
Abstract
Background and Objective
Entrectinib is a selective inhibitor of ROS1/TRK/ALK kinases, recently approved for oncology indications. Entrectinib is predominantly cleared by cytochrome P450 (CYP) 3A4, and modulation of CYP3A enzyme activity profoundly alters the pharmacokinetics of both entrectinib and its active metabolite M5. We describe development of a combined physiologically based pharmacokinetic (PBPK) model for entrectinib and M5 to support dosing recommendations when entrectinib is co-administered with CYP3A4 inhibitors or inducers.
Methods
A PBPK model was established in Simcyp® Simulator. The initial model based on in vitro–in vivo extrapolation was refined using sensitivity analysis and non-linear mixed effects modeling to optimize parameter estimates and to improve model fit to data from a clinical drug–drug interaction study with the strong CYP3A4 inhibitor, itraconazole. The model was subsequently qualified against clinical data, and the final qualified model used to simulate the effects of moderate to strong CYP3A4 inhibitors and inducers on entrectinib and M5 pharmacokinetics.
Results
The final model showed good predictive performance for entrectinib and M5, meeting commonly used predictive performance acceptance criteria in each case. The model predicted that co-administration of various moderate CYP3A4 inhibitors (verapamil, erythromycin, clarithromycin, fluconazole, and diltiazem) would result in an average increase in entrectinib exposure between 2.2- and 3.1-fold, with corresponding average increases for M5 of approximately 2-fold. Co-administration of moderate CYP3A4 inducers (efavirenz, carbamazepine, phenytoin) was predicted to result in an average decrease in entrectinib exposure between 45 and 79%, with corresponding average decreases for M5 of approximately 50%.
Conclusions
The model simulations were used to derive dosing recommendations for co-administering entrectinib with CYP3A4 inhibitors or inducers. PBPK modeling has been used in lieu of clinical studies to enable regulatory decision-making.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A PBPK model was developed, accurately predicting the in vivo pharmacokinetics of both entrectinib and its active metabolite M5. |
Dosing recommendations for co-administering entrectinib with CYP3A4 inhibitors derived from the model were 6-fold lower and 3-fold lower entrectinib doses when co-administered with a strong and a moderate CYP3A4 inhibitor, respectively, and the use of entrectinib with moderate or strong CYP3A4 inducers should be avoided. |
This PBPK modeling approach provided key support for the filing of entrectinib and dosing recommendations in the drug label. |
1 Introduction
The use of physiologically based pharmacokinetic (PBPK) modeling to predict drug concentrations in plasma and tissue has demonstrated utility for accelerating pharmaceutical development, and is now an integral part of many drug development programs [1,2,3,4,5,6,7,8]. Advancement in the discipline has been followed by increasing acceptance by regulatory authorities [5, 7,8,9,10,11,12,13], and there are now numerous examples of drug approvals supported by PBPK modeling in lieu of in vivo clinical studies [14, 15, 17, 18]. One area where PBPK modeling is now particularly widely used is the prediction of drug–drug interactions, in part because it allows quantitative predictions in complex scenarios, e.g., simultaneous induction and inhibition, multiple perpetrators, etc. [19,20,21,22]. Understanding the clinical consequences of such interactions has been further facilitated by the development of models which simultaneously predict effects on multiple pharmacologically-active species [23,24,25,26,27,28].
PBPK modeling has traditionally been regarded as a bottom–up approach whereby in vitro–in vivo extrapolation (IVIVE) techniques are used within a mechanistic framework to predict plasma and tissue concentrations from physicochemical and in vitro data. This contrasts with a top–down approach whereby empirical models are generated to describe observed in vivo data. However, both approaches are recognized to have limitations, and use of a middle–out strategy combining elements of both represents an alternative [7, 29,30,31,32]. Such hybrid multilevel models combine prior information on the system and drug with analysis of observed data, for example, by using clinical data to optimize IVIVE model parameters. Generation of a middle–out model offers potential advantages, but is not without challenges [30]. While there are now numerous examples where a middle–out modeling strategy has been used to support high-impact regulatory activities, (e.g., drug–drug interaction dosing recommendations without the need for a corresponding clinical study [12]), success of this approach ultimately requires acceptance and endorsement by regulatory authorities.
Entrectinib is a potent and selective inhibitor of pan-TRK, ROS1, and ALK receptor tyrosine kinases. These kinases are overexpressed or dysregulated in many types of cancer, such that cancer cell growth is dependent on abnormal kinase activity [33]. Recently, entrectinib was approved for treatment of adult and pediatric patients with tumors that harbor NTRK1/2/3 or ROS1 gene rearrangements.
