
Abstract
Background and Objectives
Cobimetinib is eliminated mainly through cytochrome P450 (CYP) 3A4-mediated hepatic metabolism in humans. A clinical drug–drug interaction (DDI) study with the potent CYP3A4 inhibitor itraconazole resulted in an approximately sevenfold increase in cobimetinib exposure. The DDI risk for cobimetinib with other CYP3A4 inhibitors and inducers needs to be assessed in order to provide dosing instructions.
Methods
A physiologically based pharmacokinetic (PBPK) model was developed for cobimetinib using in vitro data. It was then optimized and verified using clinical pharmacokinetic data and itraconazole–cobimetinib DDI data. The contribution of CYP3A4 to the clearance of cobimetinib in humans was confirmed using sensitivity analysis in a retrospective simulation of itraconazole–cobimetinib DDI data. The verified PBPK model was then used to predict the effect of other CYP3A4 inhibitors and inducers on cobimetinib pharmacokinetics.
Results
The PBPK model described cobimetinib pharmacokinetic profiles after both intravenous and oral administration of cobimetinib well and accurately simulated the itraconazole–cobimetinib DDI. Sensitivity analysis suggested that CYP3A4 contributes ~78 % of the total clearance of cobimetinib. The PBPK model predicted no change in cobimetinib exposure (area under the plasma concentration–time curve, AUC) with the weak CYP3A inhibitor fluvoxamine and a three to fourfold increase with the moderate CYP3A inhibitors, erythromycin and diltiazem. Similarly, cobimetinib exposure in the presence of strong (rifampicin) and moderate (efavirenz) CYP3A inducers was predicted to decrease by 83 and 72 %, respectively.
Conclusion
This study demonstrates the value of using PBPK simulation to assess the clinical DDI risk inorder to provide dosing instructions with other CYP3A4 perpetrators.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
This study demonstrates the value of using physiologically based pharmacokinetic (PBPK) modelling to assess the clinical drug–drug interaction (DDI) risk for proposing label language. |
The simulation results suggest that weak cytochrome P450 (CYP) 3A4 inhibitors will not affect cobimetinib pharmacokinetics and moderate inhibitors could increase the cobimetinib area under the plasma concentration–time curve (AUC) by ~3- to 4-fold. Strong and moderate CYP3A4 inducers could decrease cobimetinib AUC by 83 and 72 %, respectively. These results have been used to support the dosing instructions on the label of cobimetinib. |
1 Introduction
Cobimetinib (also known as GDC-0973, RO5514041, and XL518) is a potent and highly selective small-molecule inhibitor of MEK (mitogen-activated protein kinase/extracellular signal-regulated kinases [ERK]) [1]. Inhibition of MEK is a promising strategy to control the growth of tumors that are dependent on aberrant signaling in the RAS/RAF pathway. Abnormal regulation of the RAS/RAF/MEK/ERK pathway contributes to uncontrolled proliferation, invasion, metastasis, angiogenesis, and diminished apoptosis [2, 3]. Cobimetinib in combination with vemurafenib (Zelboraf®) has been shown to improve progression-free survival in BRAF V600 mutation-positive advanced melanoma [4]. Cobimetinib has recently been approved in the US. The prescribing information and clinical pharmacology review can be found in the New Drug Application (NDA) review [5, 6].
Data from in vitro studies and a human mass balance study have indicated that hepatic metabolism plays a major role in the clearance (CL) of cobimetinib, and cytochrome P450 (CYP) 3A4 is the predominant enzyme responsible for the metabolism of cobimetinib [7]; therefore, co-administration of potent CYP3A4 inhibitors and inducers were expected to affect cobimetinib pharmacokinetics. A phase I study has been conducted to evaluate the effect of multiple-dose itraconazole, a potent CYP3A4 inhibitor, administered as an oral solution, on cobimetinib pharmacokinetics in healthy subjects (manuscript in preparation). In this study, cobimetinib 10 mg was administered orally to healthy subjects on Day 1 and continuous daily dosing of itraconazole (200 mg) was started on Day 10 and continued for the next 2 weeks. Another 10 mg single dose of cobimetinib was administered on Day 4 of the itraconazole dose. Plasma samples were collected from all subjects at predetermined timepoints to evaluate cobimetinib pharmacokinetics in the presence and absence of itraconazole. The cobimetinib mean area under the plasma concentration–time curve from time zero to infinity (AUC∞) increased by 6.7-fold and the peak plasma concentration (C max) increased by 3.2-fold when co-administrated with itraconazole, indicating that cobimetinib is a sensitive CYP3A4 substrate [5, 6].
