
Abstract
Background and Objective
Following oral administration of abiraterone acetate, the parent compound abiraterone acetate is rapidly metabolized to abiraterone. To our knowledge, bioanalytical methods to date have not been able to detect the parent compound in human plasma, and bioassay was only performed on the metabolite. A highly sensitive bioanalytical method was developed and validated to measure plasma concentrations of the parent compound. In this study, both analytes were assayed and used to evaluate the full pharmacokinetic profile of abiraterone acetate tablets.
Methods
This was an open-label, single-dose, one-period, one-treatment, pharmacokinetic study performed in 18 healthy subjects. Each subject was administered four tablets (corresponding to a total dose of 1000 mg) of abiraterone acetate. Blood samples for pharmacokinetic analysis were collected up to 60 h post-dose. Subjects’ plasma concentrations for abiraterone acetate were assayed using highly sensitive validated bioanalytical methods with a lower limit of quantitation (LLOQ) of 0.5 pg/ml for abiraterone acetate and 0.1 ng/ml for abiraterone. Safety assessments were performed throughout the study.
Results
The pharmacokinetic results for abiraterone acetate showed a mean for the maximum plasma concentration (Cmax) of 54.67 ± 68.30 pg/ml, and a median time to maximum concentrations (tmax) of 5.53 h (range 2.67–35.00 h). The means for area under the concentration-time curve (AUC) from time 0 h to infinity (AUCinf) and AUC from time zero h to the time of the last measurable abiraterone acetate concentrations (AUCt) were 386.13 ± 266.80 pg·h/ml and 460.07 ± 378.78 pg·h/ml, respectively. The apparent elimination half-life (t1/2) showed a mean of 8.98 ± 3.92 h. None of the adverse events that affected three subjects (16.7%) were related to the study drug.
Conclusion
The ability to detect the low plasma abiraterone acetate concentrations, in addition to abiraterone, resulted in a complete characterization of the pharmacokinetics of abiraterone acetate that was not possible with other analytical methods that only measured the metabolite. The development of new bioanalytical methods such as these will allow for a more thorough understanding of the pharmacokinetics of abiraterone acetate, and this, in turn, can have an impact on both future examinations into abiraterone acetate pharmacokinetic behaviour and the evaluation of its generic formulations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Highly sensitive bioanalytical methods were developed and validated to detect very low concentrations of abiraterone acetate and abiraterone in human plasma samples. The methods were successfully applied to completely characterize the pharmacokinetic profile of abiraterone acetate tablets administered to healthy subjects. |
The bioanalytical method can be applied in future pharmacokinetic evaluations of abiraterone acetate generic formulations. |
1 Introduction
Abiraterone acetate is indicated for the treatment of metastatic castration-resistant prostate cancer (CRPC) and metastatic high-risk castration-sensitive prostate cancer (CSPC). It is recommended in combination with prednisone or prednisolone [1, 2]. Both abiraterone acetate and abiraterone, an active metabolite of abiraterone acetate, are potent and are selective inhibitors of 17 α-hydroxylase/C17,20-lyase (CYP17), a key enzyme in testosterone biosynthesis [3]. By blocking CYP17, abiraterone stops androgen biosynthesis in testicular, adrenal, and prostatic tumor tissues [4].
Prostatic carcinoma that is androgen sensitive responds to treatments that decrease androgen levels [1]. Androgen deprivation therapies (ADTs), such as hormonal therapy or orchidectomy, reduce androgen synthesis in the testes but do no effect its production in the tumour or by the adrenal glands. Treatment with abiraterone acetate has been shown to decrease serum testosterone to undetectable levels when given with an ADT [2].
According to the National Institute for Health and Clinical Excellence (NICE) guideline on the diagnosis and management of prostate cancer [4], the primary treatment for metastatic prostate cancer is usually hormone therapy. Orchidectomy should be offered as an alternative to continuous hormonal therapy. When biochemical evidence of hormone-refractory metastatic prostate cancer is found, a chemotherapy regimen becomes the treatment of choice [4]. Once the disease progresses on or after the chemotherapy regimen, NICE recommends abiraterone acetate [5].
