
Abstract
Purpose
SUVN-1105 is a novel formulation of abiraterone acetate which was developed to demonstrate improved bioavailability, compared to Zytiga and Yonsa, and to reduce the dose and eliminate the food effect. A Phase 1 study was conducted to assess the bioequivalence, food effect, and comparative pharmacokinetics of SUVN-1105 to Zytiga in healthy male subjects.
Methods
The study comprised of 2 segments. Segment 1 was a single-center, 4-period crossover, open-label, fixed treatment sequence, single-dose study to evaluate the safety and pharmacokinetics of SUVN-1105 (N = 12 subjects per period). Segment 2 was a single-center, open-label, single-dose, randomized, 4-period, 4-treatment, 4-sequence crossover study to evaluate bioequivalence and comparative pharmacokinetics of SUVN-1105 against Zytiga (N = 44) under overnight fasted, modified fasted, and fed conditions.
Results
Abiraterone exposures appeared to increase proportionately with SUVN-1105 dose (200 mg vs. 250 mg) in Segment 1. In Segment 2, abiraterone exposures of 250 mg SUVN-1105 in the fasted or fed conditions were higher than those of Zytiga 1000 mg in the overnight fasted conditions. Abiraterone exposures of 250 mg SUVN-1105 decreased in the fed conditions (64% and 29% decrease in Cmax and AUC, respectively) compared to overnight fasted conditions.
Conclusions
The abiraterone exposures of 250 mg SUVN-1105 in the fasted or fed conditions fall within the abiraterone exposures of 1000 mg Zytiga in fasted and modified fasted conditions. Single doses of SUVN-1105 were safe and well-tolerated in healthy males both in the fasted and fed conditions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Prostate cancer is the fifth leading cause of cancer death and the second most commonly diagnosed cancer among men worldwide with an estimated 1.4 million new cancer cases and 375,304 deaths in 2020 [1]. An estimated 3.4 million men were living with prostate cancer in the United States (U.S.) in the year 2020. In 2023, approximately 288,300 new cases of prostate cancer and 34,700 deaths are estimated from prostate cancer in the U.S. [2]. Prostate cancer spread to other parts of the body is known as metastatic prostate cancer or advanced prostate cancer.
Abiraterone acetate is an androgen synthesis inhibitor indicated for the treatment of metastatic castration-resistant prostate cancer (mCRPC) and metastatic castration- sensitive prostate cancer (mCSPC). Abiraterone acetate is the acetyl ester prodrug that is converted in vivo to abiraterone. Abiraterone, the active metabolite, is an inhibitor of CYP17 which manifests as two enzymes, 17alpha-hydroxylase and C17, 20-lyase, that are expressed in testicular, adrenal, and prostatic tumor tissues, and are required for androgen biosynthesis. Abiraterone, by inhibiting the androgen synthesis, decreases the proliferation of testosterone-sensitive prostate cancer cells and prolongs survival in patients with metastatic prostate cancer. Abiraterone acetate was approved in the U.S. under the trade name of Zytiga® for the treatment of mCRPC in the year 2011 and for mCSPC in the year 2018, and is available in strengths of 250 mg or 500 mg tablets. The approved dose is 1000 mg administered orally once daily as 4 × 250 mg or 2 × 500 mg tablets in combination with prednisone 5 mg administered twice daily or once daily for mCRPC and mCSPC, respectively. Abiraterone acetate exists as a crystalline form in Zytiga. Zytiga has increased and highly variable exposures in the fed state with 17- and 10-fold increases in peak and systemic exposures, respectively, compared to the fasted condition demonstrating a significant positive food effect. Per the product label, Zytiga must be taken on an empty stomach. No food should be consumed for at least 2 h before and 1 h after the dose of Zytiga is taken [3].
Abiraterone acetate was also approved in the U.S. as a different drug product, Yonsa®, for the treatment of mCRPC in the year 2018, and is available at the strength of 125 mg tablets. The approved dose is 500 mg administered orally once daily as 4 × 125 mg tablets in combination with methylprednisolone 4 mg administered orally twice daily. The reduced dose of Yonsa (500 mg) compared to Zytiga (1000 mg) is due to improved bioavailability attributed to the micronized or nano-amorphous form of abiraterone acetate in Yonsa [4, 5]. Yonsa tablets can be taken with or without food, although a positive food effect is evident albeit lesser than Zytiga.
SUVN-1105 is a novel formulation of abiraterone acetate that is developed to improve aqueous solubility and bioavailability compared to Zytiga and Yonsa with further reduction in dose and elimination of food effect. This formulation can become an alternative for patients with dysphagia and may improve patient adherence. SUVN-1105 was prepared by solvent evaporation method which relies on the preparation of amorphous solid dispersion of abiraterone acetate by casting the homogenous solution of abiraterone acetate with one or more pharmaceutically acceptable excipients to form thin sheets that were milled to obtain fine granules of SUVN-1105. The granules were packed into sachets each containing 250 mg of abiraterone acetate in 8.3 g of SUVN-1105.