Physiologically based pharmacokinetic (PBPK) modeling and simulation tools have increasingly been used for quantitative prediction of pharmacokinetics and drug–drug interactions (DDIs) [8–10]. The value of a PBPK approach for DDI prediction has been recognized by regulatory agencies, as highlighted by the recent US Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidances [11, 12]. The PBPK models that have been validated with clinical pharmacokinetic and DDI data can be used to predict other unknown DDI scenarios. This approach is increasingly being used in regulatory review and supporting label statements [8, 13]. The aims of the current study were to (1) develop a PBPK model for cobimetinib and verify its performance in describing the observed cobimetinib clinical pharmacokinetics obtained after intravenous and oral administration of cobimetinib [14]; (2) simulate the DDI between itraconazole and cobimetinib, and verify the contribution of the CYP3A4 pathway (fraction of cobimetinib cleared through CYP3A4 metabolism [fmCYP3A]) to the cobimetinib CL; and (3) apply the verified cobimetinib PBPK model for assessing the effect of CYP3A4 inducers (strong, moderate) and other CYP3A4 inhibitors (moderate, weak) on cobimetinib pharmacokinetics to inform label language. Ultimately, the present study can contribute to the effort of developing and adopting a general best practice for PBPK simulation to be used in support of regulatory submissions and drug labelling.
2 Methods
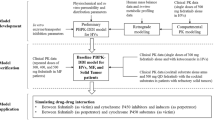
The Simcyp Population-based ADME Simulator (version 13, release 1; Sheffield, UK) with a healthy volunteer population (“Sim-Healthy Volunteers”) was used in all model development and application steps. The PBPK model for cobimetinib was built and verified using a mixed ‘bottom-up’ and ‘top-down’ approach, which maintains a mechanistic PBPK structure, using parameters from in vitro experiments (‘bottom-up’) and leveraging in vivo clinical data (‘top-down’) to bridge the gaps in our understanding of in vitro–in vivo translation. The Simcyp default PBPK models for itraconazole (ITZ) and its metabolite hydroxy-itraconazole (OH-ITZ) were modified and verified using clinical pharmacokinetic data from an itraconazole–cobimetinib DDI study and DDI data from an itraconazole–midazolam study reported by Olkkola et al. [15]. The verified cobimetinib and itraconazole models were then used to simulate the observed effect of itraconazole on cobimetinib pharmacokinetics, and to confirm the contribution of CYP3A4 [fmCYP3A4] to cobimetinib CL. Finally, the cobimetinib PBPK model was applied to simulate the effect of other inhibitors and inducers on cobimetinib pharmacokinetics. The overall model development, verification, and application process is shown in the Fig. 1.

Physiologically based pharmacokinetic model development, verification, and application. CL clearance, CYP cytochrome P450, DDI drug–drug interaction, f mCYP3A4 fraction of cobimetinib cleared through CYP3A4 metabolism, ITZ itraconazole, IV intravenous, OH-ITZ hydroxy-itraconazole, PBPK physiologically based pharmacokinetic, PK pharmacokinetic, PO oral
2.1 Cobimetinib Physiologically Based Pharmacokinetic (PBPK) Model Development and Verification
The initial PBPK model for cobimetinib was built using in vitro/in silico data (Table 1). Cobimetinib is a weak base with an acid dissociation constant (pKa) of 8.85. Cobimetinib has a logP of 3.8, and predominantly binds to albumin with experimentally determined human plasma protein binding of 94.17 % (fraction of unbound drug in plasma [f u,p] = 0.0583). The blood/plasma partition ratio (B/P) was 0.976 (Table 1). The PBPK model was then further optimized and verified using clinical pharmacokinetic data obtained from intravenous and oral administration of cobimetinib in healthy subjects. The PBPK models describing absorption, distribution, and elimination of cobimetinib are detailed below.
Absorption:
The first-order absorption model was used. The fraction absorbed after oral administration (f a = 0.88) was obtained from a human mass balance study in healthy subjects [16]. The absorption rate constant (k a) was estimated to be 0.35 h−1 based on cobimetinib pharmacokinetic data from the control arm of an itraconazole–cobimetinib DDI study in healthy subjects.
Distribution:
The full PBPK distribution model was used to describe the cobimetinib pharmacokinetic profile after intravenous administration in healthy subjects [14]. The PBPK model-predicted volume of distribution at steady state (V ss) was optimized by adding a K p (tissue/plasma partition coefficient) scalar of 3.1 in order to match the observed clinical pharmacokinetic data.
Elimination:
The results from a human mass balance study and absolute bioavailability study of cobimetinib in healthy subjects suggested that hepatic metabolism plays a predominant role in the elimination of cobimetinib, with minimal renal elimination (~3 %) [14]. Additionally, the in vitro data suggest that cobimetinib metabolic CL is predominantly through CYP3A4, with minor contribution from uridine 5′-diphospho-glucuronosyltransferase isozyme 2B7 (UGT2B7). Therefore, hepatic CL through the CYP3A4 enzyme, i.e., CYP3A4 intrinsic CL (CLint), was estimated using the Simcyp retrograde calculator based on the intravenous clearance observed in healthy subjects. A lumped additional CL representing all other non-CYP3A pathways was entered into the model to capture the observed pharmacokinetic profiles (Table 1). Cobimetinib pharmacokinetic simulations were conducted using a 10 × 10 design (ten trials with ten subjects per trial) to simulate population variability.