Studies in patients with metastatic CRPC and in healthy subjects showed that the parent compound, abiraterone acetate, is rapidly metabolized to abiraterone, an active metabolite. Following oral administration, more than 99% of the analyzed human plasma samples had abiraterone acetate concentrations below the detectable level of 0.2 ng/ml [1].
Following an oral daily dose of 1000 mg of abiraterone acetate administered to patients with metastatic CRPC, the maximum plasma concentration (Cmax) and area under the concentration-time curve (AUC) for abiraterone at steady state were approximately 226 ± 178 ng/ml and 993 ± 639 ng·h/ml, respectively. The median time for abiraterone to achieve maximum concentration (tmax) was approximately 2 h [1, 2].
Mean systemic exposure of abiraterone is higher when abiraterone acetate is administered with food. Administration of a single dose of arbiraterone acetate with a high-fat meal showed approximately tenfold and 17-fold increases in the AUCinf and Cmax, respectively, when compared to a fasted state. Therefore it is recommended to take abiraterone acetate on an empty stomach. No food should be eaten for at least 2 h before and at least 1 h after oral administration [1, 2]. In accordance with these recommendations, our study evaluated the pharmacokinetics of abiraterone acetate and its metabolite under fasting conditions.
More than 99% of abiraterone is bound to plasma proteins, alpha-1 acid glycoprotein and albumin. The apparent volume of distribution at steady state is 19,669 ± 13,358 L [1].
The rapid biotransformation of abiraterone acetate into abiraterone is likely attributed to hydrolysis by esterases, and is not mediated by the cytochrome P450 (CYP) system. Abiraterone is further metabolized in the liver into inactive metabolites, abiraterone sulfate and N-oxide abiraterone sulfate, each of which accounts for approximately 43% of the abiraterone circulating metabolite. The mean abiraterone terminal half-life (t1/2) in plasma of patients with metastatic CRPC is 12 ± 5 h. Approximately 55% of an abiraterone acetate administered dose and a corresponding 22% of abiraterone are recovered unchanged in the feces; however, only 5% is approximately recovered in the urine [1, 2].
Pharmacokinetic characterization of medicinal products is a critical factor to demonstrate bioequivalence between a generic test product and a marketed reference product. Although the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) recommend the measurement of the parent compound as a more sensitive indicator of the pharmacokinetics of a formulation, for drugs such as abiraterone acetate, where the plasma levels of the parent compound are undetectable, regulatory agencies accept evaluating bioequivalence based on the metabolite [6,7,8].
Using validated bioanalytical methods, the primary objective of this study was to evaluate the pharmacokinetics of both the parent compound, abiraterone acetate, and its metabolite, abiraterone, following a single-dose administration of four tablets of 250 mg abiraterone acetate in healthy subjects under fasting conditions. A secondary objective was to assess the safety and tolerability of abiraterone acetate tablets.
2 Methods
2.1 Study Design and Study Population
This was an open-label, single-dose, one-period, one-treatment, pharmacokinetic study conducted at Pharma Medica Research Inc., Saint Charles, MI, USA. A formulation of abiraterone acetate 250 mg tablets (Zytiga® from Janssen Biotech, Inc., USA) was used in this study.
The planned sample size for this exploratory study was 18 subjects. Following a general screening procedure conducted for volunteers, the study population consisted of 18 healthy, non-smoking male subjects, 18 years of age or older, with a body mass index (BMI) ≥ 18.0 and ≤ 33.0 kg/m2. They were judged to be healthy based on a medical history, 12-lead electrocardiogram, laboratory evaluation (biochemistry, hematology, serology, and urinalysis), physical examination, and vital signs measurements (blood pressure [BP], pulse rate [PR], respiration rate [RR], and temperature). The subjects had negative test results for urine cotinine, drugs of abuse, and pregnancy (females only), and they satisfied all the eligibility criteria as assessed on the day of screening. Subjects were included after they agreed to use effective methods of contraception and were able to tolerate venipuncture.