SUVN-1105 exhibited higher apparent solubility and dissolution rate in vitro compared to Zytiga. The comparative pharmacokinetics study in beagle dogs demonstrated improved bioavailability and a four-fold reduction in the dose of SUVN-1105 compared to Zytiga. There was no food effect observed for SUVN-1105 when tested in dogs. These findings guided us in the evaluation of SUVN-1105 against the listed drug in humans. The complete details of formulation development and preclinical studies of SUVN-1105 will be published elsewhere. This report describes the study design, conduct, and results of the clinical study that was designed to evaluate the pharmacokinetic properties of SUVN-1105 and to establish equivalence to 1000 mg Zytiga.
Materials and methods
SUVN-1105 granules and Zytiga tablets
SUVN-1105 was manufactured by Suven Life Sciences Limited. SUVN-1105 is a novel oral pharmaceutical composition that comprises of solid dispersion of amorphous abiraterone acetate. The final investigational product was granules-in-sachet containing 250 mg of abiraterone acetate for reconstitution with water. The granules were to be added to 240 mL water to produce a suspension formulation before dosing. The granules were weighed and accordingly used to achieve the required doses other than 250 mg. Zytiga tablets (250 mg strength) for the study were commercially sourced by pharmacy at Pharmaceutical Research Associates (PRA) health sciences (Lenexa, KS, U.S.).
Compliance with ethics
This Phase 1 clinical study was conducted between June 22, 2020, and May 17, 2021, at a single study center in the United States by Pharmaceutical Research Associates -Early Development Services (PRA-EDS, Lenexa, KS, U.S.). The study was conducted in accordance with the Clinical Protocol, the principles of the Declaration of Helsinki [6], and in compliance with the International Council for Harmonization (ICH) E6 (R2) Guideline for Good Clinical Practice (GCP) [7] and any applicable local laws and regulations. The study protocol, protocol amendments, and informed consent forms were reviewed and approved by an Institutional Review Board (IRB). All subjects, regardless of their eligibility for the study, signed the informed consent form before initiating any study-specific procedures.
Study population
Healthy male subjects aged 18–45 years with a body mass index (BMI) between 18 and 30 kg/m2 and a body weight of ≥ 50 kg were eligible for the study. Subjects were enrolled in the study based on their health status as defined by the absence of evidence of any clinically significant, active or chronic disease following a detailed medical and surgical history, a complete physical examination, including vital signs, 12-lead electrocardiogram (ECG) and clinical laboratory testing. Male subjects enrolling in the study and their female partner(s) must have agreed to use two forms of contraception, one of which must have been a barrier method, during the study, and for 90 days after the last drug administration. Subjects were excluded from the study if they had a history of clinically significant cardiovascular abnormalities, adrenal or pituitary problems, and abnormal vital signs at screening or received a positive test result for drugs of abuse or alcohol at the screening visit or use of any drugs known to inhibit or induce hepatic drug metabolism within 2 weeks of planned dosing. Males with female partners who were pregnant, lactating, or planning to attempt to become pregnant during the study or within 90 days after dosing of the study drug were excluded.
Study design
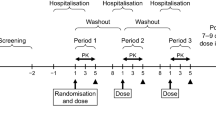
This Phase 1 study was conducted in two segments. Segment 1 was an open-label, four-period crossover, fixed treatment sequence, single-dose study to evaluate the pharmacokinetics, safety, and tolerability of SUVN-1105 in healthy male subjects (N = 12 subjects per period). No prospective calculations of statistical power were made. The sample size was selected to provide information on safety, tolerability, and pharmacokinetics following single doses of SUVN-1105.
Segment 1 was also designed to inform a suitable dose for Segment 2 and the effect of food on the pharmacokinetics of SUVN-1105. Segment 1 consists of a screening period of up to 4 weeks followed by four sequential single-day treatment periods with an intervening 7-day washout period between administrations. In Periods 1 and 2, a single dose of 200 mg and 250 mg SUVN-1105 was administered following an overnight fast (minimum 10 h), and subjects continued to fast for 4 h post-dose. In Periods 3 and 4, subjects received a single dose of 200 mg and 250 mg SUVN-1105 under fed conditions, consisting of an overnight fast of at least 10 h, consumed a high-fat breakfast 30 min before administration of study drug, and continued to fast for 4 h post-dose. An interim review of pharmacokinetic and safety data from each treatment period was conducted to determine the dose and dietary conditions for subsequent treatment periods. All subjects (N = 12 for period 1 and N = 11 for periods 2, 3, and 4) were confined in the clinic for four periods, each period extended from day -1 (admission) until day 3 (48 h post-dose). Subjects returned to the clinic for a follow-up visit 7–14 days after period 4.
