
Abstract
Sodium-glucose co-transporter 2 (SGLT2) is predominantly expressed in the S1 segment of the proximal tubule of the kidney and is the major transporter responsible for mediating renal glucose reabsorption. Dapagliflozin is an orally active, highly selective SGLT2 inhibitor that improves glycemic control in patients with type 2 diabetes mellitus (T2DM) by reducing renal glucose reabsorption leading to urinary glucose excretion (glucuresis). Orally administered dapagliflozin is rapidly absorbed generally achieving peak plasma concentrations within 2 h. Dose-proportional systemic exposure to dapagliflozin has been observed over a wide dose range (0.1–500 mg) with an oral bioavailability of 78 %. Dapagliflozin has extensive extravascular distribution (mean volume of distribution of 118 L). Dapagliflozin metabolism occurs predominantly in the liver and kidneys by uridine diphosphate-glucuronosyltransferase-1A9 to the major metabolite dapagliflozin 3-O-glucuronide (this metabolite is not an SGLT2 inhibitor at clinically relevant exposures). Dapagliflozin is not appreciably cleared by renal excretion (<2 % of dose is recovered in urine as parent). Dapagliflozin 3-O-glucuronide elimination occurs mainly via renal excretion, with 61 % of a dapagliflozin dose being recovered as this metabolite in urine. The half-life for orally administered dapagliflozin 10 mg was 12.9 h. Maximal increases in urinary glucose excretion were seen at doses ≥20 mg/day in patients with T2DM. No clinically relevant differences were observed in dapagliflozin exposure with respect to age, race, sex, body weight, food, or presence of T2DM. Pharmacodynamic changes are dependent on plasma glucose and renal function, and decreases in urinary glucose excretion were observed due to the lower filtered load (plasma glucose × glomerular filtration rate) in healthy volunteers compared to subjects with T2DM. After multiple doses of dapagliflozin, urinary glucose excretion was associated with dose-related decreases in plasma glucose parameters in subjects with T2DM. Patients with severe renal or hepatic impairment show higher systemic exposure to dapagliflozin. No clinically relevant drug interactions were observed that would necessitate dose adjustment of dapagliflozin when administered with other antidiabetic or cardiovascular medications, as well as drugs that could potentially influence dapagliflozin metabolism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
It is estimated that approximately 285 million people have diabetes worldwide and it is predicted to rise to 438 million by 2030 [1, 2]. Type 2 diabetes mellitus (T2DM) comprises around 90 % of all cases [3]. T2DM is a chronic metabolic disorder characterized by hyperglycemia and an increased risk of micro- and macrovascular complications. Current licensed antidiabetic therapies include oral agents with different mechanisms of action such as insulin-sensitizing agents (metformin and thiazolidinediones), agents that promote insulin secretion (sulfonylureas), agents that act on the incretin pathway to stimulate insulin secretion while exerting additional metabolic effects (glucagon-like peptide-1 analogs and dipeptidyl peptidase-4 inhibitors), and insulin [4]. The aim of any T2DM treatment is to improve glycemic control and to reduce the associated risk for complications. However, despite a variety of different treatment options, approximately 44 % of patients do not reach their treatment goals [5–7]. Due to the increasing global prevalence of the disease, its progressive nature (which eventually requires combination therapy in most patients), and the potential undesirable side effects of currently available therapies, there remains a need for novel treatment options for T2DM.
The novel approach of inhibiting glucose re-absorption from glomerular filtrate to induce renal glucose excretion has been investigated in T2DM over recent years. Under conditions of normoglycemia, almost all of the glucose in the glomerular filtrate is re-absorbed in the proximal tubules, mediated primarily in the S1 segment via sodium glucose co-transporter-2 (SGLT2) [8]. Phlorizin, a glucoside isolated from the bark of apple trees, is a non-selective inhibitor of SGLT1/SGLT2 and has been used in research for over 150 years. The use of phlorizin as a treatment for T2DM was limited due to low oral bioavailability and non-selectivity associated with undesirable gastrointestinal (GI) side effects. Accordingly, novel SGLT2 inhibitors were developed, with the first examples having the glucoside moiety linking via an O-linkage to a distal phenolic ring. However, these compounds proved to be susceptible to B-glucosidases in vivo, and inhibitors with a more metabolically stable C-linkage were developed, including dapagliflozin, empagliflozin, canagliflozin, and ipragliflozin [9]. Dapagliflozin is a highly selective reversible inhibitor of SGLT2 being developed for the treatment of T2DM [10]. Clinically meaningful improvements in measurements of glycemic control have been demonstrated in patients with T2DM when treated with dapagliflozin either as monotherapy [11, 12] or in combination with metformin [13] or insulin plus high doses of oral antidiabetic drugs [14]. Data pooled across 12 Phase III clinical trials have shown dapagliflozin to have an acceptable safety profile. Adverse events included an increase in incidence of genital and urinary tract infections that were related to its mechanism of action [15]. Dapagliflozin was approved for the treatment of T2DM in the European Union in November 2012. This review summarizes the clinical pharmacokinetic and pharmacodynamic properties of dapagliflozin.