None of the subjects had a known history or presence of any clinically significant diseases, including oncologic and genitourinary conditions. Within 30 days of drug administration, subjects did not use any drugs known to induce or inhibit hepatic drug metabolism, or alter gastrointestinal pH or movement. Subjects did not have any known history or presence of hypersensitivity or idiosyncratic reaction to abiraterone acetate or any other drug substances with similar activity. Any other condition that was listed as an excluding criterion or which, in the opinion of the investigator, would jeopardize the safety of any of the subjects or impact the validity of the study results, prohibited the subjects from participating in this study.
The internal review board (IRB) approved the informed consent form (ICF), which described the nature, duration, potential risks, and any benefits (i.e. monetary compensation) of the study, and was signed by all participating subjects at check-in; all subjects received a copy of the signed ICF.
2.2 Main Study Procedures
Subjects were confined in-house from at least 10 h prior to drug administration until at least 24 h post-dose. During this period, subjects fasted from at least 10 h prior to drug administration until at least 4 h after drug administration. Water was also restricted from 1 h prior to until 1 h post-dose. Other than that, standardized meals were provided throughout confinement and access to water was freely available to subjects.
2.2.1 Study Drug Administration
On the day following check-in, each subject was administered a single dose of 1000 mg of abiraterone acetate (four tablets of abiraterone acetate 250 mg), with approximately 240 ml of room temperature potable water. Attempts were made to administer the four tablets within 2 min, and the time of administration of the first tablet was considered the actual drug administration time.
2.2.2 Sample Collection, Sample Processing, and Bioanalytical Methods
Twenty-one blood samples were collected from each subject at 0 h (within 60 mins prior to dosing) and at 20 min, 40 min, 1 h, 1 h and 20 min, 1 h and 40 min, 2 h, 2 h and 20 min, 2 h and 40 min, 3 h, 3 h and 30 min, and at 4, 5, 6, 8, 12, 16, 24, 36, 48, and 60 h after the study drug administration. All attempts were made to collect the post-dose pharmacokinetic blood samples within 1 min from the scheduled sampling time point. Moreover, if the 60-h scheduled blood draw was not collected within 3 h from the scheduled time or was missed, or at least any two consecutively scheduled blood draws were missed, then the continued participation of that subject in the study would be evaluated by the study pharmacokineticists.
The pharmacokinetic plasma samples were collected by direct venipuncture, in pre-chilled, labeled, 6-ml blood collection tubes (10 ml for the 0-h sample) containing potassium ethylenediaminetetraacetic acid (K2-EDTA) as the anticoagulant. Approximately 130 ml of blood was collected from each subject over the entire study for pharmacokinetic analysis.
Blood samples were centrifuged within 45 min of collection, and were separated into two plasma aliquots. The aliquots were stored in a freezer at − 25 ± 10 °C, pending shipment. After packaging with sufficient dry ice, the aliquots were shipped to the bioanalytical facility.
Plasma samples were assayed for abiraterone acetate and abiraterone at the bioanalytical laboratory of Pharma Medica Research Inc., Ontario, Canada, using a validated liquid chromatographic tandem mass spectrometric (LC-MS/MS) method, with an LLOQ of 0.500 pg/ml for abiraterone acetate and 0.100 ng/ml for abiraterone.
2.2.2.1 Analytical Method for Abiraterone Acetate
The standard calibration range was from 0.500 to 100 pg/ml using a plasma sample volume of 0.300 ml. Plasma samples, treated with K2EDTA as the anticoagulant, were extracted under basic conditions with a mixture of organic solvents, the organic phase was dried, and the reconstituted sample was transferred for analysis. Samples were analyzed by LC-MS/MS (Shimadzu Prominence UFLC and SCIEX API 5000) and reverse phase chromatography under gradient conditions with mobile phases composed of 45–80% methanol, 10% acetonitrile, and 0.5% formic acid. Chromatographic separation was achieved using serial analytical columns (C8: 50 × 3 mm, 2.6 µm and PFP: 50 × 3 mm, 2.6 µm). Abiraterone acetate was analyzed using a positive ion scan mode and a parent-daughter mass to charge ion transition of 392–332. The retention time for abiraterone acetate was approximately 1.9 min.