Entrectinib is predominantly cleared by CYP3A4-mediated metabolism to a pharmacologically-active metabolite (M5), and both parent and metabolite are believed to contribute equally to the overall effect of entrectinib treatment [34]. Clinical drug–drug interaction studies with the potent CYP3A4 inhibitor, itraconazole, and CYP3A inducer, rifampicin, demonstrated that modulation of CYP3A enzyme activity profoundly alters the pharmacokinetics of both entrectinib and M5. However, the effects on entrectinib and M5 were quantitatively different, making it more difficult to extrapolate the observed itraconazole and rifampicin drug–drug interaction study data to other scenarios.
Here, we describe the development of a PBPK model for entrectinib and M5 where two independent methods [sensitivity analysis and nonlinear mixed effect (NLME) modeling] were used to refine estimates for key entrectinib and M5 clearance parameters. The final model was then used to define appropriate entrectinib dosing strategies with various different CYP3A4 inhibitors and inducers in order to support regulatory decision-making.
2 Methods
The overall PBPK model development, qualification, and simulation strategy is depicted schematically in Fig. 1. The initial model building focused on entrectinib only; subsequently,, M5 was incorporated. The PBPK model based on IVIVE of physiochemical, in vitro, and in vivo metabolism data was refined using sensitivity analysis and NLME modeling to optimize parameter estimates and improve model fit to data from a clinical drug–drug interaction study with the strong CYP3A4 inhibitor, itraconazole. The refined model was subsequently qualified by comparing simulated entrectinib and M5 plasma concentrations with observed data from other clinical studies in which patients with solid tumors or healthy volunteers received entrectinib dosing, including a clinical drug–drug interaction study with the strong CYP3A4 inducer, rifampin. Thereafter, the final qualified PBPK model was used to simulate the effects of several moderate-to-strong CYP3A4 inhibitors and inducers on entrectinib and M5 pharmacokinetics. The Simcyp input parameters for the final PBPK model are detailed in Table 1.

Schematic of PBPK model development, qualification, and simulation strategy. CYP cytochrome P450, DDI drug-drug interaction, NLME non-linear mixed effect, PBPK physiologically based pharmacokinetic
2.1 Model Development and Qualification
The PBPK model was established in Simcyp® Simulator (v.17.1; Certara, Princeton, USA). The model integrated available physiochemical, in vitro, and in vivo metabolism data for entrectinib and M5 (Table 1). The retrograde modeling tool was used to refine the intrinsic clearance (CLint) values obtained via in vitro to in vivo extrapolation, and a full PBPK distribution model was used with the Rodgers and Rowland method to predict tissue to plasma partition coefficients [35]. Based on insights derived from independent modeling of entrectinib absorption using the GastroPlus software tool [36], an advanced distribution, absorption, and metabolism model [37] was used to describe the kinetics in the gastrointestinal tract using the “solution formulation without precipitation” option. A built-in virtual healthy volunteer adult population was used for simulations. For simulation of dosing in the fed state, the Simcyp default gastric emptying time (1 h) was increased to 2 h to better reflect the observed timing of peak entrectinib concentrations.
2.1.1 Sensitivity Analyses
Sensitivity analyses were performed to optimize the fraction of entrectinib unbound in gut (fuGut), the fraction of entrectinib metabolized by CYP3A4 (entrectinib fmCYP3A4), the fraction of entrectinib metabolized by CYP3A4 to M5 (entrectinib fmCYP3A4[M5]), the metabolic clearance of M5, the fraction of M5 metabolized (M5 fm), and the fraction of M5 metabolized by CYP3A4 (M5 fmCYP3A4).
A matrix of 20 different values of fuGut and entrectinib fmCYP3A4 parameters were initially assessed during parent model development (fuGut: 0.5–1; entrectinib fmCYP3A4 0.737–0.831, based on the estimated 0.72 from in vitro, represented by CYP3A4 CLint of 4–7 μL/min/pmol). Predicted entrectinib exposures from each pair of parameter values were visually compared against observed data from the clinical drug–drug interaction study with itraconazole [38] (Study RXDX-101-12). Based on this initial assessment, it was determined to be most relevant to fix fuGut at 1, and a more intensive sensitivity analysis was conducted that focused on the entrectinib fmCYP3A4 over a range of 0.778–0.808 (CLint ranging between 5 and 6 μL/min/pmol).
Based on in vitro experiments [38], M5 was identified as the major metabolite, and that it was mainly formed via CYP3A4 (> 50%). In addition, CYP3A4 was the main isoform involved in M5 metabolism (70–99%). As such, during M5 model development, sensitivity analyses were performed that explored parameter value ranges for entrectinib (fmCYP3A4[M5] and M5 fm of 50–99%, and 70–99% for M5 fmCYP3A4). Metabolic clearance of M5 was explored over a 0.5- to 2-fold range relative to the metabolic clearance of entrectinib. In each case, the predicted M5 exposures from each parameter value were visually compared against observed data from a clinical drug–drug interaction study with itraconazole (Study RXDX-101-12). There was no hierarchy among the sensitivity analyses.