2.2 Itraconazole (ITZ) and Hydroxy-Itraconazole (OH-ITZ) PBPK Model Optimization and Verification
The Simcyp default PBPK models for itraconazole and its metabolite OH-ITZ were found to under-predict the itraconazole and OH-ITZ exposures observed in the DDI study. As a result, these two models were optimized by adjusting parameters, such as k a, absorption time lag (T lag), V ss, and CL, in order to describe the itraconazole and OH-ITZ exposures observed in the itraconazole–cobimetinib clinical DDI study, in which itraconazole was administered as a solution. The optimized parameters for the itraconazole and OH-ITZ PBPK model are presented in Table 2.
The optimized itraconazole and OH-ITZ PBPK models were further verified by comparing simulation results of the DDI between itraconazole and midazolam with the data reported in the literature [15]. Olkkola et al. [15] described both itraconazole and midazolam concentration–time profiles that can be used for model verification. In these verification simulations, both the f a (0.6) and k a (0.416) parameters in the itraconazole PBPK model were further modified while keeping the remaining parameters the same to describe the itraconazole pharmacokinetic profile after administration of itraconazole in capsule form [15] (Table 2). The Simcyp default midazolam PBPK model (version 13) was also modified in order to describe the midazolam pharmacokinetic profile reported by Olkkola et al. [15]. The values for the modified parameters (k a and maximum velocity [V max] for both the 1-hydroxy midazolam and 4-hydroxy midazolam formation) are listed in Electronic Supplementary Material (ESM) Table 1. The simulations for the itraconazole–midazolam DDI were conducted based on the reported study design, in which itraconazole 200 mg (capsule form) was administered as a daily oral dose for 14 days and midazolam 7.5 mg was administered as a single oral dose on Day 4 [15].
2.3 Simulation of Itraconazole–Cobimetinib Drug–Drug Interaction (DDI)
Following verification of the cobimetinib PBPK model using intravenous and oral pharmacokinetic data, and the itraconazole and OH-ITZ PBPK model using observed pharmacokinetic data from the itraconazole–cobimetinib DDI study and the DDI data from the reported itraconazole–midazolam DDI study described above, simulation of DDIs between itraconazole and cobimetinib was conducted using a Simcyp virtual population, with the study design matching the corresponding clinical DDI study in healthy subjects. The itraconazole solution (200 mg) was administered daily from Day 1 to Day 14 and a single 10 mg dose of cobimetinib was administered orally 30 min after the itraconazole dose on Day 4 following an overnight fast. The end time of the trial simulation was set to Day 70 to capture AUC∞ of cobimetinib in order to account for the long half-life of cobimetinib (130 h with itraconazole interaction). A total of ten trials with 15 subjects per trial (10 × 15) in the age range of 18–52 years (proportion of female subjects: 0.34) were simulated to evaluate the effects of itraconazole on cobimetinib exposure. The number of timepoints in the simulation tool box of Simcyp software was increased from 200 (default) to 3000 in order to accurately capture the simulated cobimetinib pharmacokinetic profiles over a period of 21–35 days, mimicking the actual DDI study design.
Sensitivity analyses were conducted to assess the impact of other key cobimetinib model parameters, fmCYP3A4 and Q gut, on the predicted DDI. The input of CYP3A4 CLint was varied to obtain a range of fmCYP3A from 0.25 to 0.95 (using the Simcyp retrograde calculator). The simulations of itraconazole–cobimetinib DDIs with different fmCYP3A values for cobimetinib were compared with the observed DDI data. Q gut is a hypothetical blood flow term used in the Simcyp PBPK models to describe the role of various processes such as passive intestinal permeability, blood flow to enterocytes, gut metabolism, etc. Changing Q gut will affect the predicted F g (fraction escaping the gut metabolism) and, in turn, the predicted magnitude of the DDI. Simulations of itraconazole–cobimetinib DDIs with different Q gut values (range from 1 to 10 L/h) were compared with the observed DDI data.
2.4 Application of the Cobimetinib PBPK Model
The verified cobimetinib PBPK model was used to predict the effect of other weak and moderate CYP3A4 inhibitors and moderate and strong inducers on cobimetinib pharmacokinetics. The end time for the trial simulation was set to Day 70 to capture the AUC∞ of cobimetinib. A total of ten trials with ten subjects per trial from the healthy volunteer population were simulated (10 × 10 design).