2 Pharmacology
2.1 Structure and Physicochemical Properties
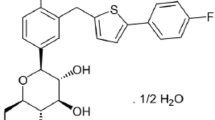
Dapagliflozin has a rationally designed chemical structure containing a C-linked glucoside, such that the aglycone component is attached to glucose by a carbon–carbon bond, which confers metabolic stability against glucosidase enzymes. The molecular weight of dapagliflozin is 408.87. It is described chemically as (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl) phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol) and its molecular formula is C21H25ClO6 (Fig. 1). Dapagliflozin is a Biopharmaceutics Classification System (BCS) Class III compound with BCS Class I-like characteristics of high, pH-independent aqueous solubility (>1 mg/mL across the physiological pH range), high in vitro permeability, and good oral bioavailability [16].

Structural formula of dapagliflozin propanediol monohydrate
2.2 Cytochrome P450 Enzymes and Transporter Proteins
In vitro studies with recombinant cytochrome (CYP) isoforms indicate that the metabolism of dapagliflozin may be catalyzed by several CYP enzymes, including CYP1A1, CYP1A2, CYP2A6, CYP2C9, CYP2D6, and CYP3A4 [10]. In humans, less than 10 % of the oral dose is eliminated via pathways involving oxidative metabolism (AstraZeneca/Bristol-Myers Squibb, data on file). Dapagliflozin is also a substrate for uridine diphosphate glucuronosyltransferases (UGT) 1A9, UGT2B4, and UGT2B7 enzymes and, in humans, dapagliflozin elimination is dependent on the formation of dapagliflozin 3-O-glucuronide mediated by UGT1A9 [17].
Dapagliflozin is a weak substrate for the permeability glycoprotein (P-gp) transporter in vitro as determined by the inhibition of the transport of digoxin, a P-gp substrate, through Caco-2 cell monolayers [17].
2.3 Non-Clinical Pharmacokinetics and Pharmacodynamics
Following oral administration, the elimination half-lives were 4.6 h in rats, 7.4 h in dogs, and 3.5 h in monkeys [17]. Time to reach peak concentrations (t max) values were 1.7 ± 2.0 h, 0.6 ± 0.4 h, and 1.9 ± 1.8 h in rats, dogs, and monkeys, respectively. Dapagliflozin was well absorbed in rats and dogs. In rats dosed with 1 mg/kg dapagliflozin, bioavailability was 84 ± 21 %, and bioavailability was 83 ± 2 % in dogs receiving a 6.6-mg/kg dose. In monkeys dosed with 6 mg/kg dapagliflozin the bioavailability was lower at 25 ± 2 %. The reason for the lower bioavailability in monkeys compared to rats and dogs is unknown at this time.
The steady-state volume of distribution (V ss) for dapagliflozin following intra-arterial or intravenous doses of 1, 6.6, and 6.0 mg/kg in rats, dogs, and monkeys was 1.6, 0.8, and 0.8 L/kg, respectively, indicating extravascular distribution [17].
In vitro, dapagliflozin competitively inhibits SGLT2 with an inhibition constant (K i) of 0.2, 3.0, and 2.3 nM for human, rat, and mouse SGLT2, respectively. Furthermore, high selectivity for SGLT2 was observed of around 1,200 versus SGLT1 for humans (based on concentration producing 50 % effect [EC50] values) and 200 in rats [10].
Inhibition of SGLT2 with dapagliflozin increases renal glucose excretion in both healthy animals and animals with T2DM accompanied by osmotic diuresis, measured as increased urine flow (Fig. 2). Studies in the Zucker diabetic fatty (ZDF) rat model indicated that chronic dapagliflozin treatment led to dose dependent reductions in endogenous glucose production resulting in decreased blood glucose levels, with a low risk for hypoglycemia [18]. Furthermore, insulin sensitivity and pancreatic beta cell function were also improved in dapagliflozin-treated ZDF rats.

Anatomical arrangement of a kidney nephron with its blood supply depicting SGLT2 inhibition with dapagliflozin leading to increased glucose excretion. SGLT2 sodium-glucose co-transporter 2. Adapted with permission from Wright, Loo and Hirayama [27]
3 Clinical Pharmacokinetics
3.1 Overview
Pharmacokinetic data for dapagliflozin (1–500 mg) were pooled and summarized across 20 studies in healthy subjects and in those with T2DM (Table 1). Dapagliflozin was rapidly absorbed with a t max of approximately 1 h. The half-life for orally administered dapagliflozin at a dose of 10 mg was 12.9 h [19].