2.2.2.2 Analytical Method for Abiraterone
The standard calibration range was from 0.100 to 200 ng/ml using a plasma sample volume of 0.200 ml. Plasma samples, treated with K2EDTA as the anticoagulant, were precipitated with a mixture of organic solvents and the diluted supernatant was transferred for analysis. Samples were analyzed by LC-MS/MS (Shimadzu Prominence UFLC and SCIEX API 4000) using reverse phase chromatography under gradient conditions using mobile phases composed of 40–70% acetonitrile, 20% methanol, 10 mM ammonium formate, and 0.1% formic acid. Chromatographic separation was achieved using a C8, 50 × 3 mm, 1.7 µm analytical column. Abiraterone was analyzed using a positive ion scan mode and a parent-daughter mass to charge ion transition of 350–156. The retention time for abiraterone was approximately 1.2 min. In accordance with the guidelines on bioanalytical methods validation [9, 10], the LC–MS/MS methods for abiraterone acetate and abiraterone were validated for the LLOQ, calibration range, precision, accuracy, recovery, selectivity, matrix effects, stability of processed samples, and stability in human plasma and in whole blood samples. The method for abiraterone acetate had recoveries of 75.6–76.3%, while the method for abiraterone had recoveries of 108.6–111.8%. Moreover, the matrix effect had a precision and accuracy of ≤ 4.1% and 95.8–96.0%, respectively, for abiraterone acetate, and ≤ 6.7% and 102.3–106.9%, respectively, for abiraterone. Table 1 presents the precision and accuracy of the validation methods and Table 2 presents the stabilities in human plasma and in whole blood samples.
2.2.3 Safety Measures and Assessment
In agreement with regulatory requirements [11], adverse events (AEs) were reported in a timely, accurate, and complete manner. The assessment of safety was based primarily on the frequency and severity of AEs, which were collected after the subjects had signed the ICF. There was no statistical evaluation of safety or tolerability. The severity and relation of AEs to the study drug were assessed by the investigator, who was on site prior to drug administration and for 4 h after the first subject was dosed. The investigator remained on call until the end of the study.
Subjects were questioned regarding their health status throughout the study. Vital signs (blood pressure (BP) and heart rate (HR)) were measured prior to abiraterone acetate administration and at 1, 2, and 5 h post dose. Temperature was measured daily during confinement, regardless of participation status while in the clinic. In addition to the aforementioned BP, HR, and temperature measurements, vital signs included respiratory rate at the end of the study. A full physical examination and clinical laboratory safety examinations (biochemistry, hematology, and urinalysis) were also performed at the end of the study. All clinical laboratory tests were performed by Quest Diagnostics, Lenexa, KS, USA.
2.3 Pharmacokinetic Parameters
The pharmacokinetic parameters were estimated for abiraterone acetate and abiraterone using a standard noncompartmental approach with Phoenix WinNonlin version 6.4 (Certara USA, Inc., Princeton, NJ, USA). Pharmacokinetic analysis was conducted using the actual times of collection of the post-dose samples. The peak concentration (Cmax) and the time to reach Cmax (tmax) were determined from individual plasma concentration-time profiles for abiraterone acetate and abiraterone. The area under the plasma concentration-time curve from 0 to 60 h (AUCt) was calculated using the linear up-log down trapezoidal method. The area under the plasma concentration-time curve from zero to time infinity (AUCinf) was calculated as AUCt + Ct/kel, where Ct is the last measurable concentration and kel is the terminal rate constant. The terminal half-life (t1/2) was calculated as 0.693/kel.