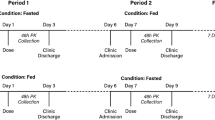
Segment 2 was an open-label, randomized, single-dose, four-period, four-treatment, four-sequence crossover study to evaluate bioequivalence and comparable pharmacokinetics of SUVN-1105 against the listed drug, Zytiga (1000 mg, 4 × 250 mg abiraterone acetate tablets), in healthy male subjects (N = 44). A sample size of 40 subjects provides at least 80% power to have a 90% CI of estimated geometric least squares (LS) mean ratio within the bioequivalence acceptance limits of 80–125%, assuming the intra-subject variability of 31% for AUC [8]. The calculation assumed that the expected ratio of means was 1.05. A total of 44 subjects were planned to be enrolled to ensure that 40 subjects completed the treatment, assuming a dropout rate of about 10%. The subjects were randomly assigned to one of the four treatment sequences according to the random table (Table 1) generated before the subjects were enrolled. Zytiga was administered as four 250 mg tablets under overnight fasted (Treatment A) or modified fasted conditions (Treatment C). Zytiga was tested under fasted and modified fasted conditions as the exposures of Zytiga under those states would represent the lower and upper limit, respectively. The modified fasting conditions employed in this study are consistent with the Zytiga product label. SUVN-1105, 250 mg was administered as a suspension under overnight fasting (Treatment B) or fed conditions (Treatment D). Under the fasted conditions, subjects received the study drug after an overnight fast for at least 10 h and fasting continued for 4 h post-dose. The modified fasting condition consisted of an overnight fast of at least 10 h followed by a high-fat breakfast 2 h before dosing, with a continued fast for at least 1-h post-dose. Subjects taking the study drug under fed conditions fasted for at least 10 h overnight, consumed a high-fat breakfast 30 min before administration of study drug, and fasted for 4 h after study drug administration. All subjects were confined in the clinic for four treatment periods, each period extended from day -1 (prior to day 1) until completion of the assessments on day 3. The minimum washout period between the consecutive doses was at least 7 days.
Blood collection
In both segments, blood samples were collected by direct venipuncture or through an indwelling catheter for the measurement of abiraterone concentrations in plasma. Blood samples of 2 mL each were collected at pre-dose, 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, and 48 h post-dose into 2 mL vacutainer tubes containing sodium fluoride and potassium oxalate. In Segment 2, blood sampling at 5 h was performed in addition to the above-mentioned timepoints. All the blood samples were centrifuged within 30 min after sampling for 10 min at 1500 × g at 4 °C to obtain plasma. The plasma samples were stored at or below −70 °C until transfer to the bioanalytical laboratory (PRA, Lenexa, KS).
Bioanalytical method
Abiraterone concentrations were analyzed in plasma using a validated liquid chromatography-tandem mass spectrometry (LC–MS/MS) method. The bioanalytical method validation, clinical sample analyses, quality control procedures, and acceptance criteria were based on the US Food and Drug Administration (FDA) Guidance for Industry—Bioanalytical Method Validation 2018 [9]. The analysts were blinded from the randomized treatment sequence during the analysis. Abiraterone and abiraterone-d4 (internal standard) were extracted from plasma samples using a liquid–liquid extraction method by employing a plasma sample volume of 0.05 mL. Chromatographic separation was achieved using a Water Acquity HSS T3 C18, 2.1 × 50 mm, 1.8 µm analytical column (Waters Corporation, MA, U.S.) at 50 °C using 10 mM ammonium formate containing 0.1% formic acid as mobile phase A, and 0.1% formic acid in acetonitrile as mobile phase B operating at isocratic conditions with flow rate of 0.800 mL/min. A triple quadrupole 5500 mass spectrometer equipped with a turbo-ion spray source was used in positive ion mode for detection. Abiraterone and abiraterone-d4 were quantified based on the multiple reaction monitoring (MRM) of mass-to-charge transitions of 350.4 to 156.2 and 354.4 to 160.2, respectively. The lower limit of quantitation for abiraterone was 0.500 ng.mL−1and the calibration curve ranged from 0.5 to 500 ng.mL−1. The assay was validated for a 20-fold dilution factor to quantitate abiraterone concentrations up to 10,000 ng.mL−1. Calibration curves were constructed using quadratic regression curve fitting, including 1/x2 as a weighting factor. Sample acquisition, data processing, and quantification were carried out by Analyst software, version 1.7.1 (AB Sciex, Concord, Canada). The intra-day accuracy and precision ranged from 102% to 105% and from 1.1 to 2.1, respectively. The inter-day accuracy and precision ranged from 102% to 105% and from 1.1 to 1.9, respectively.