3.2 Absorption
In human subjects, dapagliflozin is rapidly and extensively absorbed from the GI tract (oral bioavailability, 78 %) over a wide dose range (0.1–500 mg) [16]. Maximal peak concentrations were usually attained within 1 to 2 h following administration in the fasted state. Co-administration with food did not markedly affect the overall extent of absorption of dapagliflozin (as measured by the area under the plasma concentration-time curve [AUC]); however, food decreased the rate of absorption of dapagliflozin (prolonged t max by approximately 1 h and maximum [peak] plasma drug concentration [C max] decreased by 30–45 % compared with the fasted state) [20]. However, based on the pharmacokinetic and pharmacodynamic relationship of dapagliflozin for urinary glucose excretion up to 6 h post-dose, the food effect was not considered to be clinically meaningful since the lower concentrations of dapagliflozin with food over this time period still provide maximal glucosuria.
3.3 Distribution
The mean V ss for dapagliflozin following intravenous administration was 118 L [16]. This was greater than the estimated plasma volume, and is indicative of extravascular tissue distribution. In addition, dapagliflozin is moderately protein bound (~91 %), irrespective of T2DM or renal or hepatic function [21, 22].
3.4 Metabolism
Following a single 50-mg oral dose of [U-14C] dapagliflozin to six healthy male subjects, dapagliflozin was extensively metabolized, with 73.7 % recovered (72.0 % in urine and 1.65 % in feces) [23]. Metabolic routes included glucuronidation, dealkylation, and oxidation at various positions of the molecule to produce desmethyl dapagliflozin glucuronides [17]. Approximately 9 % of the dose underwent oxidative metabolism and around 29 % of these metabolites were also glucuronidated, while approximately 66 % were directly glucuronidated. Dapagliflozin 3-O-glucuronide was the predominant metabolite in humans accounting for 60.7 % of the dose being completely recovered in urine. Dapagliflozin 2-O-glucuronide was the only other metabolite representing >5 % of the dose (5.4 %). Dapagliflozin 3-O-glucuronide was the major drug-related component in the plasma (42 % of total drug substance), based on AUC from 0 to 12 h AUC12, while the parent drug was 39 %. No other metabolite detected in human plasma constituted >5 % of total drug substance.
In vitro data show that dapagliflozin 3-O-glucuronide is formed in both the kidney and the liver. Exposure to dapagliflozin was similarly affected by severe hepatic or renal impairment (67 and 87 % higher AUC parameters, respectively, compared with the reference population) [21, 22], suggesting the involvement of both the liver and the kidney in the metabolic clearance of dapagliflozin.
Recently, Dostalek et al. showed that UGT2B7 activity may be significantly reduced in patients with T2DM; however, UGT1A9 activity did not appear to be markedly different in patients with T2DM compared with healthy controls [24]. Consistent with these findings, an across-study analysis of the pharmacokinetics of dapagliflozin, a UGT1A9 substrate, in subjects with T2DM and normal renal function showed no meaningful differences from healthy subjects (AstraZeneca/Bristol-Myers Squibb, data on file). Furthermore, an analysis of the impact of UGT1A9 single nucleotide polymorphisms (SNPs) in 187 subjects with T2DM showed that the range of dapagliflozin clearance values in patients with UGT1A9 SNPs was contained within the range of the homozygous wild-type population. This indicates that there was no clinically meaningful impact of UGT1A9 SNP polymorphisms on dapagliflozin metabolic clearance (AstraZeneca/Bristol-Myers Squibb, data on file).
3.5 Elimination
While elimination of dapagliflozin occurs via metabolism, biliary, and/or intestinal clearance and renal clearance, the predominant mechanism is metabolic clearance. In healthy male human subjects, given a single 50-mg dose of [U-14C] dapagliflozin, 96 % of the radioactivity was recovered 312 h after dosing (75 % in urine and 21 % in feces, respectively), 76 % occurred within 24 h. Of the administered dose, 1.2 % was recovered in urine as parent drug, which was in accordance with non-radiological studies, indicating that renal clearance of parent dapagliflozin in humans is a minor route of dapagliflozin elimination [17, 19].
4 Clinical Pharmacodynamics
4.1 Inhibition of SGLT2
Approximately 162 g of glucose is filtered through the kidney each day in humans with normal renal function [25]. SGLT2, located in the proximal tubule, is responsible for reabsorbing up to the majority of the filtered glucose [26, 27]. Maximum renal glucose re-absorptive capacity (TmG) is approximately 375 mg/min. In normoglycemic individuals, the filtered glucose load does not exceed the TmG and all of the glucose is re-absorbed. As a result, virtually no glucose is excreted in the urine. If however, the filtered glucose load exceeds the TmG, that excess glucose is excreted in the urine. Above the TmG, the glucose excretion rate increases linearly with the filtered glucose load.