2.1.2 NLME Modeling to Estimate Fg and fmCYP3A4
A novel data analysis approach was also used to estimate entrectinib fraction escaping intestinal metabolism (Fg) and fmCYP3A4 parameters. A combination of NLME and PBPK modeling was used to analyze data from the clinical drug–drug interaction study with itraconazole (Study RXDX-101-12). A description of the assumptions, methodology, results, and model verification is presented elsewhere [15].
2.2 Clinical Study Data
Model qualification employed plasma concentration data from three clinical studies in which patients or healthy volunteers received entrectinib dosing (Table 2). In each study, bioanalytical samples were collected according to an intensive sampling scheme; entrectinib and M5 plasma concentrations were measured using a validated LC-MS/MS method for simultaneous determination of entrectinib and M5 (Ignyta, San Diego, CA, USA; data on file). Two different oral immediate release capsule formulations (F2A and F06) were employed, but were not differentiated during model development because the two formulations were bioequivalent [16].
2.3 Simulations with CYP3A4 Inhibitors and Inducers
The final qualified PBPK model was used to simulate the effects of moderate to strong CYP3A4 inhibitors and inducers on the pharmacokinetics of entrectinib and M5 in a virtual population of adult healthy volunteers. The perpetrator drugs and their simulated dosing regimens are detailed in Table 3. Simulations employed compound files from Simcyp (v.17.1). Simcyp parameter values for creating a virtual healthy volunteer population (e.g., physiological parameters including liver volume and blood flows, enzyme abundances) have been described previously [40]. While initial simulations employed a single 600-mg dose of entrectinib, subsequent simulations for selected perpetrators were generated using lower doses of entrectinib (100 mg and 200 mg) and dosing to steady state with a once-daily dosing regimen.
3 Results
3.1 Model Qualification
Input parameters for the final PBPK model are detailed in Table 1. Refinements to initial parameter estimates based on sensitivity analysis and NLME modeling are indicated. The two independent methods used to derive estimates for key entrectinib and M5 clearance parameters gave similar results. Based on sensitivity analyses, an entrectinib fmCYP3A4 value of 0.78 (reflected by a CYP3A4 CLint of 5.17 μL/min/pmol) was selected. A fuGut of 1 resulted in a mean estimated Fg (i.e., fraction of entrectinib escaping intestinal metabolism) of 0.60 (geometric mean 0.58). Separately, the NLME estimated an fmCYP3A4 of 0.755 (95% CI 0.697–0.804), and Fg of 0.58 (95%CI 0.460–0.718) [15]. Overall, the model predicts that the majority of an absorbed entrectinib dose is cleared by CYP3A4-mediated metabolism to the M5 metabolite, while the M5 metabolite is itself almost exclusively cleared by CYP3A4-mediated metabolism (Fig. 2).

Schematic of entrectinib and M5 clearance pathways with key parameter values. CYP cytochrome P450, UGT uridine 5'-diphospho-glucuronosyltransferase
During model development, it was noted that the observed entrectinib and M5 exposures in the clinical drug–drug interaction study with the strong CYP3A4 inducer rifampin (Part 2 of Study RXDX-101-12) were ~ 30% lower than other clinical studies employing the same entrectinib dose. As a consequence, the PBPK model initially over-estimated exposure parameters for this specific study. To improve the PBPK model fit, a study-specific lower bioavailability was incorporated by reducing the effective permeability in human value for entrectinib (Peff,man) from 1.34 × 10−4 to 0.33 × 10−4 cm/s for this study.
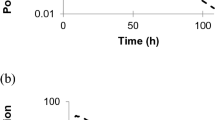
The final PBPK model showed good predictive performance for both entrectinib and M5. Predicted plasma exposures were similar to observed exposures when entrectinib was administered alone, or with the strong CYP3A4 inhibitor, itraconazole (Study RXDX-101-12 Part 1; Table 4; Fig. 3), or with the strong CYP3A inducer, rifampicin (Study RXDX-101-12 Part 2; Table 5 and Fig. 4). The 5th and 95th percentiles of the model-predicted concentrations encompassed most observed concentrations (Figs. 3, 4), while the magnitude of the drug–drug interaction effects predicted by the model were comparable with the observed results from NCA analyses. The ratios between predicted and observed changes in drug exposure (Ratiopredicted/Ratioobserved, see [11]) for co-administration of itraconazole were 1.14 (Cmax) and 0.76 (AUC) for entrectinib, and 0.33 (Cmax) and 0.52 (AUC) for M5 (Table 4). Corresponding values for co-administration of rifampicin were 0.82 (Cmax) and 0.87 (AUC) for entrectinib, and 1.00 (Cmax) and 1.36 (AUC) for M5 (Table 5). Predictive performance was also good when simulating exposures in healthy volunteers under fed and fasted conditions (Study RXDX-101-04; presented in Fig. 5), and in patients with solid tumors dosed to steady state (Study RXDX-101-02) (Figs. 6 and 7 for entrectinib and M5, respectively).

