinal permeability were the same in children and adults. The sensitivity coefficient for gastric emptying time is < 0.1.
2.4.3 Scaling Unbound Fraction
We estimated the unbound fraction of TMP and SMX in children using the default albumin ontogeny function in PK-Sim® and unbound fraction of TMP and SMX in adults [11, 43].
2.4.4 Scaling Renal Clearance
We assumed the same mechanisms of renal elimination of TMP and SMX in adults and children. Pediatric renal clearance was estimated based on adult values and the developmental changes in GFR and tubular secretion. We used the default age-dependent value for GFR in PK-Sim®. Because the transporter for tubular secretion was unknown, we used the age-dependence of tubular secretion published by Hayton [38]. We used the following equations in the scaling of renal clearance (CLR):
where CLGF(child) and CLGF(adult) are the scaled CLR due to a net effect of glomerular filtration and renal reabsorption in children and adults; fu,p(child) and fu,p(adult) are the scaled fraction unbound (plasma) in children and adults; GFRchild and GFRadult are age-specific GFRs in children and adults; CLTS(child) and CLTS(adult) are the scaled CLR due to tubular secretion in children and adults; and TSchild and TSadult are age-specific tubular secretion rates in the children and adults.
2.4.5 Scaling Hepatic Clearance
We calculated the total hepatic clearance of TMP and SMX in children as the sum of scaled values of individual clearance pathways using a physiologically based approach. The process of physiologic hepatic clearance scaling is based on the assumption that pathways of clearance are the same in adults and children, well-stirred model conditions hold, and enzyme metabolism follows first-order kinetics [44].
We used default settings for hepatic CYP3A4 and CYP2C9 ontogeny in PK-Sim®. Enzyme activity of CYP3A4 is, on average, 12% of the adult value at term, increases to 80% by the age of 1.3 years, and reaches adult activity by 5 years [45]. Enzyme activity of CYP2C9 reaches adult activity at 7 months of age. For hepatic NAT2, we derived an ontogeny function based on a maturation function of SMX clearance in a population pharmacokinetic model for SMX in children, but replaced postnatal age with postmenstrual age [46]. Using this maturation function, enzyme activity of NAT2 is 0.4% of the adult value at term, increases to 80% by 2.4 months, and reaches adult activity by 3 years of age [46]. This assumption was necessary, given the number of enzymatic and physiologic systems involved in the clearance process for which ontogeny functions remain unknown. We calculated scaled pediatric intrinsic clearance estimates from adult values using the following formulas:
where CLint CYP3A4(child)/g liver is the scaled intrinsic clearance due to CYP3A4 per gram of liver in children; CLint CYP2C9(child)/g liver is the scaled intrinsic clearance due to CYP2C9 per gram of liver in children; CLint NAT2(child)/g liver is the scaled intrinsic clearance due to NAT2 per gram of liver in children; CLint CYP3A4(adult)/g liver is the intrinsic clearance due to CYP3A4 per gram of liver in adults; CLint CYP2C9(adult)/g liver is the intrinsic clearance due to CYP2C9 per gram of liver in adults; CLint NAT2(adult)/g liver is the intrinsic clearance due to NAT2 per gram of liver in adults; OSFCYP3A4 is the ontogeny scaling factor for CYP3A4 corresponding to the age of the child; OSFCYP2C9 is the ontogeny scaling factor for CYP2C9 corresponding to the age of the child; and OSFNAT2 is the ontogeny scaling factor for NAT2 corresponding to the age of the child. We evaluated the area under the concentration–time curve (AUC) and peak plasma concentration (Cmax) assuming 10% less and 10% more contribution of CYP2C9 in an average child at age 2 months, 6 months, 1 year, 2 years, and 12 years.
2.4.6 Pediatric Dose Optimization
Using the population module in PK-Sim®, we created five virtual pediatric populations based on age (2–5 months, 5 months–1 year, > 1–6 years, > 6–12 years, and > 12–18 years), each comprising 100 children. Race and sex distributions in virtual populations were based on the general population in the USA (85% white, 15% black; 50% male) [47]. In the simulation of the virtual pediatric populations, we incorporated interpatient variability (CV%) associated with CYP3A4, CYP2C9, NAT2, and the transporter that mediates tubular secretion of TMP. The final developed pediatric PBPK model evaluated optimal dosing for the population.