Dapagliflozin is a potent, competitive, reversible, highly selective and orally active inhibitor of the human SGLT2, with a K i of 6 nM [10, 28]. There is no evidence of an upregulation in SGLT2 activity following exposure to dapagliflozin as determined by consistent percent inhibition over time [29]. TmG is reduced in the presence of dapagliflozin in individuals with T2DM [30].
4.2 Effects on Urinary Glucose Excretion
Consistent with its mechanism of action, exposure to dapagliflozin results in a dose-dependent increase in urinary glucose excretion and is dependent on the filtered load (plasma glucose × glomerular filtration rate [GFR]), suggesting a low propensity to cause hypoglycemia. Urinary glucose excretion data summarized from nine pharmacodynamic studies are shown in Fig. 3. Maximal increase in urinary glucose excretion was dose-dependent and was generally seen at doses ≥20 mg/day in subjects with T2DM [29, 31]. Similar results were seen in healthy subjects and the glucosuria was maintained over at least 24 h [31]. Following treatment with dapagliflozin (≥20 mg), glucose clearance increased to a maximum value of approximately 35 mL/min over the first 4 h (AstraZeneca/Bristol-Myers Squibb, data on file).

Scatterplot from nine studies of change from baseline (CFB) in 24-h urinary glucose versus dapagliflozin dose in healthy subjects and subjects with T2DM. T2DM type 2 diabetes mellitus
4.3 Effects on Uric Acid
Following dapagliflozin administration, urinary acid excretion transiently increased (3–7 days) and was accompanied by a reduction in serum uric acid that was sustained out to 24 weeks [19].
4.4 Thorough QTc Study
A study was performed to evaluate the potential effects of dapagliflozin on cardiac repolarization [32]. Following treatment with dapagliflozin, the upper bound of the one-sided 95 % confidence interval (CI) for time-matched, placebo-subtracted, baseline-adjusted heart-rate corrected QT (QTc) intervals was <10 ms. The change in QTc was independent of dapagliflozin concentrations. No QTc thresholds were >450 ms and no QTc increases >30 ms were observed. Thus, no clinically significant effect of dapagliflozin on the QTc interval was observed in healthy subjects at doses greater than the therapeutic dose.
5 Influence of Demographic Factors
Systemic exposures of dapagliflozin in high body weight subjects (≥120 kg, n = 91) were estimated to be 78.3 % (90 % CI 78.2–83.2) of those of reference subjects with body weight between 75 and 100 kg [19]. There was no clinically meaningful increase in exposure to dapagliflozin based on age in subjects aged ≤70 years [19]. A greater mean dapagliflozin AUC of 23.4 % (90 % CI 17.1–30.1), predicted by population modelling, was observed in a pooled analysis of clinical pharmacology studies in 82 female subjects compared with 349 males, but the C max was not increased. Sex was found to be a significant covariate in the population pharmacokinetic model: mean dapagliflozin AUC at steady state (AUCss) was 22 % higher (90 % CI 17–24) in females (n = 619) than in males (n = 634). All phase III studies included relatively balanced proportions of male and female T2DM patients so the impact of this relatively small difference in dapagliflozin systemic exposure is reflected in the safety and efficacy findings where minimal difference is observed between the sexes.
An analysis of Caucasian/White, African American/Black, and Asian races detected no meaningful differences in systemic exposures to dapagliflozin [19].
Dapagliflozin administered to healthy Japanese subjects and to patients with T2DM showed similar pharmacokinetic properties to US-based healthy subjects and patients with T2DM in clinical pharmacology studies [29, 31, 33]. Studies were performed in Japanese healthy subjects US-based subjects (N = 32: single ascending dose [SAD], dapagliflozin 2.5–50 mg), and patients with T2DM (multiple ascending dose [MAD], dapagliflozin 2.5–20 mg once daily for 14 days) [33]. In this population, dapagliflozin was rapidly absorbed, with a t max of approximately 1 h for both healthy patients and those with T2DM. As with the US-based studies systemic exposure of dapagliflozin and dapagliflozin 3-O-glucuronide measured by C max and AUC, increased proportional to dose in the Japanese population and urinary glucose also increased in a dose-dependent manner.
Two, open-label, single (N = 14) and multiple dose (N = 14) studies have also been performed in healthy Chinese subjects residing in China [34]. As with the previous studies, dapagliflozin was rapidly absorbed (median t max ≤ 1.5 h) with an elimination half-life of 10 to 12 h. Less than 1.9 % of the administered dose of dapagliflozin was excreted unchanged in the urine and minimal multiple dose accumulation was observed (≤1.13-fold). The pharmacodynamics of dapagliflozin in the Chinese subjects were comparable to that seen in the Western population.