Individual and mean plasma concentration versus time curves were plotted, and descriptive statistics for the pharmacokinetic parameters of both analytes were calculated. Descriptive statistics included number of observations, mean, standard deviation, CV, median, minimum, and maximum.
3 Results
3.1 Subjects’ Demographics
All of the 18 enrolled male subjects completed the study. The subjects had a mean age of 38.5 ± 9.3 years, and all of them had a body mass index (BMI) within the inclusion criteria, with a mean of 27.4 ± 3.6 kg/m2. Twelve of the subjects (66.7%) were black or African American, while six (33.3%) of the subjects were White, with none being Hispanic or Latino. A summary of the demographic data is presented in Table 3.
3.2 Pharmacokinetic and Statistical Analysis
None of the 18 subjects had any incidence that affected the integrity of the pharmacokinetic results. In brief, the estimated pharmacokinetic parameters for the parent compound, abiraterone acetate, after single-dose administration of abiraterone acetate, showed a mean AUCinf of 386.13 ± 266.80 pg·h/ml, and a mean AUCt of 460.07 ± 378.78 pg·h/ml. The mean Cmax, which had a mean tmax of 8.38 ± 7.44 h, was 54.67 ± 68.30 pg/ml. For the metabolite, abiraterone, the pharmacokinetic parameters showed a mean AUCinf of 512.47 ± 312.57 ng·h/ml, and a mean AUCt of 499.20 ± 305.19 ng·h/ml. The mean Cmax, which had a mean tmax of 2.51 ± 1.40 h, was 100.74 ± 50.17 ng/ml. Furthermore, the t1/2 of abiraterone acetate had a minimum of 4.12 h and a maximum of 17.50 h, which resulted in a mean of 8.98 ± 3.92 h, while the t1/2 for abiraterone had a minimum of 7.15 h and a maximum of 19.39 h, which resulted in a mean of 13.68 ± 3.46 h.
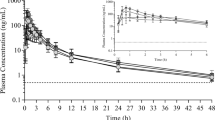
Results of the pharmacokinetic analysis for abiraterone acetate and abiraterone are presented in Table 4. The mean plasma abiraterone acetate concentration-time curve and the mean plasma abiraterone concentration-time curve from 0 to 60 h are presented in Figs. 1 and 2, respectively. The presence of two peaks in the mean concentration-time profile for abiraterone acetate can be attributed to the variability of the data. Two subjects out of 18 showed pronounced double peaks, which influenced the mean concentration-time profile for abiraterone acetate.

Mean plasma abiraterone acetate concentration-time curve linear scale (a) and log-linear scale (b) from 0 to 60 h, following abiraterone acetate single-dose administration to healthy subjects

Mean plasma abiraterone concentration-time curve linear scale (a) and log-linear scale (b) from 0 to 60 h, following abiraterone acetate single-dose administration to healthy subjects
3.3 Safety Results
The administration of abiraterone acetate was generally well tolerated by the healthy subjects participating in the study. Overall, three subjects (16.7%) reported seven AEs during the conduct of this study.
None of the AEs were related to the study drug, and none of the subjects were discontinued from the study due to an AE. All AEs were judged to be mild in severity, did not require any concomitant medications, and resolved by the end of the study. No serious AEs were reported during the conduct of this study and none of the AEs had a significant impact on the safety of the subjects or on the integrity of the study results.
4 Discussion
The rapid hydrolysis of the parent compound, abiraterone acetate, into its metabolite, abiraterone, results in undetectable concentrations of the parent compound in plasma of patients treated with abiraterone acetate [12]. Clinical studies showed that following oral administration of abiraterone acetate, plasma concentrations of the parent compound in more than 99% of the analyzed samples were below the detectable level of 0.2 ng/ml [1]. Moreover, the FDA product-specific guidance on abiraterone acetate recommends bioequivalence studies to be based on the active metabolite [6]. Therefore, pharmacokinetic analysis in clinical trials was performed on abiraterone, the metabolite responsible for clinical activity [12], rather than on its parent compound. Very few analytical methods were developed for the measurement of plasma abiraterone acetate concentrations. The analytical methods discussed in Martins et al. [13], and in Gong and Zhu [14], were developed for measuring the concentrations of both analytes in human plasma. Other analytical methods published [15] were developed to measure the concentrations of only abiraterone in human plasma.