We optimized pediatric dosing for TMP–SMX to achieve a TMP exposure of at least the lower limit of the reference adult exposure in > 80% of children and less than a safety margin of TMP in > 95% of children. The reference exposures for TMP and SMX were determined as the AUC at steady-state in adults. We calculated this as a product of dose interval and mean steady-state concentration of TMP and SMX after the recommended dose for treating MRSA (oral TMP 160 mg/SMX 800 mg, every 12 h) [48]. For TMP, the reference adult exposure is an area under the concentration-time curve at steady state (AUCss) > 20.6 mg·h/L. The safety margins of TMP (Cmax 13.6 mg/L, AUCss 141.8 mg·h/L) and SMX (Cmax 372 mg/L and AUCss 4119.4 mg·h/L) were determined based on a previous study in healthy adults, taking into account central nervous system toxicities, hepatotoxicity, and bone marrow suppression [6]. Additionally, to evaluate whether sufficient TMP and SMX concentrations are attained in target organs and tissues (skin), we extrapolated the serum concentrations to theoretical skin concentrations for a goal of greater than the MRSA MIC (TMP 2 mg/L; SMX 9.5 mg/L) for at least 50% of the dosing interval following the optimized pediatric dosing regimen as our pharmacodynamic target, since prior studies suggest that bactericidal coverage for 50% of the dosing interval is adequate to yield a positive clinical response [49,50,51,52].
3 Results
3.1 Adult Model
The adult PBPK model adequately described the observed plasma concentrations of TMP and SMX and fractions of TMP and SMX excreted unchanged in urine for all three dosing regimens: (1) TMP 5 mg/kg and SMX 25 mg/kg intravenously once, using data from Dudley et al. [34]; (2) TMP 160 mg and SMX 800 mg orally once, using data from Varoquaux et al. [7]; (3) TMP 400 mg and SMX 2000 mg orally once, using data from Kaplan et al. [35]; and (4) multiple doses of TMP 5 mg/kg and SMX 25 mg/kg orally, using data from Stevens et al. [10]. This is demonstrated by the plots overlaying simulated data from the PBPK model with plasma concentrations and the fraction of drug excreted unchanged in urine for TMP and SMX after the respective doses (Fig. 2 and ESM Online Resources 2–4). Specific physico-chemical and ADME data used for the adult TMP–SMX model are presented in Table 1.

Mean observed (dots) and simulated (lines) plasma concentrations and fraction of the drug excreted unchanged in urine for TMP (a) and SMX (b) after a single oral dose of TMP 160 mg and SMX 800 mg in healthy adults. Error bars refer to range of concentration in observed patients. SMX sulfamethoxazole, TMP trimethoprim
Weight, age, and sex were comparable between our virtual adult population and the population that provided the observed TMP and SMX concentration–time data (ESM Online Resource 5). The distribution (mean ± SD) of plasma concentrations of TMP and SMX after a single oral dose of TMP 160 mg and SMX 800 mg from the Varoquaux et al. [7] study was plotted and overlaid with simulated data from the adult population PBPK model. The mean and SD of observed plasma concentrations for TMP and SMX were acceptably simulated by the adult population PBPK model (Fig. 3). PK-Sim®-predicted intestinal permeability for TMP and SMX resulted in poor prediction of pharmacokinetic profiles. The apparent clearance, bioavailability, and volume of distribution of TMP and SMX resulting from the final adult PBPK model were 1.74 and 0.30 mL/min/kg, 96% and 98%, and 1.40 and 0.27 L/kg, respectively. All parameters were consistent with prior reported pharmacokinetic data in adult studies [6, 7, 34, 35].

Observed (dots) and simulated (lines) plasma concentration–time profiles of TMP (a) and SMX (b) following a single oral dose of TMP 160 mg and SMX 800 mg in healthy adults. Solid lines represent geometric mean of the simulated data. The shaded area represents geometric mean ± geometric SD for the simulated data. Symbols and error bars represents mean and SD for the observed data, respectively. SD standard deviation, SMX sulfamethoxazole, TMP trimethoprim
3.2 Pediatric Model
To evaluate the predictive accuracy of the pediatric PBPK model, we used 85 TMP and 89 SMX plasma concentrations from 54 participants in the POPS study. Participants received a median of eight doses (range 1–23) of TMP–SMX via oral administration at a median dose of 2.86 mg/kg (range 1.06–12.05) for TMP and 14.29 mg/kg (range 5.29–60.24) for SMX. All drug concentrations were above the limit of quantifications, with a median number of samples collected per participant of 1 (range 1–3). Table 2 summarizes the demographic characteristics of children with pharmacokinetic data by age groups. No concomitant medications were reported for these participants.