A total of 3328 plasma samples were analyzed for abiraterone by fully validated LC–MS/MS method. The results from calibration samples and quality control samples were well within the acceptable limit throughout the study sample analysis. More than 10% of a total number of study samples was selected for the assessment of incurred sample reanalysis (ISR) and 99.5% of samples were within the specification of absolute relative difference ≤ 20%.
Pharmacokinetic and statistical analyses
The plasma pharmacokinetic parameters of abiraterone were estimated using non-compartmental methods using Phoenix WinNonlin software, version 8.1 (Pharsight Corp., Certara Company, Princeton, New Jersey). The plasma concentrations of abiraterone below the quantifiable limit (BQL) were set to 0 in the computation of mean values. Actual sampling times, rather than scheduled sampling times were used in all computations involving sampling times. The estimated pharmacokinetic parameters in both the segments included maximum plasma concentration (Cmax), time to reach Cmax (Tmax), area under the concentration–time curve from time zero to time of last quantifiable concentration (AUClast) or AUC from time zero extrapolated to infinity (AUCinf), elimination half-life (t1/2), observed plasma concentration at 24 h post-dose (C24h), apparent oral clearance (CL/F) and apparent oral volume of distribution (V/F). AUC was calculated using the linear up/log down method. The diagnostic parameters used to evaluate the integrity of the pharmacokinetic parameters were the percentage of AUCinf due to extrapolation from the last quantifiable concentration observed to infinity (AUC% ext) and the goodness-of-fit statistic for the log-linear terminal elimination phase of the concentration–time profile, the adjusted R squared (adj Rsq). AUC% ext should not be more than 20% and adj Rsq should not be less than 0.8. Pharmacokinetic parameters derived from such profiles are not included in the pharmacokinetic summary.
In Segment 2, the bioequivalence of SUVN-1105 (Test) compared to Zytiga (Reference) under overnight fasted conditions was assessed using the ratio and 90% CIs of the geometric least-square means of Cmax, C24h, AUClast and AUCinf for abiraterone. A linear mixed effects model, where treatment, sequence, and period were considered as fixed effects and subjects nested within the sequence as random effect, was performed on the natural log-transformed pharmacokinetic parameters. Estimates on the original scale of measurement were obtained by exponentiating point estimates on the natural log scale. Bioequivalence was concluded when 90% CIs of the geometric LS mean ratios were entirely within the confidence limits of 80–125%. Similarly, the effect of food on the relative oral bioavailability of SUVN-1105 granules and Zytiga was assessed following a single oral administration.
A comparable pharmacokinetics could be established if abiraterone exposures of 250 mg SUVN-1105 granules are at least equivalent or higher than the lower limit (abiraterone exposures of 1000 mg Zytiga under overnight fasted conditions) and are equivalent or lower than the upper limit (abiraterone exposures of 1000 mg Zytiga under modified fasted condition).
Safety assessments
In both the segments, safety and tolerability assessments consisted of physical examination, clinical laboratory tests, vital signs, 12-lead ECG, and Adverse Events (AEs) monitoring. A complete physical examination was performed at screening only. A brief physical examination was performed at admission to each treatment period and follow-up visit. Clinical laboratory tests including clinical chemistry, hematology, coagulation, and urinalysis were conducted at screening, on day -1, and at 48 h post-dose of each treatment period, and follow-up visit. Vital signs viz. systolic and diastolic blood pressure, heart rate, respiration rate, and body temperature, were recorded after subjects had been in the supine position for at least 5 min wherever applicable. Vital signs were recorded at screening, on day -1, and at 2, 24, and 48 h post-dose on each treatment period, and follow-up visit. A standard 12-lead ECG was performed during screening, and each treatment period on day -1, pre-dose, 2 h post-dose, and at follow-up visit. AEs were recorded from the date of subject consent, throughout the study and up to 30 days after the last dose. AEs were classified according to the Medical Dictionary for Regulatory Activities (MedDRA) version 24.0. The severity of the AEs was rated as mild, moderate, severe, life-threatening, and death, and the relationship between the AEs and the study drug was indicated as none, unlikely, possibly, likely or definitely.
Results
Subject disposition and baseline characteristics
The subject disposition and baseline characteristics of subjects in Segment 1 and Segment 2 are summarized in Table 2. A total of 12 subjects were enrolled in Segment 1 and included in the pharmacokinetic and safety sets. One subject (8.3%) withdrew consent and was discontinued from the study after Period 1. A total of 11 subjects (91.7%) completed Segment 1. A total of 44 subjects were enrolled in Segment 2 and included in the pharmacokinetic and safety sets. Four subjects (9.1%) were discontinued from the study. Two subjects discontinued due to AEs, one subject withdrew consent, and one subject was discontinued as per physician’s decision. More details about the discontinued subjects were provided in the safety assessments section below. A total of 40 subjects (90.9%) completed Segment 2.
