
Abstract
Mucopolysaccharidosis type II (MPS II) is a rare, pediatric, neurometabolic disorder due to the lack of activity of the lysosomal hydrolase iduronate 2-sulfatase (IDS), normally degrading heparan sulfate and dermatan sulfate within cell lysosomes. The deficit of activity is caused by mutations affecting the IDS gene, leading to the pathological accumulation of both glycosaminoglycans in the lysosomal compartment and in the extracellular matrix of most body districts. Although a continuum of clinical phenotypes is described, two main forms are commonly recognized—attenuated and severe—the latter being characterized by an earlier and faster clinical progression and by a progressive impairment of central nervous system (CNS) functions. However, attenuated forms have also been recently described as presenting some neurological involvement, although less deep, such as deficits of attention and hearing loss. The main treatment for the disease is represented by enzyme replacement therapy (ERT), applied in several countries since 2006, which, albeit showing partial efficacy on some peripheral organs, exhibited a very poor efficacy on bones and heart, and a total inefficacy on CNS impairment, due to the inability of the recombinant enzyme to cross the blood–brain barrier (BBB). Together with ERT, whose design enhancements, performed in the last few years, allowed a possible brain penetration of the drug through the BBB, other therapeutic approaches aimed at targeting CNS involvement in MPS II were proposed and evaluated in the last decades, such as intrathecal ERT, intracerebroventricular ERT, ex vivo gene therapy, or adeno-associated viral vector (AAV) gene therapy. The aim of this review is to summarize the main clinical aspects of MPS II in addition to current therapeutic options, with particular emphasis on the neurological ones and on the main CNS-targeted therapeutic approaches explored through the years.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
About two-thirds of MPS II patients present with the severe form of the disease, which affects all organs including the CNS, with a significant impairment of cognitive and behavioral functions. |
Up until now, several therapeutic strategies targeting the CNS have been developed, including cell-based therapies, gene therapy, and genome-editing therapies, as well as brain-targeting ERT approaches. Some of them are still under clinical trial, while some have been approved by the regulatory agencies for CNS treatment of MPS II. |
All approaches so far evaluated for MPS II present both advantages and limitations, and thus far no ideal therapeutic solution is available for the neurological disease, although among the therapies under development, the most promising appear to be fusion proteins. |
1 Introduction
Mucopolysaccharidosis type II, also known as Hunter disease, first described more than 100 years ago, is a rare genetic condition, transmitted as an X-linked trait, and belonging to the wide group of the lysosomal storage disorders (LSD) [1]. Together with a general somatic involvement affecting most organ systems, a distinctive trait of about two-thirds of the patients is heavy neurological impairment, mainly characterized by a significant developmental delay and behavioral difficulties, with hyperactivity and impulsivity, once defined as aggression [2]. From the retrospective clinical data collected during the last decades by the international registry known as the Hunter Outcome Survey (HOS) [3, 4], we know that early signs of the disease may appear around 1–1.5 years of age, especially in the severe forms. However, children are not commonly diagnosed precociously, due to the overlapping signs and symptoms with other pediatric neurological disorders, misleading the correct diagnosis [5]. This causes a significant delay also in the application of the available therapeutic options, reducing their potential efficacy.
Although it remains a therapeutic option in some countries, hematopoietic stem cell transplantation (HSCT) has not shown great efficacy in MPS II [6, 7]. For a long time the high risk/benefit ratio has discouraged HSCT application in the disease. However, the development of a new preconditioning regimen prior to HSCT in the last 2 decades has reduced the mortality rate, as well as risks of infection and graft-versus-host disease [8]. A lowering of the age at transplant has led to a better outcome in some more recently described cases [9, 10].
Together with symptomatic therapy, the most applied therapeutic strategy since 2006 is enzyme replacement therapy (ERT), consisting of weekly intravenous (i.v.) injection of the recombinant functional enzyme. However, ERT, showing some efficacy at the somatic level, has been revealed to have poor efficacy or be totally ineffective in all central nervous system (CNS) manifestations [2, 11].
Hence, the challenge to find an efficient treatment for the brain impairment in MPS II, as well as for other neuronopathic pediatric disorders sharing similar problems of drug impenetrability in the brain, remained mostly unanswered, albeit several brain-targeted therapeutic strategies have been proposed and tested, both preclinically and clinically, throughout the years. Finding a brain-targeted path to reach and treat the neurological implications of the syndrome would represent a hope for many other pediatric diseases where the CNS is involved, leading the way to other drugs that could be delivered using the same or similar pathways or strategies.