We observed good model predictability when comparing predicted and observed concentration–time data. Because pediatric participants in the POPS study had different dosing regimens, we generated a 90% prediction interval of TMP and SMX plasma concentrations for each individual dosing regimen. The number of observations and subjects in each age group and the number of observations outside the 90% prediction interval are summarized in Table 3. The developed PBPK model characterized 67–87% and 71–91% of the opportunistic pharmacokinetic data across age groups for TMP and SMX, respectively. Predictions were poorest in the 5-month to 1-year age group.
When evaluating different contributions of CYP2C9, the differences in AUC and Cmax were < 1% except in the 2-month-old group where the difference was 10%. This suggests the initial model assumption of equal contribution predicted concentrations in pediatric patients reasonably well.
Following the optimized age-based dosage regimens (oral, every 12 h), approximately 90% of subjects had an AUC of at least the lower limit of TMP exposure in adults, and < 1% of subjects had a Cmax or AUCss above the safety margin (Fig. 4a and Table 4). The PBPK model predicted therapeutic TMP concentrations at the target organs and tissues were above the MRSA MIC (2 mg/L) for at least 50% of the dosing interval in ≥ 84% of patients (Table 4). For SMX, 63–90% of subjects had AUC greater than the reference exposure (Fig. 4b and Table 5). One of the subjects in the > 1–6 years old group (1%) had Cmax or AUCss values above the safety margin. The PBPK model predicted that therapeutic SMX concentrations at the target organs and tissues were more than the MRSA MIC required to inhibit the growth of 90% of organisms (MIC90) (9.5 mg/L) for at least 50% of the dosing interval in ≥ 98% of patients (Table 5).

Simulated total drug exposure (AUCss) of TMP (a) and SMX (b) with age-based TMP–SMX dosage regimens. Dotted lines demonstrate target and toxicity thresholds. AUCss area under the concentration–time curve at steady state, SMX sulfamethoxazole, TMP trimethoprim
4 Discussion
We developed a pediatric population PBPK model to predict TMP and SMX exposure using an established workflow for scaling PBPK models from adults to children, and evaluated the model using sparse and opportunistic pediatric concentration versus time data. To our knowledge, this is the first report of using this approach to characterize the pharmacokinetics of a primarily renally eliminated drug such as TMP–SMX.
Our developed adult population PBPK model adequately characterized the pharmacokinetics of TMP and SMX. The model-predicted clearance, volume of distribution, and bioavailability were comparable to previously reported values, and the model adequately captured the concentration–time data of these studies [6, 7, 34, 35]. The final pediatric model adequately characterized the opportunistic pharmacokinetic data collected in children: 67–87% of the observed data were predicted by the population PBPK model for TMP and 71–91% were predicted by the population PBPK model for SMX. Observed values may be different from predicted values since opportunistic data were collected as part of routine care, which may be associated with higher degrees of unspecified error than prototypical pharmacokinetic studies dedicated to a single drug. However, rather than being purely opportunistic, our samples were collected at set times relative to dosing to collect the most optimal pharmacokinetic data possible. Furthermore, PBPK models do not account for residual variability. Further deviation from the predicted values may be due, in part, to the validity of underlying assumptions regarding the type and speed of maturation of the involved clearance pathways in addition to limitations from the opportunistic nature of the data. The primary route of TMP elimination is renal, with 75–85% of the parent drug being eliminated unchanged in the urine [11, 48, 53]. Only 10–20% of TMP is metabolized via CYP2C9 and CYP3A4 to inactive metabolites [29]. SMX undergoes hepatic metabolism through both the CYP2C9 pathway and N-acetylation by NAT-2 [29]. The most prominent hepatic metabolite, N-acetylsulfamethoxazole, is mainly excreted in the urine [11, 48, 53]. However, the exact combination of renal elimination mechanisms, glomerular filtration, tubular secretion, and tubular reabsorption ultimately responsible for the clearance of both TMP and SMX is unknown. Similarly, the assumption of equal contribution of liver enzymes to metabolism holds for older age ranges but may not as adequately reflect metabolism in the youngest infants. This is due to the different ontogeny curves for these enzymes. Our model may also be further limited by the overarching assumption of physiologic scaling—that pediatric pathways are the same as adults [54]. When compared to a recently published pediatric population pharmacokinetic study of TMP and SMX, our PBPK model-predicted clearance, volume of distribution, and bioavailability for TMP were similar [46].