Pharmacokinetics of SUVN-1105
In Segment 1, following a single oral administration of 200 mg or 250 mg SUVN-1105 under fasted conditions, the median Tmax was 0.75 h post-dose. The median Tmax following administration of SUVN-1105 under fed condition occurred at 0.5 h and 1.5 h for 200 mg and 250 mg, respectively. The concentrations of abiraterone increased with dose and were higher when SUVN-1105 was administered under fasted conditions relative to fed conditions, for both 200 mg and 250 mg doses. Cmax decreased by approximately 63% and AUClast and AUCinf decreased by approximately 32% following administration of 200 mg SUVN-1105 with food. Similarly, Cmax decreased by approximately 73% and AUClast and AUCinf decreased approximately by 37% following administration of 250 mg SUVN-1105 with food.
The mean abiraterone concentrations were quantifiable until the last collection timepoint of 48 h for all treatment groups. The geometric mean t1/2 was comparable across all four treatment groups and ranged from 10.35 h to 13.56 h. The geometric mean C24h was in the range of 2.00 to 3.31 ng.mL−1 across the treatment groups and appeared to increase slightly with the dose and did not appear to be affected by food. CL/F and V/F of abiraterone were comparable between two doses of SUVN-1105 under fasted conditions and appeared to increase slightly with food.
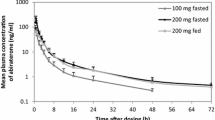
Cmax increased by 39% and AUClast and AUCinf increased by approximately 35% with a 25% increase in the dose of SUVN-1105 from 200 to 250 mg under fasted conditions. Similarly, Cmax increased by 17% and AUClast and AUCinf increased by 30% with a 25% increase in dose from 200 to 250 mg under fed conditions. Based on these observations, 250 mg SUVN-1105 was selected to test the bioequivalence or comparable pharmacokinetics, to Zytiga 1000 mg, in Segment 2. The mean plasma concentration–time profiles for abiraterone following oral administration of SUVN-1105 under fasted and fed conditions are depicted in Fig. 1. The pharmacokinetics parameters of abiraterone are summarized in Table 3.

Plasma concentration (Geometric mean) vs. time curves of abiraterone following administration of single oral doses of SUVN-1105, 200 mg in fasted (filled light grey circles) and fed (filled light grey diamonds) conditions, and 250 mg in fasted (filled dark grey circles) and fed (filled dark grey diamonds) conditions. The error bars are computed as the geometric mean times or divided by the geometric SD factor. The dashed line indicates the level of lower limit of quantitation (0.5 ng.mL−1) for abiraterone. The plasma concentration vs. time curve up to 6 h is shown in the inset
In Segment 2, following a single-dose oral administration of 250 mg SUVN-1105 under fasted or fed conditions and Zytiga 1000 mg in fasted or modified fasted conditions, the median abiraterone Tmax was similar for SUVN-1105 in fasted or fed groups (0.75 h) and was slightly prolonged for Zytiga 1000 mg in fasted (1.5 h) and modified fasted (2.5 h) conditions relative to SUVN-1105 treatments.
The geometric mean abiraterone Cmax and AUCinf were highest with Zytiga 1000 mg in modified fasted conditions followed by 250 mg SUVN-1105 in fasted, 250 mg SUVN-1105 in fed, and Zytiga 1000 mg in overnight fasted conditions. The geometric mean t1/2 was comparable across all four treatment groups and ranged from approximately 11.7 h to 14.5 h. The geometric mean abiraterone CL/F and V/F were highest with Zytiga 1000 mg, overnight fasted, followed by 250 mg SUVN-1105, fed, 250 mg SUVN-1105, overnight fasted and Zytiga 1000 mg, modified fasted. The mean plasma concentration–time profiles for abiraterone following oral administration of 250 mg SUVN-1105 under fasted and fed conditions and Zytiga 1000 mg in fasted and modified fasted conditions are depicted in Fig. 2. The geometric mean pharmacokinetic parameters of abiraterone are summarized in Table 4. The geometric mean abiraterone C24h appeared to be similar for 250 mg SUVN-1105 in fasted, 250 mg SUVN-1105 in fed, and Zytiga 1000 mg in fasted conditions, and approximately sevenfold higher for Zytiga 1000 mg in modified fasted conditions (Fig. 3).

Plasma concentration (Geometric mean) vs. time curves of abiraterone following administration of single oral doses of SUVN-1105, 250 mg in fasted (filled light grey circles) and fed (filled dark grey circles) conditions, and Zytiga 1000 mg in fasted (filled light grey diamonds) and modified fasted (filled dark grey diamonds) conditions. The error bars are computed as the geometric mean times or divided by the geometric SD factor. The dashed line indicates the level of lower limit of quantitation (0.5 ng.mL−1) for abiraterone. The plasma concentration vs. time curve up to 6 h is shown in the inset

Box plot of geometric mean abiraterone C24h following the administration of a single oral dose of SUVN-1105 250 mg in fasted and fed conditions, and Zytiga 1000 mg in fasted and modified fasted (MF) conditions
The concentrations of abiraterone were higher when SUVN-1105 was administered under fasted conditions relative to fed conditions. Cmax decreased by approximately 64% and AUClast and AUCinf decreased by approximately 29% following administration of 250 mg SUVN-1105 with food.