In the present review, we report the main clinical features of the disease, including the neurological ones, and summarize the most significant brain-targeted approaches evaluated in recent years for MPS II, their technical advantages/limitations, and the positive outcomes or difficulties encountered in their preclinical or clinical application.
2 MPS II: The Disease and its Clinical Features
Mucopolysaccharidosis type II (MPS II), or Hunter disease, is a rare pediatric metabolic disorder caused by the deficit of the lysosomal hydrolase iduronate 2-sulfatase (IDS). Hunter disease is inherited as an X-linked trait; therefore, only male subjects are affected, though a few cases of female patients have also been reported in literature, mostly due to an unbalanced inactivation of the X chromosome during the lyonization process in the female carriers [12,13,14].
IDS is an enzyme required to degrade the glycosaminoglycans (GAG) heparan sulfate and dermatan sulfate; hence, its deficit causes the accumulation of these two macromolecules, both within the endolysosomal compartment and in the extracellular matrix. Such pathological deposits progressively impair cellular functioning, leading to cell death in most tissues and organs.
MPS II belongs to the cluster of mucopolysaccharidoses (MPSs), a subgroup of the lysosomal storage disorders (LSD) that overall includes 12 different syndromes so far (including the recently discovered MPS-plus [15] and Usher syndrome type IV [16, 17]). The disease has a reported incidence that varies in different geographic areas and from one country to another, ranging from 0.38 per 100,000 live newborns in Brazil to 1.09 per 100,000 live newborns in Portugal, being in general much lower in European countries compared with East Asian ones [1]. It is also likely that the aforementioned incidence rates may be underestimated at the moment, since the diagnosis of MPS II remains difficult and in some countries underconsidered [18]. Except for the very few suspected cases highlighted by newborn screenings, which sometimes include MPS II [19,20,21,22], diagnosis of Hunter commonly starts with clinical evidence of early signs or symptoms known to be associated to the disorder or in general to mucopolysaccharidoses. However, this may be challenging if symptoms are subtly expressed [23], thus contributing to a delayed diagnosis. Next, evaluation of urinary GAG content is performed, followed by the analysis of some lysosomal enzyme activities, enabling a differential diagnosis between MPS II and the other MPSs. The detected enzyme deficit finally drives the molecular identification of the causal gene variant [24]. In recent years, the application of the next generation sequencing approach has allowed the process to be reversed and shortened by applying a genome-targeted (gene panels) [25, 26] or untargeted (whole genome or whole exome sequencing) [27] analysis to the DNA of the subjects with suspected MPS II. This eliminates the intermediate steps, reducing costs and accelerating a patient’s diagnosis.
Although MPS II generally occurs on a continuum of clinical phenotypes, commonly two main forms are recognized—attenuated and severe—albeit an intermediate form has also been reported in some cases. The severe phenotype, present in about two-thirds of the diagnosed patients [28], has an earlier onset than the other two forms, before 2 years of age [29], and also on average an earlier exitus, commonly around the second decade of life. The attenuated form presents with a later onset, around 3.5–4 years of age, and a longer life expectancy, commonly mid-adulthood, with patients possibly reaching their 50s or 60s in some cases [30,31,32]. Clinical peripheral involvement is equally compromised in all forms, with almost all of the organs progressively involved [33], although in the attenuated cases the progression is described as slower. Atypical as well as late-onset presentations have been rarely described [34, 35]. IDS enzyme activity is rather defective in all subjects, commonly undetectable at biochemical evaluation, and this does not help the differential diagnosis and the following prognosis of the neurological impairment, which characterizes the severe form and which will be discussed in detail in the next section. The disease has different signs and symptoms, although the most common traits are coarse facial features, skeletal deformities and joint stiffness, growth retardation, cardiorespiratory impairment [36, 37] including a diffuse valvulopathy [38], hearing loss, and organomegaly. Clinical diagnosis is mainly based on the typical facial features, liver and spleen enlargement, dysostosis multiplex, growth retardation, and joint stiffness often observed by orthopedists at the time of the first evaluation [39]. Neurological involvement is also observed in about two-thirds of the cases [2, 40, 41], commonly in the severe forms of the disease. In MPS II patients, death commonly occurs because of cardiorespiratory failure [1, 42].
From 2005 to 2022, an international survey collecting data on patients with a confirmed diagnosis of MPS II was carried out, known as the Hunter Outcome Survey (HOS). Aims of the project were to better describe the natural history of the disease and to monitor the long-term ERT efficacy, as most of the enrolled subjects were under enzyme treatment [3].