Simulated and observed entrectinib (a, b) and M5 (c, d) plasma concentrations following a single 100-mg dose of entrectinib alone (a, c) or co-administered with the CYP3A4 inhibitor, itraconazole (b, d). Black and gray lines median model-predicted concentrations with 5th and 95th percentiles; circles observed individual concentrations from Study RXDX-101-12 Part 1. Main panel linear Y-axis; inset log scale Y-axis. CYP cytochrome P450

Simulated and observed Entrectinib (a, b) and M5 (c, d) plasma concentrations following a single 600-mg dose of entrectinib alone (a, c) or co-administered with the CYP3A inducer, ifampin (b, d). Black and gray lines median model-predicted concentrations with 5th and 95th percentiles; circles observed individual concentrations from Study RXDX-101-12 Part 2. Main panel linear Y-axis; inset log scale Y-axis. CYP cytochrome P450

Simulated and observed entrectinib plasma concentrations following administration of entrectinib under fasted 400 mg (a) and 600 mg (b) or fed 600 mg (c) conditions. Black and gray lines median model-predicted concentrations with 5th and 95th percentiles; circles observed individual concentrations from Study RXDX-101-04. Main panel linear Y-axis; inset log scale Y-axis

Simulated and observed entrectinib plasma concentrations in adults with NTRK1/2/3+ or ROS1+ solid tumors after a single dose of entrectinib (a) and at steady state (b). Black and gray lines median model-predicted concentrations with 5th and 95th percentiles; circles observed individual concentrations from Study RXDX-101-03. Main panel linear Y-axis; inset log scale Y-axis

Simulated and observed M5 plasma concentrations in adults with NTRK1/2/3+ or ROS1+ solid tumors after a single dose of entrectinib (a) and at steady state (b). Black and gray lines median model-predicted concentrations with 5th and 95th percentiles; circles observed individual concentrations from Study RXDX-101-02. Main panel linear Y-axis; inset log scale Y-axis
3.2 Simulations with CYP3A4 Perpetrators
The final qualified PBPK model was used to simulate the effects of various moderate to strong CYP3A4 inhibitors and inducers on entrectinib and M5 pharmacokinetics in a virtual population of adult healthy volunteers. Ratios of simulated entrectinib AUCinf values in the presence and absence of the perpetrator, and corresponding 95% confidence intervals, are summarized in Fig. 8. The model predicted that co-administration of various moderate CYP3A4 inhibitors (verapamil, erythromycin, clarithromycin, fluconazole, and diltiazem) would result in average increases in entrectinib exposure between 2.2- and 3.1-fold (Fig. 8a). Corresponding average increases for M5 were predicted to be approximately 2-fold (Fig. 8b). The model predicted that co-administration of various moderate CYP3A4 inducers (efavirenz, carbamazepine, phenytoin) would result in average decrease in entrectinib exposure between 45 and 79% (Fig. 8a), while corresponding average decreases for M5 were predicted to be approximately 50% (Fig. 5b). Simulations of repeat dosing with entrectinib produced predicted interactions of similar magnitudes. For example, median AUC interaction ratios after a single dose of entrectinib co-administered with itraconazole were 4.58 and 1.40 for entrectinib and M5, respectively, while the corresponding values from repeat dosing to steady state were 5.06 and 1.86, respectively (data not shown).

Proportional changes in simulated entrectinib (a) and M5 (b) AUCinf exposure parameters from co-administration of various CYP3A4 inhibitors and inducers. Symbols geometric mean ratios, error bars upper and lower 95% confidence intervals. AUCinf area under the concentration-time curve from time zero to infinity, CYP cytochrome P450, sim simulated, obs observed, PK pharmacokinetic
Based on the magnitude of the simulated interactions, 3-fold and 6-fold lower entrectinib doses (i.e., 200 mg and 100 mg) are required to mitigate the effects of moderate and strong CYP3A4 inhibitors, respectively. To confirm the appropriateness of these dose adjustments, 100 mg and 200 mg entrectinib co-administered with strong and moderate CYP3A4 inhibitors, respectively, were also simulated (Table 6). These confirmed that simulated entrectinib and M5 exposures using the recommended dose adjustments were comparable to those from dosing with 600 mg entrectinib alone.