Pediatric drug trials are challenging. Once validated, our PBPK model offers a potential alternative to data collection for population pharmacokinetic model development for both blood and other compartments. Our model leverages existing knowledge of drug disposition and physiology and extrapolates it across different life stages [21]. It is therefore less dependent on prospectively collected population-specific pharmacokinetic data. We previously published our successful pediatric PBPK model for clindamycin [22], which showed that opportunistic pharmacokinetic data were well-suited for PBPK model development (which is more reliant on prior knowledge than clinical data). However, to truly confirm this hypothesis, it was important to replicate this result with other drugs. Taken together, our prior clindamycin model and this TMP–SMX model support the validity of this approach across the two main drug elimination routes, and create a basis for an efficient, minimally invasive method of pharmacokinetic study in children.
The optimal dosing identified using this modeling approach is in the range of the current treatment guidelines from the Infectious Diseases Society of America (IDSA) [55]. Not surprisingly, our model predicted that higher body weight-normalized doses were needed in children (than in adults) to achieve similar exposures [48]. This was further reinforced when comparing our results to dosing recommendations from a recent population pharmacokinetic model in children up to 21 years of age [46]: for the same dosing regimen of TMP 8 g/kg/day divided every 12 h, exposures were lower than reference adult exposures for children < 6 years old (AUCss 19.0–19.2 mg·h/L) but were comparable at ages 6–21 years (19.5–22.8 mg·h/L).
Our model is primarily limited by assumptions made during its development, including renal clearance maturation in children, and that both glomerular filtration and tubular secretion are responsible for renal elimination of TMP while glomerular filtration and tubular reabsorption are responsible for renal elimination of SMX. Further, we applied previously described ontogeny functions for glomerular filtration and tubular secretion, the latter of which had to be generalized given that the actual transporters for both TMP and SMX at the renal tubules are unknown. We are also limited by racial considerations. We chose a racial distribution reflective of our sampling population; however, there are known differences in hepatic metabolism in the Asian race compared with Caucasians [56]. More robust studies may be helpful to evaluate the differences in TMP–SMX metabolism between races. We are currently performing a prospective clinical trial to validate our model-predicted dosing and further optimize the PBPK model.
5 Conclusion
The developed pediatric population PBPK model adequately characterized the pharmacokinetics of TMP and SMX, and supports age- and body weight-based dosing regimens to treat MRSA. We have shown success in developing models that predict both hepatically metabolized and renally excreted drugs. Once properly validated, this approach offers an alternative to the use of sparse and opportunistic data for population pharmacokinetic model development. Due to the scarcity of pediatric data, leveraging adult data and scaling a model to children can diminish the need for more complex pharmacokinetic trials. PBPK models can also be used to determine age-specific dosing parameters, accounting for developmental changes. Our findings support the generalizability of using opportunistic pharmacokinetic data to evaluate the predictive accuracy of pediatric PBPK models across different elimination routes and provide a path towards more widespread use of this modeling tool in pediatric drug development. Future PBPK model development should continue to incorporate developmental changes and use opportunistic pediatric data to improve model predictability and optimize pediatric dosing.
References
Alonso Campero R, Bernardo Escudero R, Del Cisne Valle Alvarez D, González de la Parra M, Namur Montalvo S, Burke Fraga V, et al. Bioequivalence of two commercial preparations of trimethoprim/sulfamethoxazole: a randomized, single-dose, single-blind, crossover trial. Clin Ther. 2007;29:326–33.