3 Neurological Aspects of MPS II
3.1 Pathogenesis of Neurological Disease
CNS disease comprehension has always been particularly difficult in MPS II. Since in the vast majority of the patients, no or extremely low levels of IDS enzyme activity can be detected, independently from the clinical form of the disease, it remains unclear which other primary or secondary molecular alterations, apart from the common enzyme deficit, could drive the neuronopathic prognosis in severe patients. Focusing our attention on the primary effect of the IDS deficit, i.e., the GAG accumulation progressively involving cells, tissues, and organs and causing their malfunctioning, may provide some pathogenetic information. However, it does not seem to be sufficient to explain the entire pathological scenario [43]. For a few years, we have known that elevated levels of brain heparan sulfate (HS) and partially catabolized HS fragments are commonly identified in MPS patients with significant central nervous system involvement [44]. However, several questions remain open. What determines the higher HS level in the brain of neuronopathic patients considering the poor genotype–phenotype correlation? When does this pathological accumulation start? What is the primary metabolic alteration? Hence, there is a connection between CNS involvement and HS accumulation, which, however, cannot explain the neuronopathic drift and its origin. Other secondary deposits, as cholesterol, as well as alterations involving basic processes such as autophagy and mitophagy, essential for correct cell functioning, together with morphogen signaling, have been identified as altered in MPS II and in other LSD [43]. Bhalla et al. [45] in 2020 reported high levels of the marker of neuronal degeneration neurofilament light chain (Nf-L) in cerebrospinal fluid (CSF) and serum of MPS II neuronopathic patients; moreover, Nf-L levels correlated with CSF HS levels. The same authors identified high levels of lysosomal lipids such as gangliosides GM3 and GD3, bis(monoacylglycerol)BMP, and glucosylceramides in CSF. In addition, we know that the neurodevelopment of the children appears mostly normal in their first year of life, but it remains unclear when the neurological drift begins. To help the understanding of the origin of CNS impairment, several in vivo and in vitro models have been proposed and analyzed. Two brain areas of the MPS II mouse model were analyzed a few years ago from our group by an RNA-Seq approach [46], which identified in the model versus the wild type, differentially expressed genes involved in mitochondrial impairment and oxidative stress and some possible connections with chronic neurodegenerative diseases, as Alzheimer and Parkinson’s diseases. Differential alterations between the severe and the attenuated forms, which could be extremely informative on the primary metabolic alterations driving toward a neuronopathic prognosis, are currently mainly conducted in reprogrammed human-induced pluripotent stem cells. They mostly derived from primary somatic cells of the patients [47, 48], which can be subsequently differentiated neurogenically [49,50,51]. This strategy could help delve deeper MPS II CNS pathogenesis, being at the same time an easy model for the testing of new CNS-targeted therapeutic molecules.
3.2 Neurological Signs and Symptoms
Together with a wide range of somatic symptoms, at least two-thirds of MPS II patients present a deep progressive neurological impairment, mainly identified with a developmental delay of cognitive skills and significant behavioral problems [28]. In patients with Hunter syndrome many genomic variants have been identified, most of which are private, being half of them present only in a single family. Thus, although each patient is hemizygote, carrying only one variant, the analysis so far conducted have highlighted a limited genotype–phenotype correlation. In the experience of our laboratory, even in cases where the molecular diagnosis was successfully conducted in the early phases of the disorder, it is very difficult to understand a possible prognosis for the patients. This is different in the cases of large deletions/insertions or recombinational events, for the vast majority associated with a severe clinical phenotype, with rapid neuronopathic evolution (Zanetti et al.; personal communication). A high level of CSF GAG has been reported in MPS II patients with neurocognitive impairment [52], while in the attenuated forms the level was reported as intermediate between severe MPS II patients and surrogate-normal children.
Thus, to better define how the disease will evolve, it is important to strictly monitor the patients with suspected Hunter disorder using a multidisciplinary approach, which, for the neurological form, will be extremely difficult to conduct due to important differences in the timing of its development and progression [53] and considering that attenuated forms may also present some neurological involvement, although less intense, such as deficits of attention and hearing loss [54]. According to a recent paper, among all mucopolysaccharidoses, “MPS II has the greatest uncertainty about the neurocognitive progression of the disease” [53]. As in other neuronopathic MPSs, in MPS II, neurologic development is also initially normal, and then it declines toward devastating impairment.