4 Discussion
A PBPK model of entrectinib and its active metabolite M5 was developed by integrating in vitro, non-clinical, and clinical data. The PBPK model based on IVIVE was refined using a sensitivity analysis and NLME modeling (described in detail elsewhere [15]) to optimize parameter estimates of the fraction metabolized by CYP3A4 and the fraction escaping gut metabolism. The two separate approaches were used in parallel, and both gave very similar parameter estimates (Fg: 0.6 vs. 0.58; fmCYP3A4: 0.78 vs. 0.75). As well as demonstrating the utility of a NLME modeling approach as a tool to refine key parameter estimates, concordance increased confidence in the two key determinant parameters of the pharmacokinetics and drug–drug interaction liability of entrectinib. Parameter estimates were further corroborated by independent data from a human ADME study in which entrectinib disposition in humans in vivo was investigated by the administration of a single dose of radiolabeled entrectinib to healthy volunteers (unpublished data). Based on the radiolabel recovered in excreta, it was estimated that, on average, up to 73% of the administered entrectinib dose was cleared by metabolism to M5, while the corresponding parameter in the final PBPK model was 70% (Fig. 2). The consistency with a completely independent clinical data source thereby provides additional confidence in the robustness of the PBPK model.
The final PBPK model showed good predictive performance for both entrectinib and M5, and met commonly-used predictive performance acceptance criteria when compared with observed clinical data [11, 12, 39]. Considering the drug–drug interactions with itraconazole and rifampicin, the ratios of predicted AUCs were all within 2-fold of the observed ratio (i.e. calculated Ratiopredicted/Ratioobserved > 0.5 and < 2.0), while, in many cases, the ratios of AUC and Cmax were within 25% of the observed ratio (i.e., calculated Ratiopredicted/Ratioobserved > 0.8 and < 1.25). It is notable that predictions of the effect of itraconazole on M5 were less accurate, underpredicting the magnitude of the effect on AUC while overpredicting the effect on Cmax. While this suggests that there is still potential to improve this aspect of the model, it was not considered to compromise the value of the model for supporting dosing recommendations.
The PBPK model, which describes both entrectinib parent and M5 metabolite pharmacokinetics, has particular utility since M5 is pharmacologically active, and consequently both parent and metabolite are believed to contribute to the overall efficacy of entrectinib treatment. Therefore, the model provides a useful quantitative tool with which to evaluate alternative dosing strategies under circumstances where the pharmacokinetics of both entrectinib and M5 are altered. However, the dosing recommendations for co-administering entrectinib with CYP3A4 inhibitors or inducers focus principally on entrectinib exposure, primarily because entrectinib is the principal circulating species in vivo (M5 plasma exposures are typically ≤½ those of entrectinib under normal dosing conditions). Consequently, as metabolite exposures are well below those of the parent, it is expected that M5 makes a smaller contribution than entrectinib to the pharmacological effects of entrectinib treatment. This is supported by analyses of the exposure versus response relationships, which showed that using parameters representing the sum of entrectinib and M5 exposures together yielded no additional insight over use of entrectinib exposure alone [41]. Furthermore, the concurrent use of CYP3A inhibitors and inducers with entrectinib both lead to a decrease in the metabolite:parent ratio. While itraconazole use increases both observed entrectinib and M5 exposure, the proportional change is greater for entrectinib (approximately 5.8-fold) than M5 (approximately 2.6-fold), and the average metabolite:parent ratio decreases. Conversely, rifampicin use decreases both entrectinib and M5 exposure, the proportional change is smaller for entrectinib (approximately 66%) than M5 (approximately 92%), and the average metabolite:parent ratio decreases. Therefore, in each scenario, the contribution of the M5 metabolite to the pharmacological effect of treatment will be decreased rather than increased. As a consequence, it is appropriate to place most importance on entrectinib exposures when making dosing recommendations.