In Martins et al. [13], a liquid chromatography (LC) tandem mass spectrometry (MS/MS) has been developed for the quantification of abiraterone acetate and abiraterone in human plasma. The method was validated and it was intended to be applied for investigating the PK of both analytes. To our knowledge, pharmacokinetic analyses performed to date were for abiraterone [16,17,18].
In Gong and Zhu [14], a β-cyclodextrin sensitized spectrofluorimetric method was developed with a limit of quantitation (LOQ) for abiraterone acetate of 22.4 ng/ml, and 21.8 ng/ml for abiraterone. Bellivile et al. [19] also used fluorescence detection with an HPLC. The calibration was in the range of 1.75–50 ng/ml. For both of these studies, the LOQ for abiraterone acetate was above the levels indicated in the FDA-approved abiraterone acetate label [1], and accordingly could not detect abiraterone acetate concentrations in human plasma. Therefore the β-cyclodextrin spectrofluorimetric method was applied to determine abiraterone acetate in the tablet formulation. However, both studies were successfully applied to determine abiraterone in human plasma [14, 19].
For our exploratory study, a validated HPLC–MS/MS was developed and successfully used to measure both analytes concentrations in human plasma. The LLOQ for abiraterone acetate was 0.5 pg/ml, and the calibration range was up to 100 pg/ml, while for abiraterone the LLOQ was 0.1 ng/ml, and the calibration range was up to 200 ng/ml. To our knowledge these LLOQs are the lowest published to date. Bioanalytical methods’ calibration ranges for abiraterone varied between different studies. For both Reddy et al. [20] and Gurav et al. [17] the bioanalytical methods had an LLOQ of 0.2 ng/ml, while for Wani et al. [18] the method had an LLOQ of 0.1 ng/ml, and its calibration range was up to 50 ng/ml. Other published bioanalytical methods for abiraterone had an LLOQ of 1 ng/ml [16, 21], with a calibration range up to 500 ng/ml [16].
In our study, the results of the pharmacokinetic analysis for abiraterone were comparable to those already published. In a phase I dose-escalation pharmacokinetic study of abiraterone acetate in healthy men, following a 1000-mg daily dose of abiraterone acetate, the results for abiraterone showed a mean AUCinf of 617 ± 249 ng·h/ml, a mean Cmax of 112 ± 36.7 ng/ml and a median tmax of 1.75 h, with a minimum and a maximum of 1.0 and 3.0 h, respectively. The half-life was 12.7 ± 1.91 h [22]. In another phase I dose-escalation pharmacokinetic study, following a 1,000-mg dose of abiraterone acetate to patients with CRPC under fasting conditions, the mean tmax was 1.8 ± 0.4 h for abiraterone, and the mean t1/2 was 14.4 ± 7.7 h [3]. Moreover, the FDA-approved label for abiraterone acetate [1] indicated that following oral administration of abiraterone acetate in patients with metastatic CRPC under fasting conditions, abiraterone reaches Cmax in approximately 2 h and the mean t1/2 is 12 ± 5 h.