6 Pharmacokinetics/Pharmacodynamics in Specific Populations
6.1 Renal Impairment
Renal impairment is a common complication of T2DM. Expression of SGLT2 is restricted to the kidney and the filtered load is a product of plasma glucose concentration and the GFR. In addition, dapagliflozin’s major metabolite is mainly cleared via the kidney. Therefore, the effect of renal impairment on the pharmacokinetics and pharmacodynamics of dapagliflozin were evaluated (Table 2) [22]. A single 50-mg dose of dapagliflozin was administered to healthy patients and those with T2DM (38 in total) with either normal kidney function or mild, moderate, or severe renal impairment based on estimated creatinine clearance (mild 51–80 mL/min; moderate 30–50 mL/min; severe <30 mL/min). Subsequently, once-daily 20-mg multiple doses were assessed in patients with T2DM. In those with reduced kidney function, plasma concentrations of dapagliflozin and dapagliflozin 3-O-glucuronide incrementally increased. Steady-state C max values were 4, 6, and 9 % higher for dapagliflozin, and for dapagliflozin 3-O-glucuronide were 20, 37, and 52 % higher in patients with mild, moderate and severe renal impairment, respectively compared with normal function. Furthermore, AUC from 0 to the last dosing interval (AUCτ) was similarly higher. Formation of dapagliflozin 3-O-glucuronide in kidney microsomes was three-fold higher than in liver and 109-fold higher than in the intestine.
In renally impaired subjects, pharmacodynamic effects were attenuated compared with those with normal renal function. In diabetic subjects with moderate to severe renal impairment, dapagliflozin pharmacodynamics are consistent with the observation of reduced efficacy in this patient population, likely due to reduced filtered load as a consequence of decline in GFR [22]. With decreasing renal function, at the same levels of plasma glucose, as GFR decreases, less glucose is delivered to the kidney and less is filtered into the tubule. Thus, with less glucose in the tubule to be re-absorbed, the efficacy of dapagliflozin is reduced. Steady-state renal glucose clearance was reduced by 42, 83, and 84 % in subjects with mild, moderate, or severe renal impairment, respectively. Both the kidney and liver significantly contribute to the metabolism of dapagliflozin, resulting in higher systemic exposure with declining kidney function.
6.2 Hepatic Impairment
The pharmacokinetics of dapagliflozin were evaluated in an open-label study in 24 subjects with mild, moderate, or severe hepatic impairment (Child-Pugh classification), following a single 10-mg oral dose [21]. Dapagliflozin mean C max values were 12, 12, and 40 % higher in subjects with mild, moderate, or severe hepatic impairment, respectively, and mean AUC with the last concentration extrapolated to infinity (AUC∞) values were 3, 36, and 67 % higher, respectively, than in healthy subjects. Furthermore, dapagliflozin 3-O-glucuronide mean C max values were 4 % higher, 58 % higher, and 14 % lower, respectively, in those with mild, moderate, or severe hepatic impairment, and AUC∞ values were 6, 100, and 30 % higher, respectively, both compared with healthy controls. These values were also correlated with the renal function (as assessed by calculated creatinine clearance) of each group. The findings indicate that systemic exposure to dapagliflozin is correlated with the degree of hepatic impairment and single 10-mg doses of dapagliflozin are generally well tolerated. However, in patients with severe hepatic impairment the benefit:risk ratio should be individually assessed as the long-term safety and efficacy of dapagliflozin have not been specifically studied in this population. There is no dose adjustment recommended for patients with mild to moderate hepatic impairment and a lower starting dose of 5 mg is recommended for those patients with severe hepatic impairment [19].
7 Drug Interactions
Interaction studies were performed with drugs that had the potential to alter the pharmacokinetics of dapagliflozin. For example, drugs that were metabolized by CYP enzymes or excreted by human organic anion transporter (hOAT)-3/human organic cation transporter (hOCT)-1/hOCT-2, as well as medications potentially affecting UGT1A9. Where possible, prototype drugs were chosen to reflect medications commonly administered to patients with T2DM. A summary of the results of the drug–drug interaction studies is shown in Table 3.
7.1 Antidiabetic Medications
Co-administration of dapagliflozin with the oral antidiabetic drugs pioglitazone, metformin, glimepiride, or sitagliptin (single doses) was evaluated in three open-label, randomized, crossover studies in healthy subjects [35]. Neither the C max nor AUC of dapagliflozin were affected by the co-administered drugs (Table 3). In addition, the effect of the antidiabetic agent, voglibose, on dapagliflozin pharmacokinetics was also investigated [36]. Single oral doses of dapagliflozin (10 mg) were administered to 22 Japanese patients with T2DM in the presence and absence of voglibose (0.2 mg three times daily) and dapagliflozin pharmacokinetics were unaffected by the presence of voglibose. Likewise, dapagliflozin had no clinically meaningful effect on the exposure to pioglitazone, metformin, glimepiride, sitagliptin, or voglibose.