The statistical analyses and comparison of SUVN-1105 and Zytiga PK exposures (Cmax, AUClast, and AUCinf) are summarized in Table 5. The point estimate for the ratio of geometric LS means of abiraterone for SUVN-1105, 250 mg overnight fasted relative to Zytiga 1000 mg, overnight fasted was 3.94 for Cmax, 1.79 for AUClast and 1.73 for AUCinf, with the 90% CIs not contained within the interval of 0.80 to 1.25. Administration of 250 mg SUVN-1105 in fed conditions led to a slight increase of 1.43 for Cmax, 1.27 for AUClast, and 1.23 for AUCinf relative to Zytiga 1000 mg, overnight fasted. The upper bound of the 90% CIs was not contained within the interval of 0.80 to 1.25.
The point estimate for the ratio of geometric LS means of abiraterone for SUVN-1105, 250 mg overnight fasted relative to Zytiga 1000 mg, modified fasted was 0.262 for Cmax, 0.163 for AUClast, and 0.164 for AUCinf. Administration of 250 mg SUVN-1105 in fed conditions led to decreases in point estimates with a value of 0.095 for Cmax, 0.115 for AUClast, and 0.117 for AUCinf, relative to Zytiga 1000 mg, modified fasted condition. For all the above comparisons, the 90% CIs were well below the lower bound of the interval of 0.80 to 1.25.
Administration of Zytiga 1000 mg, modified fasted conditions led to a large increase in exposures as shown in the point estimates with a value of 15 for Cmax, 11 for AUClast, and 10.5 for AUCinf, relative to Zytiga 1000 mg overnight fasted. For all the above comparisons, the 90% CIs were well above the upper bound of the interval of 0.80 to 1.25 (Table 6).
Safety assessments
In Segment 1, a total of 12 subjects were enrolled in the study and included in safety analyses. All 12 subjects were administered SUVN-1105 at a single dose of 200 mg (periods 1 and 3) or 250 mg (periods 2 and 4) following an overnight fast (periods 1 and 2), or under fed conditions (periods 3 and 4). One subject (8.3%) withdrew consent and was discontinued from the study prior to period 2 dosing. The remaining 11 subjects (91.7%) completed Segment 1 as per the protocol. Treatment-emergent adverse events (TEAEs) were reported in 1 of 12 subjects (8.3%) who received 200 mg SUVN-1105 in fasted conditions (period 1). No adverse events were reported by any subjects during other periods (period 2 to period 4). All TEAEs were considered by the investigator to be of mild intensity and not related to the treatment. Adverse events reported in period 1 were Hordeolum, musculoskeletal pain, and headache. No deaths, SAEs, or discontinuations due to AEs were reported during Segment 1. There were no clinically significant abnormalities in physical examinations, laboratory parameters, vital signs, and ECG. The summary of TEAEs by SOC or preferred term in Segment 1 is provided in Table 7.
In Segment 2, a total of 44 subjects were enrolled and included in the safety analyses. The subjects were randomized to 1 of 4 treatment sequences (ABCD, BADC, CDBA, or DCAB) and received single oral doses of treatment in each period per protocol. A total of 42 (95.5%) subjects received Zytiga 1000 mg in overnight fasted conditions (Treatment A), 43 subjects (97.7%) received 250 mg SUVN-1105 in overnight fasted conditions (Treatment B), 40 subjects (90.9%) received Zytiga 1000 mg in modified fasted conditions (Treatment C) and 41 subjects (93.2%) received 250 mg SUVN-1105 in fed state (Treatment D). Four subjects (9.1%) discontinued during the study. Of these 4 subjects, 2 subjects discontinued due to AEs, SARS-Cov-2 positive and dysrhythmia, 1 subject (2.3%) withdrew consent and 1 subject (2.3%) was unable to comply with study procedures since period 3, and was discontinued as per physician’s decision. A total of 40 subjects (90.9%) completed the study.
A total of 13 subjects (29.55%) experienced at least one TEAE with 4 subjects (9.5%) on Treatment A (Zytiga 1000 mg, overnight fasted), 4 subjects (9.3%) on Treatment B (SUVN-1105, 250 mg overnight fasted), 5 (12.5%) subjects on Treatment C (Zytiga 1000 mg, modified fasted), and 6 subjects (14.6%) on Treatment D (SUVN-1105, 250 mg fed). The overall incidence of TEAEs was similar across all the treatments. Most AEs were considered mild (12 subjects, 27.3%) or moderate (1 subject, 2.3%) intensity; none were severe or life-threatening. One subject on Treatment A, experienced TEAEs of moderate intensity (arrhythmia and palpitations) and was discontinued from the study, and the events resolved without treatment. The events were considered by the investigator as unlikely to be related to the treatment. There were no deaths or SAEs reported during the study.