Lindenberg M, Kopp S, Dressman JB. Classification of orally administered drugs on the World Health Organization Model list of Essential Medicines according to the biopharmaceutics classification system. Eur J Pharm Biopharm. 2004;58:265–78.
Dahan A, Miller JM, Amidon GL. Prediction of solubility and permeability class membership: provisional BCS classification of the world’s top oral drugs. AAPS J. 2009;11:740–6.
Schwartz DE, Zeigler WH. Assay and pharmacokinetics of trimethoprim in man and animals. Postgrad Med J. 1969;45:Supp 32–7.
Karpman E, Kurzrock EA. Adverse reactions of nitrofurantoin, trimethoprim and sulfamethoxazole in children. J Urol. 2004;172:448–53.
Stevens RC, Laizure SC, Williams CL, Stein DS. Pharmacokinetics and adverse effects of 20-mg/kg/day trimethoprim and 100-mg/kg/day sulfamethoxazole in healthy adult subjects. Antimicrob Agents Chemother. 1991;35:1884–90.
Varoquaux O, Lajoie D, Gobert C, Cordonnier P, Ducreuzet C, Pays M, et al. Pharmacokinetics of the trimethoprim-sulfamethoxazole combination in the elderly. Br J Clin Phamarcol. 1985;20:575–81.
Chin TW, Vandenbroucke A, Fong IW. Pharmacokinetics of trimethoprim-sulfamethoxazole in critically ill and non-critically ill AIDS patients. Antimicrob Agents Chemother. 1995;39:28–33.
Walker SE, Paton TW, Churchill DN, Ojo B, Manuel MA, Wright N. Trimethoprim-sulfamethoxazole pharmacokinetics during continuous ambulatory peritoneal dialysis (CAPD). Perit Dial Int. 1989;9:51–5.
Stevens RC, Laizure SC, Sanders PL, Stein DS. Multiple-dose pharmacokinetics of 12 milligrams of trimethoprim and 60 milligrams of sulfamethoxazole per kilogram of body weight per day in healthy volunteers. Antimicrob Agents Chemother. 1993;37:448–52.
BACTRIM—sulfamethoxazole and trimethoprim tablet package insert. Philadelphia: AR Scientific, Inc; 2013.
Yeldandi V, Strodtman R, Lentino J. In-vitro and in-vivo studies of trimethoprim-sulphamethoxazole against multiple resistant Staphylococcus aureus. J Antimicrob Chemother. 1988;22:873–80.
Close S, McBurney CR, Garvin CG, Chen DC, Martin SJ. Trimethoprim-sulfamethoxazole activity and pharmacodynamics against glycopeptide-intermediate Staphylococcus aureus. Pharmacotherapy. 2002;22:983–9.
Martinez M, Papich M, Drusano G. Dosing regimen matters: the importance of early intervention and rapid attainment of the pharmacokinetic/pharmacodynamic target. Antimicrob Agents Chemother. 2012;56:2795–805.
Wilfert CM, Gutman LT. Pharmacokinetics of trimethoprim-sulfamethoxazole in children. Can Med Assoc J. 1975;112:73–6.
Rylance GW, George RH, Healing DE, Roberts DG. Single dose pharmacokinetics of trimethoprim. Arch Dis Child. 1985;60:29–33.
Laughon MM, Benjamin DK Jr, Capparelli EV, Kearns GL, Berezny K, Paul IM, et al. Innovative clinical trial design for pediatric therapeutics. Expert Rev Clin Pharmacol. 2011;4:643–52.
Zimmerman K, Gonzalez D, Swamy GK, Cohen-Wolkowiez M. Pharmacologic studies in vulnerable populations: Using the pediatric experience. Semin Perinatol. 2015;39:532–6.
Kern SE. Challenges in conducting clinical trials in children: approaches for improving performance. Expert Rev Clin Pharmacol. 2009;2:609–17.
Zhao P, Zhang L, Grillo JA, Liu Q, Bullock JM, Moon YJ, et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther. 2011;89:259–67.
Maharaj AR, Edginton AN. Physiologically based pharmacokinetic modeling and simulation in pediatric drug development. CPT Pharmacomet Syst Pharmacol. 2014;3:e150.
Hornik CP, Wu H, Edginton AN, Watt K, Cohen-Wolkowiez M, Gonzalez D. Development of a pediatric physiologically-based pharmacokinetic model of clindamycin using opportunistic pharmacokinetic data. Clin Pharmacokinet. 2017;56:1343–53.