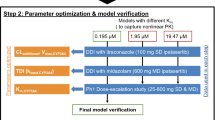
2.4.1 Simulation of the Effect of Weak and Moderate Cytochrome P450 (CYP) 3A Inhibitors
Simulations were conducted to predict the effects of weak (fluvoxamine) and moderate (diltiazem and erythromycin) CYP3A inhibitors on cobimetinib pharmacokinetics using the validated cobimetinib PBPK model. The Simcyp default PBPK models for erythromycin, diltiazem, and fluvoxamine were used in these simulations. The inhibitors were administered daily for 21 days and a single dose of cobimetinib (60 mg) was administered at the same time as the inhibitor on Day 7 (Table 3). The effect of dose staggering on the DDI was also simulated using a similar design (daily dosing of inhibitor for 21 days), but with cobimetinib administered 4 h before the inhibitor dose on Day 7.
2.4.2 Simulation of the Effect of Moderate and Strong CYP3A Inducers
Simulations were conducted to predict the effects of moderate (efavirenz) and strong (rifampicin) inducers of CYP3A on cobimetinib pharmacokinetics using the verified cobimetinib PBPK model. The Simcyp default PBPK model for rifampicin was used for the simulation. An efavirenz PBPK model reported in the literature was used for the simulation as an efavirenz PBPK model was not available in the Simcyp model library [17, 18]. The inducers were administered daily for 21 days and a single dose of cobimetinib (60 mg) was co-administered with an inducer on Day 7.
3 Results
3.1 Cobimetinib PBPK Model Development and Verification
The cobimetinib PBPK model was developed and verified using in vitro and in vivo clinical data. The final model parameters are presented in Table 1. The observed and simulated mean plasma concentration–time profiles of cobimetinib after (a) intravenous infusion (2 mg) and (b) oral administration (10 mg) of cobimetinib in healthy subjects are shown in Fig. 2. The multi-exponential disposition pharmacokinetic profile of cobimetinib after intravenous administration was captured by the full PBPK distribution model. The first-order absorption model with the optimized parameters f a and k a was able to describe the observed cobimetinib pharmacokinetic profiles after oral administration.

Observed and simulated mean plasma concentration–time profiles of cobimetinib following a intravenous infusion of cobimetinib 2 mg and b oral administration of cobimetinib 10 mg
3.2 Optimization and Verification of the Itraconazole and OH-ITZ PBPK Models
The optimized PBPK model parameters for itraconazole and OH-ITZ are presented in Table 2. As shown in Fig. 3, the optimized itraconazole and OH-ITZ models are able to capture the observed itraconazole and OH-ITZ pharmacokinetic profiles from the itraconazole–cobimetinib DDI study.

Observed and simulated plasma concentration–time profiles of itraconazole and OH-ITZ using optimized PBPK model following administration of itraconazole 200 mg in solution, once daily for 14 days. a Shows the profile over a period of 14 days and b shows the profile on day 4 of the itraconazole dose. ITZ itraconazole, OH-ITZ hydroxy-itraconazole
Further verification of the optimized itraconazole model was performed by simulating the DDI between itraconazole and midazolam as reported by Olkkola et al. [15]. It has been previously shown that f a and k a after oral administration of itraconazole are formulation specific [19–21]. As a result, minor changes to the absorption parameters (f a and k a) in the itraconazole model were required to capture the itraconazole pharmacokinetic profile from the capsule formulation (whereas the solution formulation was administered in the itraconazole–cobimetinib DDI study). The simulated itraconazole pharmacokinetic profile (ESM Figure 1) and the midazolam pharmacokinetic profile in the presence and absence of itraconazole are comparable with the observed data (ESM Figure 2). The model-predicted midazolam C max ratio (3.6) and AUC ratio (12.01) were comparable with the reported values (C max ratio of 3.4 and AUC ratio of 10.8) (ESM Table 2).
3.3 Simulation of the DDI Between Itraconazole and Cobimetinib
The verified cobimetinib and itraconazole PBPK models were used for itraconazole–cobimetinib DDI simulation. The comparison between simulated and observed cobimetinib PK in the presence and absence of ITZ are presented in Fig. 4. The model-predicted cobimetinib AUC ratio (6.7; 90 % CI 4.2–12.9) was comparable with the observed AUC ratio of 6.7 (90 % CI 5.6–8.0). Similarly, the model-predicted cobimetinib C max ratio of 2.5 (90 % CI 1.7–4.4) was comparable with the observed ratio of 3.2 (90 % CI 2.7–3.7). The model-predicted median time to C max (t max) of cobimetinib was 3.11 h (range 1.71–4.71 h) and 3.63 h (range 2.16–5.96 h) in the absence and presence of itraconazole, respectively, which is slightly less than the observed prolongation of t max (from a median of 2 h [range 1–6 h] to a median of 4 h [range 2–8 h]). Considering the high variability of cobimetinib pharmacokinetics as a CYP3A4 substrate, overall the model reasonably well captured cobimetinib t max, C max, and AUC changes in the presence of itraconazole.