AEs reported for 3 of 44 subjects (6.8%) were considered by the investigator as likely related to SUVN-1105. The treatment-related AEs occurred in 1 subject (2.3%) on Treatment B: 250 mg SUVN-1105, overnight fasted (nausea) and in 2 subjects (4.9%) on Treatment D: 250 mg SUVN-1105, fed (fatigue and headache in 1 subject, and somnolence in 1 subject, 2.4% for each event). The majority of TEAEs in subjects treated with Zytiga were considered by the investigator as unlikely related or unrelated. The Somnolence reported by 1 of 44 subjects (2.3%) was considered by the investigator as possibly related to Zytiga, and the headache reported by 1 of 44 subjects (2.3%) was considered as likely related to Zytiga, both subjects were on Treatment C (Zytiga 1000 mg, modified fasted). The summary of TEAEs by SOC or preferred term in Segment 2 is provided in Table 8.
Discussion
SUVN-1105 is a novel formulation of abiraterone acetate that is developed to improve aqueous solubility and bioavailability compared to Zytiga and Yonsa with further reduction in dose and elimination of food effect. A single dose of 50 mg SUVN-1105 administered to dogs resulted in approximately 2- and 1.4-fold higher AUC and Cmax, respectively, compared to abiraterone exposures following a single dose of 250 mg Zytiga (data on file). Therefore, it was anticipated that a dose range of 200 to 250 mg of SUVN-1105 may be equivalent to 1000 mg Zytiga in humans and 200 mg was selected as a starting dose for period 1 in Segment 1. In period 1, the abiraterone exposures of 200 mg SUVN-1105 were 2.88-fold and 1.19-fold higher, respectively, for Cmax and AUCinf, than the reported abiraterone exposures of Zytiga 1000 mg (Geometric mean abiraterone Cmax was 70 ng.mL−1 and AUCinf was 409 ng.h.mL−1) under fasted conditions [4]. The dose of SUVN-1105 was increased to 250 mg, the maximum allowable daily dose, in period 2 to obtain the abiraterone exposures. SUVN-1105, 250 mg is the maximum allowable daily dose, because the amounts of few excipients cross the Inactive Ingredient Database (IID) limits. Abiraterone exposures, Cmax and AUCinf, of 250 mg SUVN-1105 were 4.70-fold and 1.82-fold higher under fasted conditions, and were 1.27-fold and 1.16-fold higher in the fed state, respectively, than the abiraterone exposures of 1000 mg Zytiga under fasted conditions. Based on these results, 250 mg SUVN-1105 was selected as a suitable dose to evaluate in Segment 2. The exposures of SUVN-1105 appeared to increase proportionately with an increase in dose from 200 to 250 mg.
In Segment 2, 250 mg SUVN-1105 under overnight fasted conditions showed a significant increase in abiraterone exposures with approximately 394% in Cmax and 173% in AUCinf compared to Zytiga 1000 mg in overnight fasted conditions. In fed conditions, 250 mg SUVN-1105 showed a decrease in abiraterone exposures with approximately 64% in Cmax and 29% in AUCinf compared to 250 mg SUVN-1105 in overnight fasted conditions. Although the abiraterone exposures, following administration of 250 mg SUVN-1105, decreased in the fed state compared to the fasted state, the exposures were higher, with 143% increase in Cmax and 123% increase in AUCinf, than the abiraterone exposures observed with Zytiga 1000 mg under overnight fasted conditions. The results from Segment 2 showed that the exposures of abiraterone following administration of 250 mg SUVN-1105, either in a fasted or fed state, fall between the lower and upper limit defined for Zytiga in the fasted and modified fasting conditions, respectively. For C24h, the point estimate was similar and close to unity for 250 mg SUVN-1105 in overnight fasted (0.983) and fed (0.924) conditions, relative to Zytiga 1000 mg in overnight fasted conditions.
Zytiga 1000 mg under modified fasted conditions showed a very large food effect leading to an approximately 1500% increase in peak exposures, 1050–1100% increase in systemic exposures, and 690% increase in C24h compared to Zytiga 1000 mg under overnight fasted conditions.