The final PBPK model has been used to derive dosing recommendations for co-administering entrectinib with CYP3A4 inhibitors or inducers. Based on the magnitude of the simulated interactions, 3-fold and 6-fold lower entrectinib doses (i.e., 200 mg and 100 mg) are required to mitigate the effects of moderate and strong CYP3A4 inhibitors, respectively. The appropriateness of the recommended dose adjustments was confirmed by further simulations of 100 mg and 200 mg entrectinib co-administered with CYP3A4 inhibitors. When considering the concomitant use of moderate and strong CYP3A inducers, the magnitude of the simulated interactions suggests that 2-fold and 4-fold higher entrectinib doses (i.e., 1200 mg and 2400 mg) would be required to mitigate the effects of enzyme induction. However, clinical use of entrectinib doses > 600 mg is not considered appropriate given the safety profile of entrectinib. While the recommended dose of 600 mg is well tolerated, doses above 600 mg produced dose-limiting toxicities in dose-finding studies [33, 42, 43]. Modeling of the exposure versus response relationship demonstrated that the likelihood of a patient experiencing a ≥ Grade 3 adverse event was markedly higher at exposures above those typically produced by 600 mg dosing [41]. Use of high doses of entrectinib would therefore carry potential safety risks for individuals, and in this context it is more prudent to recommend that use of entrectinib with moderate or strong CYP3A4 inducers be avoided rather than attempt a dose adjustment.
5 Conclusions
A PBPK model of entrectinib and its active metabolite M5 was developed, and has been shown to accurately predict the pharmacokinetics of both entrectinib and M5 in vivo. This model has been used to derive dosing recommendations for co-administering entrectinib with CYP3A4 inhibitors or inducers. A 6-fold lower entrectinib dose (i.e., 100 mg) is recommended when co-administered with a strong CYP3A4 inhibitor, and a 3-fold lower entrectinib dose (i.e., 200 mg) is recommended when co-administered with a moderate CYP3A4 inhibitor, but use of entrectinib with moderate or strong CYP3A4 inducers should be avoided. The PBPK modeling has been used in lieu of clinical studies to enable regulatory decision-making.
References
Jones H, Rowland-Yeo K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst Pharmacol. 2013;2:e63.
Jones HM, Chen Y, Gibson C, Heimbach T, Parrott N, Peters SA, Snoeys J, Upreti VV, Zheng M, Hall SD. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2015;97(3):247–62.
Jamei M. Recent advances in development and application of physiologically-based pharmacokinetic (pbpk) models: a transition from academic curiosity to regulatory acceptance. Curr Pharmacol Rep. 2016;2:161–9.
Templeton IE, Chen Y, Mao J, Lin J, Yu H, Peters S, Shebley M, Varma MV. Quantitative prediction of drug-drug interactions involving inhibitory metabolites in drug development: how can physiologically based pharmacokinetic modeling help? CPT Pharmacomet Syst Pharmacol. 2016;5(10):505–15.
Zhuang X, Lu C. PBPK modeling and simulation in drug research and development. Acta Pharm Sin B. 2016;6(5):430–40.
Miller NA, Reddy MB, Heikkinen AT, Lukacova V, Parrott N. Physiologically based pharmacokinetic modelling for first-in-human predictions: an updated model building strategy illustrated with challenging industry case studies. Clin Pharmacokinet. 2019;58(6):727–46.
Peters SA, Dolgos H. Requirements to establishing confidence in physiologically based pharmacokinetic (PBPK) models and overcoming some of the challenges to meeting them. Clin Pharmacokinet. 2019;58(11):1355–71.
Taskar KS, Pilla Reddy V, Burt H, et al. Physiologically-based pharmacokinetic models for evaluating membrane transporter mediated drug-drug interactions: current capabilities, case studies, future opportunities, and recommendations. Clin Pharmacol Ther. 2020;107(5):1082–115. https://doi.org/10.1002/cpt.1693.
Zhao P, Rowland M, Huang SM. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin Pharmacol Ther. 2012;92(1):17–20.
Rowland M, Lesko LJ, Rostami-Hodjegan A. Physiologically based pharmacokinetics is impacting drug development and regulatory decision Making. CPT Pharmacomet Syst Pharmacol. 2015;4(6):313–5.
Wagner C, Pan Y, Hsu VJA, et al. Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin Pharmacokinet. 2015;54(1):117–112.
Shebley M, Sandhu P, Emami Riedmaier AM, et al. Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin Pharmacol Ther. 2018;104(1):88–110.
Grimstein M, Yang Y, Zhang XJ, et al. Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. Food and Drug Administration’s Office of Clinical Pharmacology. J Pharm Sci. 2019;108(1):21–5.
de Zwart L, Snoeys J, De Jong JJ, et al. Ibrutinib dosing strategies based on interaction potential of CYP3A4 perpetrators using physiologically based pharmacokinetic modeling. Clin Pharmacol Ther. 2016;100(5):548–57.
Cleary Y, Gertz M, Djebli N, et al. Mechanistic Modeling approaches to simultaneously estimate FMCYP3A and FG of CYP3A substrates from clinical DDI study data. Clinical Pharmacology & Therapeutics. 2020;107(S1);S27.