The contribution of CYP3A4 to the total CL of cobimetinib (fmCYP3A4) was verified by conducting a sensitivity analysis. The fmCYP3A4 value of 0.78 best described the observed DDI between itraconazole and cobimetinib (ESM Table 3). The corresponding intestine bioavailability (F g) was predicted to be 0.46, which was close to the calculated value from the absolute bioavailability and mass balance studies [16].
The effect of the Q gut parameter on the predicted DDI (C max and AUC ratio) is presented in ESM Figure 3a, b. The Q gut value (1.45 L/h) in the cobimetinib PBPK model was predicted in Simcyp based on the apparent passive permeability (P app) with verapamil as a reference compound (Table 1). When the Q gut value was varied from 0.10 to 2, the predicted AUC and C max ratios are close to the observed values, whereas when it was varied from 2 to 10, the predicted AUC and C max ratio are significantly below the observed value (Fig. 4; ESM Figure 3).

Observed and simulated plasma concentration–time profiles of cobimetinib following oral administration of itraconazole 200 mg solution administered once daily for 14 days and cobimetinib 10 mg on day 4. a, c cobimetinib alone or with co-administration of itraconazole on semi-log scale; b, d cobimetinib alone or with co-administration of itraconazole on linear scale (first 48 h shown). ITZ itraconazole
3.4 Application of the Cobimetinib PBPK Model
3.4.1 Simulation of the Effect of Weak and Moderate CYP3A Inhibitors
Results of the simulated effect of moderate and weak CYP3A inhibitors on cobimetinib pharmacokinetics are summarized in Table 3 and illustrated in Fig. 5. The model-simulated cobimetinib mean C max and AUC ratios are 2.15 and 4.35, 1.85 and 3.26, and 1.02 and 1.03, respectively with a moderate inhibitor erythromycin, a moderate inhibitor diltiazem, and a weak inhibitor fluvoxamine. Dose staggering of cobimetinib with respect to inhibitor administration appeared not to have a significant effect on cobimetinib exposure (Table 3).

Observed and simulated cobimetinib AUC and C max ratios with various CYP3A4 inhibitors and inducers. Geometric mean ratio (90 % confidence interval) of AUC or C max of victim drug (cobimetinib) in the presence of inhibitor/inducer to AUC or C max in the absence of inhibitor/inducer. AUC area under the plasma concentration–time curve, C max peak plasma concentration, CYP cytochrome P450, DDI drug–drug interaction
3.4.2 Simulation of the Effect of Moderate and Strong CYP3A Inducers
Results of the simulated effect of moderate and weak CYP3A inducers on cobimetinib pharmacokinetics are shown in Fig. 5. The model-simulated cobimetinib mean AUC ratios were 0.17 and 0.28 when interacting with the strong inducer rifampin and the moderate inducer efavirenz, respectively. Similarly, the model-simulated cobimetinib C max ratios in the presence and absence of rifampicin and efavirenz are 0.37 and 0.36, respectively.
4 Discussion
Cobimetinib is absorbed almost completely following oral administration, and is eliminated predominantly through hepatic metabolism along with limited renal excretion. The in vitro data suggest that the CYP3A4 isozyme plays a predominant role in the metabolic CL of cobimetinib. The clinical DDI study with itraconazole showed that cobimetinib is a sensitive CYP3A substrate (AUC ratio 6.7 and C max ratio 3.2). Given this magnitude of interaction with a strong CYP3A inhibitor, we investigated the effect of weak and moderate CYP3A4 inhibitors and moderate and strong CYP3A4 inducers on cobimetinib pharmacokinetics using a PBPK modeling approach.
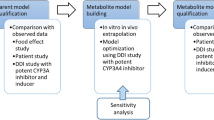
The cobimetinib PBPK model was initially built using in vitro and physicochemical data. Further model optimization and verification were carried out by leveraging the learning from clinical data obtained from absolute bioavailability and mass balance studies. This combined ‘bottom-up’ and ‘top-down’ approach leverages the use of all existing cobimetinib in vitro ADME (absorption, distribution, metabolism, and excretion) information and clinical data, which builds confidence regarding using the PBPK model for the prediction of unknown scenarios. The refined PBPK model was able to describe the cobimetinib pharmacokinetic profile observed in pharmacokinetic and DDI studies, setting up the first step toward to the prediction of DDIs between cobimetinib and other CYP3A4 inhibitor and/or inducers. The sensitivity analysis on the parameters fmCYP3A4 and F g (through Q gut) increased confidence in DDI prediction for cobimetinib with other inhibitors and inducers. The final cobimetinib PBPK model was applied to predict the effect of other CYP3A4 inhibitors and inducers on cobimetinib pharmacokinetics to inform dosing instructions.