7.2 Cardiovascular Medications
Coronary heart disease, peripheral vascular disease, and stroke are the most common causes of mortality in patients with T2DM. Due to the potential for co-administration of dapagliflozin with cardiovascular medications, pharmacokinetic interactions between dapagliflozin and simvastatin, valsartan, warfarin, digoxin, and hydrochlorothiazide were evaluated [19, 37]. In an open-label, five-period, five-treatment crossover study, the effects of dapagliflozin 20 mg, simvastatin 40 mg, or valsartan 320 mg were assessed in 24 healthy subjects, while in another open-label, randomized, two-period, two-treatment study in 30 healthy subjects, the effects of steady-state dapagliflozin on the pharmacokinetics of warfarin 25 mg or digoxin 0.25 mg were assessed. No clinically meaningful effect on the pharmacokinetics of dapagliflozin was observed with simvastatin, valsartan, warfarin, or digoxin. AUC values for simvastatin, its acid, and valsartan were increased by 19, 30, and 6 %, respectively, by dapagliflozin, and the C max of valsartan was decreased by approximately 6 %. These effects were not considered clinically meaningful. No clinically meaningful effect of dapagliflozin was observed on exposure to simvastatin, valsartan, warfarin, digoxin, or hydrochlorothiazide. The pharmacodynamics of warfarin were also unaffected by dapagliflozin.
The potential interaction of dapagliflozin with bumetanide, a loop diuretic used to manage edema associated with congestive heart failure and hypertension (common co-morbidities of T2DM) was also assessed [38]. Forty-two healthy subjects received either bumetanide 1 mg/day, dapagliflozin 10 mg/day, or the drugs in combination for 7 days. Then all subjects were given both agents for a further 7 days. No pharmacokinetic interaction was observed between dapagliflozin and bumetanide. The addition of bumetanide in the setting of prior dapagliflozin treatment had similar effects on fluid and electrolyte balance to the effects of bumetanide alone. Likewise, the addition of dapagliflozin in the setting of prior bumetanide treatment had similar effects on fluid and electrolyte balance to the effects of dapagliflozin alone. Because of the potential for increased risk of volume depletion, dapagliflozin is not recommended for use in patients who are receiving loop diuretics [19].
7.3 Other Concomitant Medications
Dapagliflozin is primarily metabolized to its major inactive metabolite, dapagliflozin 3-O-glucuronide, via the UGT1A9 pathway. Therefore, the potential for drug–drug interaction on pharmacokinetics and pharmacodynamics between dapagliflozin and rifampin (a pleiotropic drug-metabolizing enzyme inducer) or mefenamic acid (a strong UGT1A9 inhibitor) was evaluated in two studies in healthy subjects [39]. In the first study, 14 subjects were dosed with single doses of dapagliflozin 10 mg alone and with rifampin 600 mg once daily for 6 days. Rifampin reduced AUC∞ to dapagliflozin by 22 %. In the second study, 16 subjects received the same single doses of dapagliflozin alone or in combination with mefenamic acid 250 mg every 6 h for 5 days. Mefenamic acid increased dapagliflozin AUC∞ by 51 %. Mean urinary glucose excretion (standard deviation) over the 24 h following dapagliflozin administration was 50.6 (10.7) g in the first study and 43.5 (9.2) g in the second study. A small (∼10 %) decrease in urinary glucose excretion was observed over the first 24 h following co-administration of dapagliflozin with rifampin (−5.2 g/24 h; 95 % CI −9.17 g/24 h to −1.18 g/24 h) and a small (∼18 %) increase with mefenamic acid (7.97 g/24 h; 95 % CI 3.17 g/24 h to 12.77 g/24 h). The effects of rifampin and mefenamic acid on the pharmacokinetics of dapagliflozin or dapagliflozin-mediated urinary glucose excretion were not considered clinically meaningful.
8 Summary
The worldwide burden of mortality and morbidity from T2DM is expected to grow over the coming decades, emphasizing the need for novel antidiabetic agents that are safe and effective and have minimal interaction with other drugs used to manage T2DM in these patients. Dapagliflozin is an oral, selective, and reversible inhibitor of SGLT2 shown to improve glycemic control and is accompanied by modest weight loss.
The comprehensive pharmacokinetic and pharmacodynamics evaluation of dapagliflozin demonstrates that it has high systemic exposure over a wide dose range (0.1–500 mg) and its pharmacokinetic properties are unchanged upon repeated dosing. Dapagliflozin is rapidly absorbed after oral administration, with maximum C max values generally achieved within 2 h of administration. The oral bioavailability is 78 %. The mean plasma half-life is 12.9 h and the high mean V ss of 118 L indicates extravascular distribution. Dapagliflozin gives sustained inhibition of urinary glucose re-absorption over 24 h post-dose with 10 mg, the clinical dose, confirming that this dose is suitable for once-daily administration.