Meneses-Lorente G, Bentley D, Guerini E, et al. Characterization of the pharmacokinetics of entrectinib and its active m5 metabolite in healthy volunteers and patients with solid tumors. Clin Pharmacol Ther. 2020;107(S1):S27.
Pilla Reddy V, Bui K, Scarfe GD, et al. Physiologically based pharmacokinetic modeling for olaparib dosing recommendations: bridging formulations, drug interactions, and patient populations. Clin Pharmacol Ther. 2019;105(1):229–41.
Umehara K, Huth F, Jin YH, et al. Drug-drug interaction (DDI) assessments of ruxolitinib, a dual substrate of CYP3A4 and CYP2C9, using a verified physiologically based pharmacokinetic (PBPK) model to support regulatory submissions. Drug Metab Pers Ther. 2019. https://doi.org/10.1515/dmpt-2018-0042.
Rowland Yeo K, Jamei M, Yang JGT, et al. Physiologically based mechanistic modelling to predict complex drug-drug interactions involving simultaneous competitive and time-dependent enzyme inhibition by parent compound and its metabolite in both liver and gut - the effect of diltiazem on the time-course of exposure to triazolam. Eur J Pharm Sci. 2010;39(5):298–309.
Guo J, Zhou D, Li Y, Khanh BH. Physiologically based pharmacokinetic modeling to predict complex drug-drug interactions: a case study of AZD2327 and its metabolite, competitive and time-dependent CYP3A inhibitors. Biopharm Drug Dispos. 2015;36(8):507–19.
Asaumi R, Toshimoto K, Tobe YK, et al. Comprehensive PBPK model of rifampicin for quantitative prediction of complex drug-drug interactions: CYP3A/2C9 induction and OATP inhibition effects. CPT Pharmacomet Syst Pharmacol. 2018;7(3):186–96.
Yoshida K, Maeda K, Konagaya A, et al. Accurate estimation of in vivo inhibition constants of inhibitors and fraction metabolized of substrates with physiologically based pharmacokinetic drug-drug interaction models incorporating parent drugs and metabolites of substrates with cluster newton method. Drug Metab Dispos. 2018;46(11):1805–16.
Marsousi N, Samer CF, Fontana P, et al. Coadministration of ticagrelor and ritonavir: toward prospective dose adjustment to maintain an optimal platelet inhibition using the PBPK approach. Clin Pharmacol Ther. 2016;100(3):295–304.
Cleary Y, Gertz M, Morcos PNL, et al. Model-based assessments of CYP-mediated drug-drug interaction risk of alectinib: physiologically based pharmacokinetic modeling supported clinical development. Clin Pharmacol Ther. 2018;104(3):505–14.
Gu H, Dutreix S, Rebello ST, et al. Simultaneous physiologically based pharmacokinetic (PBPK) modeling of parent and active metabolites to investigate complex CYP3A4 drug-drug interaction potential: a case example of midostaurin. Drug Metab Dispos. 2018;46(2):109–21.
Zhou D, Podoll T, Xu YG, et al. Evaluation of the drug-drug interaction potential of acalabrutinib and its active metabolite, ACP-5862, using a physiologically-based pharmacokinetic modeling approach. CPT Pharmacometrics Syst Pharmacol. 2019;8(7):489–99.
Posada MM, Morse BL, Turner PK, Kulanthaivel P, Hall SD, Dickinson GL. Predicting clinical effects of CYP3A4 modulators on abemaciclib and active metabolites exposure using physiologically based pharmacokinetic modeling [published online ahead of print, 2020 Feb 20]. J Clin Pharmacol. 2020. https://doi.org/10.1002/jcph.1584.
Van den Bergh A, Snoeys J, De Zwart L, et al. Pharmacokinetic drug-drug interaction of apalutamide, part 2: investigating interaction potential using a physiologically based pharmacokinetic [published online ahead of print, 2020 Apr 27]. Clin Pharmacokinet. 2020. https://doi.org/10.1007/s40262-020-00881-3.
Li R, Barton HA, Yates PD, Ghosh A, Wolford AC, Riccardi KA, Maurer TS. A ‘“middle-out”’ approach to human pharmacokinetic predictions for OATP substrates using physiologically-based pharmacokinetic modeling. J Pharmacokinet Pharmacodyn. 2014;41:197–209.
Tsamandouras N, Rostami-Hodjegan A, Aarons L. Combining the “bottom up” and “top down” approaches in pharmacokinetic modelling: fitting PBPK models to observed clinical data. Br J Clin Pharmacol. 2015;79(1):48–55.
Tylutki Z, Polak S, Wiśniowska B. Top-down, bottom-up and middle-out strategies for drug cardiac safety assessment via modeling and simulations. Curr Pharmacol Rep. 2016;2(4):171–7.