Accurate prediction of the itraconazole and OH-ITZ pharmacokinetic profiles is important for the simulation of DDIs between itraconazole and cobimetinib. Optimization of the itraconazole and OH-ITZ PBPK models was necessary as the default model (Simcyp Version 13.1) under-predicted the exposures of both itraconazole and OH-ITZ observed in the DDI study. The under-prediction of pharmacokinetics, especially the accumulation of itraconazole and OH-ITZ after multiple doses, has also been observed and reported by other researchers [22]. Furthermore, the typical ‘bottom-up’ PBPK model development for itraconazole and OH-ITZ is challenging due to the limited and/or largely variable in vitro data reported. In this study, the itraconazole and OH-ITZ default PBPK models were optimized by adjusting several parameters (k a, Tlag, V ss, CL) based on the observed pharmacokinetic data from the itraconazole–cobimetinib DDI study and information from the literature. The optimized models were able to simulate the observed itraconazole and OH-ITZ data from our DDI study, which validated its use for the itraconazole–cobimetinib DDI simulation and provided confidence in the fmCYP3A estimation. Further development of an itraconazole and OH-ITZ PBPK model is necessary for a broad application of the model in itraconazole DDI prediction.
The retrospective PBPK simulation of the observed CYP-mediated DDIs has been recognized as a useful tool for determining the contribution of the CYP in the total CL of a victim drug (fraction of dose metabolized by CYP [fmCYP]) and for predicting the DDI with other untested scenarios [23–27]. Sensitivity analysis on the key parameters, such as fmCYP3A and F g (through Q gut), in the retrospective simulations of itraconazole–cobimetinib DDI, provided a better understanding of the effect of these parameters on the predicted DDI. The fmCYP3A4 (0.78) and F g (0.46) estimated based on the DDI data using retrospective model simulation is consistent with the findings in other clinical studies, including a human mass balance study and absolute bioavailability study [16], which in turn provided confidence in using the PBPK model for the prediction of DDIs for other scenarios for cobimetinib as a victim drug. In the cobimetinib DDI study, itraconazole was administered for 2 weeks due to the significant accumulation of itraconazole and its inhibitory metabolite (e.g., OH-ITZ), and also to minimize the potential safety risk. To test the effect of a longer duration of itraconazole administration, additional simulations were performed using the final PBPK model with itraconazole doses for 3 and 4 weeks. The simulated cobimetinib AUC ratios were 7.28 and 7.54 for the 3- and 4-week itraconazole dosing scenarios, respectively, and there was a C max ratio of 2.54 in both scenarios, which is not significantly different from 2 weeks of itraconazole doses. This result indicated that a near-maximum inhibition effect has been achieved with the current study design. In addition, since cobimetinib follows linear pharmacokinetics, the effect of itraconazole on cobimetinib pharmacokinetics is not expected to be different for single- or multiple-dose cobimetinib scenarios (e.g., steady state).
Finally, the verified cobimetinib PBPK model was used to predict the effect of weak (fluvoxamine) and moderate (erythromycin and diltiazem) CYP3A4 inhibitors, and moderate (efavirenz) and strong (rifampicin) CYP3A4 inducers on cobimetinib pharmacokinetics. Weak CYP3A4 inhibitors did not appear to have any effect on cobimetinib pharmacokinetics, which is consistent with observations from a population pharmacokinetic analysis [28]. Moderate CYP3A4 inhibitors such as erythromycin and diltiazem increased cobimetinib exposure by three to fourfold. Similarly, the weak inducers were not expected to affect cobimetinib pharmacokinetics based on the lack of effect of vemurafenib, a weak CYP3A inducer [28]. Given that cobimetinib is metabolized predominantly by CYP3A, higher variability in the predicted DDI with CYP3A perpetrators is expected, as expressed by the wide confidence intervals around the predicted AUC and C max ratios of cobimetinib (Fig. 5), which is generally consistent with other known CYP3A substrates [29, 30]. However, the accuracy of the predicted variability is yet to be verified. Nevertheless, The PBPK model allowed prediction of DDIs under different scenarios, such as cobimetinib administered either concomitantly or 4 h before the dose of the inhibitor, to test the effect of dose staggering on the DDI. Cobimetinib dose staggering did not appear to reduce the simulated effect of CYP3A inhibition caused by moderate and strong inhibitors significantly. This is not surprising given that a near-maximum inhibition effect on cobimetinib pharmacokinetics has been reached with a significantly accumulated itraconazole and OH-ITZ concentration after a total of 3 weeks of itraconazole daily dosing (to cover the long elimination half-life of cobimetinib). Results from these analyses have informed clinical DDI risk and label language [5], and it can also be used to guide dose regimen design and adjustment in combination with other prediction tools to inform clinicians when using cobimetinib with other drugs [31, 32].