Maharaj AR, Barrett JS, Edginton AN. A workflow example of PBPK modeling to support pediatric research and development: case study with lorazepam. AAPS J. 2013;15:455–64.
Leong R, Vieira ML, Zhao P, Mulugeta Y, Lee CS, Huang SM, et al. Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin Pharmacol Ther. 2012;91:926–31.
Gonzalez D, Melloni C, Poindexter BB, Yogev R, Atz AM, Sullivan JE, et al. Simultaneous determination of trimethoprim and sulfamethoxazole in dried plasma and urine spots. Bioanalysis. 2015;7:1137–49.
Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95:1238–57.
Rodgers T, Leahy D, Rowland M. Tissue distribution of basic drugs: accounting for enantiomeric, compound and regional differences amongst beta-blocking drugs in rat. J Pharm Sci. 2005;94:1237–48.
Rodgers T, Leahy D, Rowland M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci. 2005;94:1259–76.
Ribera E, Pou L, Fernandez-Sola A, Campos F, Lopez RM, Ocaña I, et al. Rifampin reduces concentrations of trimethoprim and sulfamethoxazole in serum in human immunodeficiency virus-infected patients. Antimicrob Agents Chemother. 2001;45:3238–41.
Cribb A, Spielberg S, Griffin G. N4-hydroxylation of sulfamethoxazole by cytochrome P450 of the cytochrome P450C subfamily and reduction of sulfamethoxazole hydroxylamine in human and rat hepatic microsomes. Drug Metab Dispos. 1995;23:406–14.
Kagaya H, Miura M, Niioka T, Saito M, Numakura K, Habuchi T, et al. Influence of NAT2 polymorphisms on sulfamethoxazole pharmacokinetics in renal transplant recipients. Antimicrob Agents Chemother. 2012;56:825–9.
McDonagh EM, Boukouvala S, Aklillu E, Hein DW, Altman RB, Klein TE. PharmGKB summary: very important pharacogene information for N-acetyltransferase 2. Pharmacogenet Genom. 2014;24:409–25.
Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–82.
Dudley MN, Levitz RE, Quintiliani R, Hickingbotham JM, Nightingale CH. Pharmacokinetics of trimethoprim and sulfamethoxazole in serum and cerebrospinal fluid of adult patients with normal meninges. Antimicrob Agents Chemother. 1984;26:811–4.
Kaplan SA, Weinfeld RE, Abruzzo CW, McFaden K, Jack ML, Weissman L. Pharmacokinetic profile of trimethoprim-sulfamethoxazole in man. J Infect Dis. 1973;128(Suppl):547–55.
Medina JR, Miranda M, Hurtado M, Dominguez-Ramirez AM, Ruiz-Segura JC. Simultaneous determination of trimethoprim and sulfamethoxazole in immediate-release oral dosage forms by first-order derivative spectroscopy: application to dissolution studies. Int J Pharm Pharm Sci. 2013;5:505–10.
Paixao P, Gouveia L, Morais J. Prediction of the in vitro permeability determined in Caco-2 cells by using artificial neural networks. Eur J Pharm Sci. 2010;41:107–17.
Hayton WL. Maturation and growth of renal function: dosing renally cleared drugs in children. AAPS PharmSci. 2000;2:E3.
Achour B, Barber J, Rostami-Hodjegan A. Expression of hepatic drug-metabolizing cytochrome p450 enzymes and their intercorrelations: a meta-analysis. Drug Metab Dispos. 2014;42:1349–56.
Weber W, Vatsis K. Individual variability in p-aminobenzoic acid N-acetylation by human N-acetyltransferase (NAT1) of peripheral blood. Pharmacogenetics. 1993;3:209–12.
Edginton AN, Schmitt W, Willmann S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin Pharmacokinet. 2006;45:1013–34.
Willmann S, Höhn K, Edginton A, Sevestre M, Solodenko J, Weiss W, et al. Development of a physiologically-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J Pharmacokinet Pharmacodyn. 2007;34:401–31.
Deng F, Dong C, Liu Y, Yu Y. Study on the interaction between trimethoprim and human serum albumin by spectroscopic and molecular modeling methods. Spectrosc Lett. 2013;46:13–20.