Abiraterone C24h was comparable between subjects administered 250 mg SUVN-1105 in fasted or fed conditions (2.8 to 3.0 ng.ml−1) and Zytiga 1000 mg in fasted conditions (3.0 ng.ml−1), whereas C24h are higher in subjects administered Zytiga 1000 mg in modified fasted conditions (21.4 ng.ml−1; Fig. 3). In general, maintaining abiraterone trough concentrations above 8.4 ng.ml−1 has been associated with testosterone suppression and improved progression-free survival compared to lower levels [10]. Further investigation in prostate cancer patients is warranted to evaluate the pharmacodynamic effects of SUVN-1105 upon repeat dosing which would enable the direct comparison of trough concentrations between 250 mg SUVN-1105 and Zytiga.
In terms of safety and tolerability, single oral doses of 250 mg SUVN-1105, in the fasted and fed conditions, were generally well-tolerated in healthy male subjects. All the AEs were mild to moderate in severity and the events were considered unrelated to SUVN-1105 or Zytiga. There were no deaths or serious AEs reported during the study. There was no meaningful difference between Zytiga 1000 mg and 250 mg SUVN-1105 treatments in the incidence, severity, or attribution to study drug for reported AEs during the study under overnight fasted, fed (SUVN-1105) and modified fasted (Zytiga) conditions.
Conclusion
Abiraterone exposures of 250 mg SUVN-1105, in fasted or fed condition, fall within the abiraterone exposures of 1000 mg Zytiga under fasted and modified fasted conditions. Single doses of SUVN-1105 were safe and well-tolerated in healthy males both in the fasted and fed conditions. The SUVN-1105 formulation has eliminated the food effect observed with Zytiga and may improve adherence in patients with dysphagia.
Data availability
All the data supporting the findings of this study are available within this paper.
Code availability
Not Applicable.
References
Wang L, Lu B, He M, Wang Y, Wang Z, Du L (2022) Prostate cancer incidence and mortality: global status and temporal trends in 89 countries from 2000 to 2019. Front Public Health 10:811044. https://doi.org/10.3389/fpubh.2022.811044
National cancer institute, surveillance, epidemiology, and end results program (SEER), Cancer Stat Facts: prostate cancer (2023). https://seer.cancer.gov/statfacts/html/prost.html
Zytiga. Highlights of prescribing information. https://www.janssenlabels.com/package-insert/product-monograph/prescribing-information/ZYTIGA-pi.pdf. Accessed 18 Nov 2023
Goldwater R, Hussaini A, Bosch B, Nemeth P (2017) Comparison of a novel formulation of abiraterone acetate vs. the originator formulation in healthy male subjects: two randomized, open-label crossover studies. Clin Pharmacokinet 56(7):803–813. https://doi.org/10.1007/s40262-017-0536-2
Papangelou A, Olszanski AJ, Stein CA, Bosch B, Nemeth P (2017) The effect of food on the absorption of abiraterone acetate from a fine particle dosage form: a randomized crossover trial in healthy volunteers. Oncol Ther 5:161–170. https://doi.org/10.1007/s40487-017-0054-2
International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (2018) Guideline for good clinical principles. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2_Step_4_2016_1109.pdf
ICH Harmonized Tripartite Guideline, Guideline for Good clinical practice, E6 (R1), Current Step 4 version, dated 10 June 1996
Center for drug evaluation and research (2011) NDA 202–379, Clinical pharmacology and biopharmaceutics review(s). p 29. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202379Orig1s000ClinPharmR.pdf. Accessed 25 Sep 2023
US Food Drug Administration. Guidance for industry: bioanalytical method validation. Rockville, MD: Food and Drug Administration; 2018. Available from: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf
Carton E, Noe G, Huillard O, Golmard L, Giroux J, Cessot A, Saidu NE, Peyromaure M, Zerbib M, Narjoz C, Guibourdenche J, Thomas A, Vidal M, Goldwasser F, Blanchet B, Alexandre J (2017) Relation between plasma trough concentration of abiraterone and prostate-specific antigen response in metastatic castration-resistant prostate cancer patients. Eur J Cancer 72:54–61. https://doi.org/10.1016/j.ejca.2016.11.027
Acknowledgements
The authors acknowledge the principal investigators and biostatistician from PRA-EDS (Lenexa, KS, USA) for their support in the conduct of the study reported in this manuscript.
Funding
This study was sponsored by Suven Life Sciences Ltd.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors are employees of Suven Life Sciences Ltd.
Ethical approval
The study was conducted in compliance with the International Ethical Guidelines for Biomedical Research Involving Human Subjects, Good Clinical Practice Guidelines, and the Declaration of Helsinki. The protocols were reviewed and approved by the institutional review board.
Informed consent
All subjects provided written informed consent. The study was conducted at PRA-EDS (Lenexa, KS, USA).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nirogi, R., Ravula, J., Benade, V. et al. Bioequivalence, food effect and comparative pharmacokinetics of SUVN-1105, a novel granule formulation of abiraterone acetate, to Zytiga in healthy male subjects. Cancer Chemother Pharmacol 93, 253–264 (2024). https://doi.org/10.1007/s00280-023-04629-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-023-04629-1