5 Conclusions
This study demonstrated the value of using PBPK model simulation to assess clinical DDI risk, which can be useful in proposing label language. A cobimetinib PBPK model developed and verified using existing clinical pharmacokinetic and DDI data was used to predict the effect of moderate and strong inducers and weak and moderate inhibitors of CYP3A4 on cobimetinib pharmacokinetics. The simulation results suggest that weak CYP3A4 inhibitors will not affect cobimetinib pharmacokinetics and moderate inhibitors could increase the cobimetinib AUC by ~3- to 4-fold and C max by ~twofold. Strong and moderate CYP3A4 inducers could decrease the cobimetinib AUC by 83 and 72 %, respectively, and the C max by ~63 %. Dose staggering appeared to have no significant effect on the magnitude of DDIs predicted for cobimetinib. These results were used to support the label language used for cobimetinib.
References
Rice KD, Aay N, Anand NK, Blazey CM, Bowles OJ, Bussenius J, et al. Novel carboxamide-based allosteric MEK inhibitors: discovery and optimization efforts toward XL518 (GDC-0973). ACS Med Chem Lett. 2012;3(5):416–21.
Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22.
Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–310.
Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76.
US Food and Drug Administration. Cobimetinib (Cotellic) presecribing information. Silver Spring: US Food and Drug Administration; 2015. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/206192s000lbl.pdf.
US Food and Drug Administration. Cobimetinib (COTELLIC) clinical pharmacology and biopharmaceutics review: summary basis of approval. Silver Spring: US Food and Drug Administration; 2015. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206192Orig1s000ClinPharmR.pdf. Accessed 5 May 2016.
Choo EF, Belvin M, Boggs J, Deng Y, Hoeflich KP, Ly J, et al. Preclinical disposition of GDC-0973 and prospective and retrospective analysis of human dose and efficacy predictions. Drug Metab Dispos. 2012;40(5):919–27.
Zhao P, Zhang L, Grillo JA, Liu Q, Bullock JM, Moon YJ, et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther. 2011;89(2):259–67.
Rostami-Hodjegan A, Tamai I, Pang KS. Physiologically based pharmacokinetic (PBPK) modeling: it is here to stay! Biopharm Drug Dispos. 2012;33(2):47–50.
Rostami-Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov. 2007;6(2):140–8.
European Medicines Agency (EMA)—Committee for Human Medicinal Products (CHMP). Guideline on the investigation of drug interactions. London: EMA; 2013.
US Food and Drug Administration. Guidance for industry: drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations (Draft Guidance). Rockville: US Department of Health and Human Services; 2012.
US Food and Drug Administration. Ibrutinib (IMBRUCIVIA) prescribing information. Sunnyvale: Phamacyclics, Inc.; 2015.
Musib L, Choo E, Deng Y, Eppler S, Rooney I, Chan IT, et al. Absolute bioavailability and effect of formulation change, food, or elevated pH with rabeprazole on cobimetinib absorption in healthy subjects. Mol Pharm. 2013;10(11):4046–54.
Olkkola KT, Backman JT, Neuvonen PJ. Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;55(5):481–5.
Takahashi RH, Choo EF, Ma S, Wong S, Halladay J, Deng Y, et al. Absorption, metabolism, excretion, and the contribution of intestinal metabolism to the oral disposition of [14C]cobimetinib, a MEK inhibitor, in humans. Drug Metab Dispos. 2016;44(1):28–39.
Rekic D, Roshammar D, Mukonzo J, Ashton M. In silico prediction of efavirenz and rifampicin drug-drug interaction considering weight and CYP2B6 phenotype. Br J Clin Pharmacol. 2011;71(4):536–43.
Xu C, Quinney SK, Guo Y, Hall SD, Li L, Desta Z. CYP2B6 pharmacogenetics-based in vitro-in vivo extrapolation of efavirenz clearance by physiologically based pharmacokinetic modeling. Drug Metab Dispos. 2013;41(12):2004–11.
Waterhouse TH, Redmann S, Duffull SB, Eccleston JA. Optimal design for model discrimination and parameter estimation for itraconazole population pharmacokinetics in cystic fibrosis patients. J Pharmacokinet Pharmacodyn. 2005;32(3–4):521–45.
Fotaki N, Klein S. Mechanistic understanding of the effect of PPIs and acidic carbonated beverages on the oral absorption of itraconazole based on absorption modeling with appropriate in vitro data. Mol Pharm. 2013;10(11):4016–23.
Heykants J, Van Peer A, Van de Velde V, Van Rooy P, Meuldermans W, Lavrijsen K, et al. The clinical pharmacokinetics of itraconazole: an overview. Mycoses. 1989;32(Suppl 1):67–87.
Ke AB, Zamek-Gliszczynski MJ, Higgins JW, Hall SD. Itraconazole and clarithromycin as ketoconazole alternatives for clinical CYP3A inhibition studies. Clin Pharmacol Ther. 2014;95(5):473–6.