No clinically meaningful differences in dapagliflozin pharmacokinetics were evident in adult subjects, regardless of T2DM, age, race, gender, body weight, or mild renal/hepatic impairment. As such, no dose adjustment is necessary in these patients.
The structure of dapagliflozin includes a C-linked glucoside, which confers metabolic stability. It is extensively metabolized by UGT1A9 in the liver and kidneys to give a glucuronidated metabolite, dapagliflozin 3-O-glucuronide, which does not inhibit SGLT2 at a dapagliflozin dose of 10 mg. Dapagliflozin 3-O-glucuronide does not meaningfully contribute to the glucose-lowering effects of dapagliflozin in humans.
In vitro metabolism data and clinical drug interaction studies indicate that dapagliflozin has limited potential to cause drug–drug interactions or to be meaningfully affected when co-administered with other drugs. A number of studies evaluated dapagliflozin administration with drugs commonly prescribed to patients with T2DM, including antidiabetic agents, cardiovascular medications, and drugs associated with UGT1A9, and no interactions were observed that would necessitate dose adjustment for any of the drugs studied.
In clinical trials, dapagliflozin has clearly demonstrated clinically relevant glycemic control together with a favorable safety and tolerability profile. The mechanism of action of dapagliflozin is different from, and complimentary to, those of currently available antidiabetic agents and is independent of insulin. Dapagliflozin represents a valuable addition to the agents used to manage T2DM.
References
International Diabetes Foundation. The global burden. International Diabetes Foundation Web site. http://www.diabetesatlas.org/book/export/html/36. Accessed 4 Mar 2013.
World Diabetes Foundation. Diabetes facts. World Diabetes Foundation Web site. http://www.worlddiabetesfoundation.org/composite-35.html. Accessed 4 Mar 2013.
World Health Organisation. Global strategy on diet, physical activity and health. World Health Organisation Web site. http://www.who.int/dietphysicalactivity/publications/facts/obesity/en/. Accessed 4 Mar 2013.
American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care. 2013;36:S11–66.
Braga MF, Casanova A, Teoh H, et al. Poor achievement of guidelines-recommended targets in type 2 diabetes: findings from a contemporary prospective cohort study. Int J Clin Pract. 2012;66:457–64.
Hermans MP, Brotons C, Elisaf M, et al. Optimal type 2 diabetes mellitus management: the randomised controlled OPTIMISE benchmarking study: baseline results from six European countries. Eur J Prev Cardiol. 2012. doi:10.1177/2047487312449414.
Wong ND, Glovaci D, Wong K, et al. Global cardiovascular disease risk assessment in United States adults with diabetes. Diab Vasc Dis Res. 2012;9:146–52.
Marsenic O. Glucose control by the kidney: an emerging target in diabetes. Am J Kidney Dis. 2009;53:875–83.
Kim Y, Babu AR. Clinical potential of sodium-glucose cotransporter 2 inhibitors in the management of type 2 diabetes. Diabetes Metab Syndr Obes. 2012;5:313–27.
Meng W, Ellsworth BA, Nirschl AA, et al. Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J Med Chem. 2008;51:1145–9.
Ferrannini E, Ramos SJ, Salsali A, et al. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2010;33:2217–24.
List JF, Woo V, Morales E, et al. Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care. 2009;32:650–7.
Bailey CJ, Gross JL, Pieters A, et al. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:2223–33.
Wilding JP, Norwood P, T’joen C, et al. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers: applicability of a novel insulin-independent treatment. Diabetes Care. 2009;32:1656–62.
Ptaszynska A, Johnsson K, Apanovitch A, et al. Safety of dapagliflozin in clinical trials for T2DM. Diabetes. 2012;61(suppl 1):A258–9.
Boulton DW, Kasichayanula S, Keung CF, et al. Simultaneous oral therapeutic and intravenous (1)(4)C-microdoses to determine the absolute oral bioavailability of saxagliptin and dapagliflozin. Br J Clin Pharmacol. 2013;75:763–8.
Obermeier M, Yao M, Khanna A, et al. In vitro characterization and pharmacokinetics of dapagliflozin (BMS-512148), a potent sodium-glucose cotransporter type II inhibitor, in animals and humans. Drug Metab Dispos. 2010;38:405–14.
Zinker B, Ma X, Liu H, et al. Chronic dapagliflozin treatment reduces elevated hepatic glucose production and enhances pancreatic insulin content in male ZDF rats. (Abstract 1031-P). Diabetes. 2011;60(suppl 1):A283.