The proper evaluation of a drug’s pharmacokinetics is important as it is known that this is what drives the pharmacological activity of the drug. Pharmacokinetic parameters such as Cmax and AUC provide a means to evaluate the rate and extent of absorption of a drug’s formulation. The Cmax of the metabolite is not as sensitive as the Cmax of the parent compound in detecting differences in the formulation [7]. Moreover, the concentration-time profile of the parent compound reflects changes in the performance of the formulation more than the metabolite concentration-time profile, which is more sensitive to the metabolite formation [8]. Therefore, for bioequivalence studies, it is recommended to measure the parent compound in the biological fluids rather than the metabolite. However, in exceptional cases where the bioanalytical methods are not precise and accurate enough to detect low levels of the parent compound in human plasma, it is acceptble to use the metabolite data as a substitute for bioequivalence [7, 8]. Accordingly, the FDA guidance on abiraterone acetate recommends bioequivalence studies to be done on abiraterone [1, 6]. In our study we were able to successfully quantify both the parent compound and its metabolite in human plasma. This is a more suitable indicator of the drug’s pharmacokinetic behaviour than measuring just the metabolite alone. Therefore, in order to gain a deeper insight into the bioequivalence of future abiraterone acetate formulations, regulatory agencies should consider the need to quantify abiraterone acetate as the primary analyte in assessing generic formulations.
5 Conclusion
With the development and validation of an assay to quantify both abiraterone and abiraterone acetate, a more accurate and complete characterization of the pharmacokinetics of abiraterone acetate formulations will be possible. The lower LLOQ allowed a better understanding of the pharmacokinetics of abiraterone acetate, which was not possible with other analytical methods that measured only the levels of the metabolite. Therefore this advancement may stimulate changes in the evaluation of abiraterone acetate bioequivalence to include the measurement of the parent compound. This will help in the implementation of further investigations into the pharmacokinetics of abiraterone acetate, and consequently enhance future abiraterone acetate formulations.
References
Janssen Biotech, Inc. Highlights of prescribing information. Zytiga® (abiraterone acetate) tablets. U.S. FDA revised 02/2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/202379s024lbl.pdf. Accessed 1 Mar 2018.
Janssen-Cilag International NV. Summary of product characteristics Zytiga 250 mg tablets. EMA updated: 11/2017. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002321/WC500112858.pdf. Accessed 30 Mar 2018. Accessed 01 Mar 2018.
Ryan CJ, Smith MR, Fong L, Rosenberg JE, Kantoff P, Raynaud F, Martins V, Lee G, Kheoh T, Kim J, Molina A, Small E. Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. J Clin Oncol. 2010. https://doi.org/10.1200/JCO.2009.24.1281.
National Institute for Health and Care Excellence. Prostate cancer: diagnosis and management, clinical guideline. 2014. https://www.nice.org.uk/guidance/CG175?UNLID=. Accessed 1 Mar 2018.
National Institute for Health and Care Excellence. Abiraterone for castration-resistant metastatic prostate cancer previously treated with a docetaxel-containing regimen. 2016. https://www.nice.org.uk/guidance/ta259. Accessed 1 Mar 2018.
United States Department of Health and Human Services, Food and Drug Administration. Draft guidance on abiraterone acetate. 2016. https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm333001.pdf. Accessed 1 Mar 2018.
European Medicines Agency, Committee for Medicinal Products for Human Use. Guideline on the investigation of bioequivalence. 2010. https://www.ema.europa.eu/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf. Accessed 6 Jan 2019.
United States Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Guidance for industry: bioequivalence studies with pharmacokinetic endpoints for drugs submitted under an ANDA: draft guidance. 2013. https://www.fda.gov/downloads/drugs/guidances/ucm377465.pdf. Accessed 26 Mar 2018.
United States Department of Health and Human Services, Food and Drug Administration. Guidance for industry: bioanalytical method validation: draft guidance. 2013. https://www.fda.gov/downloads/drugs/guidances/ucm070107.Pdf. Accessed 26 Mar 2018.
European Medicines Agency. Guideline on bioanalytical method validation: draft guidance. 2011. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf. Accessed 26 Mar 2018.
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonised tripartite guideline: clinical safety data management: definitions and standards for expedited reporting: E2A. 1994. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E2A/Step4/E2A_Guideline.pdf. Accessed 23 Feb 2018.
Benoist GE, Hendriks RJ, Mulders PF, Gerritsen WR, Somford DM, Schalken JA, van Oort IM, Burger DM, van Erp NP. Pharmacokinetic aspects of the two novel oral drugs used for metastatic castration-resistant prostate cancer: abiraterone acetate and enzalutamide. Clin Pharmacokinet. 2016. https://doi.org/10.1007/s40262-016-0403-6.