Templeton I, Ravenstijn P, Sensenhauser C, Snoeys J. A physiologically based pharmacokinetic modeling approach to predict drug-drug interactions between domperidone and inhibitors of CYP3A4. Biopharm Drug Dispos. 2015;37(1):15–27.
Yamazaki S, Johnson TR, Smith BJ. Prediction of drug–drug interactions with crizotinib as the CYP3A substrate using a physiologically based pharmacokinetic model. Drug Metab Dispos. 2015;43(10):1417–29.
Marsousi N, Daali Y, Rudaz S, Almond L, Humphries H, Desmeules J, et al. Prediction of metabolic interactions with oxycodone via CYP2D6 and CYP3A inhibition using a physiologically based pharmacokinetic model. CPT Pharmacometrics Syst Pharmacol. 2014;3:e152.
Nordmark A, Andersson A, Baranczewski P, Wanag E, Stahle L. Assessment of interaction potential of AZD2066 using in vitro metabolism tools, physiologically based pharmacokinetic modelling and in vivo cocktail data. Eur J Clin Pharmacol. 2014;70(2):167–78.
Chen J, Liu D, Zheng X, Zhao Q, Jiang J, Hu P. Relative contributions of the major human CYP450 to the metabolism of icotinib and its implication in prediction of drug-drug interaction between icotinib and CYP3A4 inhibitors/inducers using physiologically based pharmacokinetic modeling. Expert Opin Drug Metab Toxicol. 2015;11(6):857–68.
Han K, Jin JY, Marchand M, Eppler S, Choong N, Hack SP, et al. Population pharmacokinetics and dosing implications for cobimetinib in patients with solid tumors. Cancer Chemother Pharmacol. 2015;76(5):917–24.
de Jong J, Skee D, Murphy J, Sukbuntherng J, Hellemans P, Smit J, et al. Effect of CYP3A perpetrators on ibrutinib exposure in healthy participants. Pharmacol Res Perspect. 2015;3(4):e00156.
Bakken GV, Rudberg I, Molden E, Refsum H, Hermann M. Pharmacokinetic variability of quetiapine and the active metabolite N-desalkylquetiapine in psychiatric patients. Ther Drug Monit. 2011;33(2):222–6.
Tod M, Nkoud-Mongo C, Gueyffier F. Impact of genetic polymorphism on drug–drug interactions mediated by cytochromes: a general approach. AAPS J. 2013;15(4):1242–52.
Linares OA, Fudin J, Boston RC, Daly AL. Integrating clinical pharmacokinetics, pharmacogenetics, and quantitative cytochrome P450 polymorphic gene drug–drug interactions (DDIs). Pain Physician. 2015;18(3):E439–41.
US Food and Drug Administration. Itraconazole (SPORANOX) clinical pharmacology and biopharmaceutics review: summary basis of approval. Silver Spring: US Food and Drug Administration; 1999. http://www.accessdata.fda.gov/drugsatfda_docs/nda/99/20-966_SPORANOX%20INJECTION%2010MG%20PER%20ML_BIOPHARMR.PDF. Accessed 5 May 2016.
Author contributions
NRB, TJ, and YC participated in model design; NRB, TJ, SE, LM, and YC collected data and ran simulations; NRB, TJ, YC, and JYJ performed data analysis and wrote the manuscript; NRB, TJ, LM, SE, MD, YC, and JYJ reviewed the manuscript and approved it for submission.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Compliance with Ethical Standards
This study was funded by Genentech (a member of the Roche group). All authors (NRB, TJ, LM, SE, MD, YC, and JYJ) were employees of Genentech when this work was carried out. They have no other conflicts of interest to declare.
Electronic supplementary material
Below is the link to the electronic supplementary material.
40262_2016_412_MOESM1_ESM.tif
Supplementary Figure 1: Observed and simulated plasma concentration-time profiles of itraconazole following administration of 200 mg as capsule formulation daily for 4 days
40262_2016_412_MOESM2_ESM.tif
Supplementary Figure 2: Observed and simulated plasma concentration-time profiles of midazolam with and without co-administration of 200 mg of itraconazole capsule QD for 4 days
40262_2016_412_MOESM3_ESM.tif
Supplementary Figure 3: The effect of Qgut on the predicted DDI between cobimetinib and itraconazole in healthy subjects (a) Cmax ratio, (b) AUC ratio
Rights and permissions
About this article

Cite this article
Budha, N.R., Ji, T., Musib, L. et al. Evaluation of Cytochrome P450 3A4-Mediated Drug–Drug Interaction Potential for Cobimetinib Using Physiologically Based Pharmacokinetic Modeling and Simulation. Clin Pharmacokinet 55, 1435–1445 (2016). https://doi.org/10.1007/s40262-016-0412-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0412-5