Forxiga: Summary of Product Characteristics. AstraZeneca and Bristol-Myers Squibb. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002322/WC500136026.pdf. Accessed 8 Apr 2013.
Kasichayanula S, Liu X, Zhang W, et al. Effect of a high-fat meal on the pharmacokinetics of dapagliflozin, a selective SGLT2 inhibitor, in healthy subjects. Diabetes Obes Metab. 2011;13:770–3.
Kasichayanula S, Liu X, Zhang W, et al. Influence of hepatic impairment on the pharmacokinetics and safety profile of dapagliflozin: an open-label, parallel-group, single-dose study. Clin Ther. 2011;33:1798–808.
Kasichayanula S, Liu X, Benito MP, et al. The influence of kidney function on dapagliflozin exposure, metabolism, and efficacy in healthy subjects and in patients with type 2 diabetes mellitus. Br J Clin Pharmacol. 2013;76(3):432–44. doi:10.1111/bcp.12056.
Kasichayanula S, Yao M, Vachharajani M, et al. Disposition and Mass Balance of [14C]-dapagliflozin after single oral dose in healthy male volunteers. AAPS J. 2008;10(S2).
Dostalek M, Court MH, Hazarika S, et al. Diabetes mellitus reduces activity of human UDP-glucuronosyltransferase 2B7 in liver and kidney leading to decreased formation of mycophenolic acid acyl-glucuronide metabolite. Drug Metab Dispos. 2011;39:448–55.
Abdul-Ghani MA, DeFronzo RA. Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus. Endocr Pract. 2008;14:782–90.
Vallon V, Platt KA, Cunard R, et al. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol. 2011;22:104–12.
Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;91:733–94.
Hummel CS, Lu C, Liu J, et al. Structural selectivity of human SGLT inhibitors. Am J Physiol Cell Physiol. 2012;302:C373–82.
Komoroski B, Vachharajani N, Feng Y, et al. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin Pharmacol Ther. 2009;85:513–9.
DeFronzo RA, Hompesch M, Kasichayanula S, et al. Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care. 2013 [Epub ahead of print].
Komoroski B, Vachharajani N, Boulton D, et al. Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Ther. 2009;85:520–6.
Carlson GF, Tou CK, Parikh S, et al. Evaluation of the effect of dapagliflozin on cardiac repolarization: a thorough QT/QTc study. Diabetes Ther. 2011;2:123–32.
Kasichayanula S, Chang M, Hasegawa M, et al. Pharmacokinetics and pharmacodynamics of dapagliflozin, a novel selective inhibitor of sodium-glucose co-transporter type 2, in Japanese subjects without and with type 2 diabetes mellitus. Diabetes Obes Metab. 2011;13:357–65.
Yang L, Li H, Li H, et al. Pharmacokinetic and pharmacodynamic properties of single- and multiple-dose of dapagliflozin, a selective inhibitor of SGLT2, in healthy Chinese subjects. Clin Ther. 2013;35(8):1211–22.e2.
Kasichayanula S, Liu X, Shyu WC, et al. Lack of pharmacokinetic interaction between dapagliflozin, a novel sodium-glucose transporter 2 inhibitor, and metformin, pioglitazone, glimepiride or sitagliptin in healthy subjects. Diabetes Obes Metab. 2011;13:47–54.
Imamura A, Kusunoki M, Ueda S, et al. Impact of voglibose on the pharmacokinetics of dapagliflozin in Japanese patients with type 2 diabetes. Diabetes Ther. 2013;4(1):41–9.
Kasichayanula S, Chang M, Liu X, et al. Lack of pharmacokinetic interactions between dapagliflozin and simvastatin, valsartan, warfarin, or digoxin. Adv Ther. 2012;29:163–77.
Wilcox CS, Liu X, Kasichayanula S, et al. Evaluation of interactions of dapagliflozin and bumetanide. Presented at: American Society of Nephrology, Denver (2010).
Kasichayanula S, Liu X, Griffen SC, et al. Effects of rifampin and mefenamic acid on the pharmacokinetics and pharmacodynamics of dapagliflozin. Diabetes Obes Metab. 2013;15:280–3.
Acknowledgments
Medical writing assistance was provided by Ray Ashton and Karen Pemberton, PhD of PPSI (a PAREXEL company) and was funded by AstraZeneca and Bristol-Myers Squibb. All authors are employees of Bristol-Myers Squibb.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kasichayanula, S., Liu, X., LaCreta, F. et al. Clinical Pharmacokinetics and Pharmacodynamics of Dapagliflozin, a Selective Inhibitor of Sodium-Glucose Co-transporter Type 2. Clin Pharmacokinet 53, 17–27 (2014). https://doi.org/10.1007/s40262-013-0104-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-013-0104-3