Edginton AN, Schmitt W, Voith B, Willmann S. A mechanistic approach for the scaling of clearance in children. Clin Pharmacokinet. 2006;45:683–704.
Willmann S, Lippet J, Sevestre M, Solodenko, Fois F, Schmitt W. PK-Sim®: a physiologically based pharmacokinetic ‘whole-body’ model. Biosilico. 2003;1:121-4.
Autmizguine J, Melloni C, Hornik CP, Dallefeld S, Harper B, Yogev R, et al. Population pharmacokinetics of trimethoprim-sulfamethoxazole in infants and children. Antimicrob Agents Chemother. 2018;62:e01813–7.
ACS Demographic and Housing Estimates—2011–2015 American Community Survey 5-year estimates. U.S. Census Bureau. https://factfinder.census.gov/faces/tableservices/jsf/pages/productview.xhtml. Accessed 5 Sept 2018.
Kremers P, Duvivier J, Heusghem C. Pharmacokinetic studies of co-trimoxazole in man after single and repeated doses. J Clin Pharmacol. 1974;14:112–7.
Cenizal MJ, Skiest D, Luber S, Bedimo R, Davis P, Fox P, et al. Prospective randomized trial of empiric therapy with trimethoprim-sulfamethoxazole or doxycycline for outpatient skin and soft tissue infections in an area of high prevalence of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2007;51:2628–30.
Mendes RE, Moet GJ, Janechek MJ, Jones RN. In vitro activity of telavancin against a contemporary worldwide collection of Staphylococcus aureus isolates. Antimicrob Agents Chemother. 2010;54:2704–6.
Thomson KS, Goering RV. Activity of tedizolid (TR-700) against well-characterized methicillin-resistant Staphylococcus aureus straings of diverse epidemiological origins. Antimicrob Agents Chemother. 2013;57:2892–5.
Craig WA. Interrelationship between pharmacokinetics and pharmacodynamics in determining dosage regimens for broad-spectrum cephalosporins. Diagn Microbiol Infect Dis. 1995;22:89–96.
Siber GR, Gorham CC, Ericson JF, Smith AL. Pharmacokinetics of intravenous trimethoprim-sulfamethoxazole in children and adults with normal and impaired renal function. Rev Infect Dis. 1982;4:566–78.
Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology—drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349:1157–67.
Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis. 2011;52:285–92.
Yamaori S, Yamazaki H, Iwano S, Kiyotani K, Matsumura K, Saito T, et al. Ethnic differences between Japanese and Caucasians in the expression levels of mRNAs for CYP3A4, CYP3A5, and CYP3A7: lack of co-regulation of the expression of CYP3A in Japanese livers. Xenobiotica. 2005;35:69–83.
Benjamin D. Pharmacokinetics of understudied drugs administered to children per standard of care (PTN_POPS) [ClinicalTrials.gov identifier NCT01431326]. US National Institutes of Health, ClinicalTrials.gov. https://clinicaltrials.gov. Accessed 23 Dec 2018
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was funded by the National Institutes of Health (1R01-HD076676-01A1; M.C.W.). Funding acknowledgements: NIGMS/NICHD (National Institute of General Medical Sciences/National Institute of Child Health and Human Development) 2T32GM086330-06; Clinical and Translational Science Awards (CTSA).
Conflict of interest
Stephen J. Balevic receives salary and research support from the National Institutes of Health (5R01-HD076676-04, HHSN275201000003I, the Rheumatology Research Foundation’s Scientist Development Award, and the Thrasher Research Fund. Andrea N. Edginton receives support for research from the National Institutes of Health (1R01-HD076676-01A1 [PI: Cohen-Wolkowiez]). Elizabeth J. Thompson, Huali Wu, Anil Maharaj, Marjan Cobbaert, Anthony P. Cunningham, Christoph P. Hornik, and Michael Cohen-Wolkowiez have nothing to disclose.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Thompson, E.J., Wu, H., Maharaj, A. et al. Physiologically Based Pharmacokinetic Modeling for Trimethoprim and Sulfamethoxazole in Children. Clin Pharmacokinet 58, 887–898 (2019). https://doi.org/10.1007/s40262-018-00733-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-018-00733-1