Rostami-Hodjegan A. Reverse translation in PBPK and QSP: going backwards in order to go forward with confidence. Clin Pharmacol Ther. 2018;103(2):224–32.
Drilon A, Siena S, Ou SI, et al. Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK Inhibitor entrectinib: combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017;7(4):400–9. https://doi.org/10.1158/2159-8290.CD-16-1237.
Meneses-Lorente G, Fowler S, Guerini E, et al. In vitro and clinical investigations to determine the drug-drug interaction potential of entrectinib. In: Under Press in Investigational New Drugs.
Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions [published correction appears in J Pharm Sci. 2007 Nov;96(11):3153-4]. J Pharm Sci. 2006;95(6):1238–57. https://doi.org/10.1002/jps.20502.
Parrott N, Stillhart C, Lindenberg M, et al. Physiologically based absorption modelling to explore the impact of food and gastric pH changes on the pharmacokinetics of entrectinib. AAPS J. 2020;22(4):78. https://doi.org/10.1208/s12248-020-00463-y.
Jamei M, Turner D, Yang J, et al. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009;11(2):225–37. https://doi.org/10.1208/s12248-009-9099-y.
Meneses-Lorente G, Fowler S, Guerini E, et al. In vitro and clinical investigations to determine the drug-drug interaction potential of entrectinib, a small molecule inhibitor of neurotrophic tyrosine receptor kinase (NTRK). Invest New Drugs. 2021. https://doi.org/10.1007/s10637-021-01156-9.
Sager JE, Yu J, Ragueneau-Majlessi I, Isoherranen N. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab Dispos. 2015;43(11):1823–37. https://doi.org/10.1124/dmd.115.065920.
Jamei M, Dickinson GL, Rostami-Hodjegan A. A framework for assessing inter-individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: A tale of “bottom-up” vs “top-down” recognition of covariates [published correction appears in Drug Metab Pharmacokinet. 2009;24(5):488]. Drug Metab Pharmacokinet. 2009;24(1):53–75. https://doi.org/10.2133/dmpk.24.53.
Mercier F, Djebli N, González-Sales M, Meneses-Lorente G, Jaminion F, Phipps A, Frey N. Exposure–response analysis of entrectinib supports the recommended dose in patients with advanced/metastatic solid tumors. Clinical Pharmacology & Therapeutics; Vol 107 Supplement S1;S50
Drilon A, Siena S, Dziadziuszko R, et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21(2):261–70.
Doebele RC, Drilon A, Paz-Ares L, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21(2):271–82.
Acknowledgements
The clinical studies reported in this manuscript were funded by F. Hoffmann-La-Roche (formerly Ignyta Inc., a member of the Roche Group). The modeling analyses were also funded by F. Hoffmann-La-Roche.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The clinical studies reported in this manuscript were funded by F. Hoffmann-La-Roche (formerly Ignyta Inc., a member of the Roche Group). The modeling analyses were also funded by F. Hoffmann-La-Roche. Administrative support was provided by Ashfield Medcomms, an Ashfield Health company, and was funded by F. Hoffmann-La Roche Ltd.
Conflicts of Interest
G.M-L. is an employee of Roche Products Ltd. N.D. E.G. Y.C., and A.P. are employees and stockholders of F. Hoffmann-La Roche Ltd. F.M., V.B., N.P., N.F., and S.F. are employees of Roche Innovation Center Basel, F. Hoffmann-La Roche Ltd, Basel, Switzerland. L.Y. is a former employee of the Roche Innovation Center, Little Falls, NJ, USA.
Availability of Data and Material
Qualified researchers may request access to individual patient level data through the clinical study data request platform (https://vivli.org/). Further details on Roche's criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
Code Availability
Not applicable.
Authors' Contributions
All authors were involved in interpretation of the data, revising the manuscript critically for important intellectual content, approved the final version, and agree to be accountable for the work. Additionally, the authors contributed as follows: S.F. performed the data analysis; V.B. contributed to the conception and planning of the work that led to the manuscript; N.B. drafted the manuscript content.
Ethics Approval
All studies were approved by the relevant ethics committees, and were conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines.
Consent to Participate
All subjects provided written informed consent prior to enrollment in the clinical studies.
Consent for Publication
Not applicable.
Rights and permissions
About this article

Cite this article
Djebli, N., Buchheit, V., Parrott, N. et al. Physiologically-Based Pharmacokinetic Modelling of Entrectinib Parent and Active Metabolite to Support Regulatory Decision-Making. Eur J Drug Metab Pharmacokinet 46, 779–791 (2021). https://doi.org/10.1007/s13318-021-00714-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-021-00714-z