Martins V, Asad Y, Wilsher N, Raynaud F. A validated liquid chromatographic–tandem mass spectroscopy method for the quantification of abiraterone acetate and abiraterone in human plasma. J Chromatogr B. 2006. https://doi.org/10.1016/j.jchromb.2006.06.010.
Gong A, Zhu X. β-Cyclodextrin sensitized spectrofluorimetry for the determination of abiraterone acetate and abiraterone. J Fluoresc. 2013. https://doi.org/10.1007/s10895-013-1261-3.
Pushpa Latha E, BushraY Sayeda, Navaya Sri O, Juveriya N, Anusha M, Mani Sagar G, Samuel Shine S. A review on analytical methods for estimation of abiraterone acetate. WJPMR. 2016;2(2):23–5.
Benoist GE, van der Meulen E, Lubberman FJE, Gerritsen WR, Smilde TJ, Schalken JA, Beumer JH, Burger DM, van Erp NP. Analytical challenges in quantifying abiraterone with LC–MS/MS in human plasma. Biomed Chromatogr. 2017. https://doi.org/10.1002/bmc.3986.
Gurav S, Punde R, Farooqui J, Zainuddin M, Rajagopal S, Mullangi R. Development and validation of a highly sensitive method for the determination of abiraterone in rat and human plasma by LC–MS/MS-ESI: application to a pharmacokinetic study. Biomed Chromatogr. 2012;26(6):761–8.
Wani TA. Highly sensitive ultra-performance liquid chromatography–tandem mass spectrometry method for the determination of abiraterone in human plasma. Anal Methods. 2013. https://doi.org/10.1039/C3AY26611G.
Belleville T, Noé G, Huillard O, Thomas-Schoemann A, Vidal M, Goldwasser F, Alexandre J, Blanchet B. A HPLC-fluorescence method for the quantification of abiraterone in plasma from patients with metastatic castration-resistant prostate cancer. J Chromatogr B. 2015. https://doi.org/10.1016/j.jchromb.2015.03.001.
Reddy S, Thomas L, Ramanna L, Mukhopadhyay A, Thangam S. Estimation of abiraterone in human plasma by liquid chromatography tandem mass spectrometry. Acta Pharm Sci. 2018. https://doi.org/10.23893/1307-2080.APS.05605.
van Nuland M, Hillebrand MJX, Rosing H, Schellens JHM, Beijnen JH. Development and validation of an LC–MS/MS method for the simultaneous quantification of abiraterone, enzalutamide, and their major metabolites in human plasma. Ther Drug Monit. 2017. https://doi.org/10.1097/FTD.0000000000000387.
Acharya M, Bernard A, Gonzalez M, Jiao J, De Vries R, Tran N. Open-label, phase I, pharmacokinetic studies of abiraterone acetate in healthy men. Cancer Chemother Pharmacol. 2012. https://doi.org/10.1007/s00280-012-1865-3.
Acknowledgements
The authors acknowledge the assistance provided by Ashraf El Sheikh Ali in the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Pharma Medica Research Inc.
Conflict of interest
Mohammed Bouhajib and Zia Tayab are employees of Pharma Medica Research Inc. They do not have any other conflicts of interest to declare.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The study protocol and informed consent form were approved by Salus Institutional Review Board (IRB), Texas, USA.
Informed consent
Informed consent was obtained from all individual participants included in the study, prior to any activity in the trial.
Rights and permissions
About this article

Cite this article
Bouhajib, M., Tayab, Z. Evaluation of the Pharmacokinetics of Abiraterone Acetate and Abiraterone Following Single-Dose Administration of Abiraterone Acetate to Healthy Subjects. Clin Drug Investig 39, 309–317 (2019). https://doi.org/10.1007/s40261-019-00752-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-019-00752-1