Together with neurocognitive decline, the main features that are most commonly described are the following: behavioral difficulties with hyperactivity/impulsivity, seizures [2, 41], communicating hydrocephalus, sleep apnea, and spinal cord compression. A study conducted in 2011 on 36 Italian patients described a very complex scenario of magnetic resonance imaging (MRI) features, where perivascular spaces enlargement (89%), white matter abnormalities (WMAs; 97%), subarachnoid space enlargement (83%), and IIIrd-ventricle dilatation (100%) were very often associated [55]. In the same study, about half of the patients showed spinal stenosis. Subarachnoid spaces and ventricle enlargement, WMAs, and spinal stenosis were seen to progress despite ERT and might represent potential disease severity biomarkers. Spinal cord compression is often described in these children, commonly treated by surgical decompression [56]. Seizures are a less common sign (about 40% of the patients); they are usually of tonic–clonic type, but can also be focal and absence seizures, and respond to standard anticonvulsant treatments [2, 57]. Hyperactive/impulsive behavior is reported, in the absence of therapy, in about 35–40% of the cases [58], and it may be worsened by sleep disturbances [59, 60]. A recent paper underlined that behavioral and sleep manifestations are early clinical markers of CNS involvement in the neuronopathic forms of the disease [58].
The treatment of this devastating set of neurological manifestations has always represented a challenge for the scientific and medical community as well as for the pharmaceutical companies. Hunter disease is one of the many neurological and neurometabolic disorders for which difficulties in reaching the brain region impede effective use of drugs that have already been developed. The blood–brain barrier (BBB), a selectively permeable barrier between the CNS and the systemic circulation [61], works as an efficient shield to protect the brain from most forms of attack, such as toxins and pathogens. It also represents an insurmountable barrier for most drugs, which are unable to reach the correct brain target unless they are small, highly lipophilic molecules [57, 62]. Specifically for the enzymes, because of their size, molecular instability and chemical-physical properties, the delivery of these molecules represents a particularly difficult challenge [63].
4 Therapeutic Approaches, an Overview
Until the introduction of hematopoietic stem cell transplantation (HSCT) and then of enzyme replacement therapy (ERT), the management of MPS II was mainly focused on relieving the signs and symptoms of the disease. Current symptomatic therapeutic approaches are aimed at treating symptoms that have remained unresolved following the advent of ERT, such as those related to the neurological system (seizure, communicating hydrocephalus, spinal cord compression, and carpal tunnel syndrome), to the skeletal system (dysplasia and limited range of motion), and to the cardiac apparatus (mainly valvulopathies) [59].
Attempts have been made to use bone marrow transplantation (BMT) and hematopoietic stem cell transplantation (HSCT) in these patients; in most cases a poor efficacy in these children was observed, principally on the neurological side. This is very different from what happened with a related disease, MPS I or Hurler disease, where transplant is considered to be a standard of care [56]. This represented a strong step back in the treatment of Hunter disease, since allogeneic HSCT might provide healthy cells able to cross the blood–brain barrier, thus supplying functional enzymes to treat the CNS disease through the mechanism of cross-correction [64], and in most cases, this needs to be performed only once in life [56]. It is supposed that this significantly different outcome between the two disorders may be due to the significant diagnostic delay of MPS II versus MPS I, allowing for irreversible damage in the brain, which therefore cannot benefit much from the transplant [56]. The inclusion of MPS II in a newborn screening program will potentially change this landscape, as the sooner any therapies can be applied, the better the patients’ outcome will be. An early application of HSCT may also result in a better outcome [9, 10], although the procedure remains quite risky in these patients.
Since it is due to the deficit of a lysosomal hydrolase, iduronate 2-sulfatase, MPS II was one of the first disorders for which a protocol of enzyme substitution, able to provide the functional hydrolase, was developed. Enzyme replacement therapy consists of the intravenous administration of the functional recombinant version of IDS enzyme and has been available since 2006. In 2012, an additional recombinant enzyme, idursulfase beta, was approved for MPS II treatment [65]. This followed previous preclinical and clinical evaluations conducted in two other LSD, where strategies of enzyme substitution were proposed starting several years before, in 1973 for Fabry disease [66] and in 1974 for Gaucher disease [67], in both cases using enzymes isolated from the human placenta.
Clinical data collected in these 17 years of application of ERT protocol have clearly shown, since the analysis of the first data, that the administration of the recombinant enzyme exerts some benefits at the somatic level, whereas it has no efficacy in all CNS manifestations [68]. Indeed, the effectiveness of ERT for MPS II patients is significantly limited by its inability to address the neurological manifestations of the disease, as the recombinant enzyme does not cross the blood–brain barrier (BBB) at therapeutic concentrations. This is due to its large molecular size, 76 kDa, as only small hydrophobic molecules (< 500 daltons) can cross the BBB and reach an effective concentration in the brain [69], and to its chemical-physical properties. Moreover, a general inability of the lysosomal enzymes to reach the brain region due to a progressive decrease of cation-independent mannose 6-phosphate receptor (CI-M6PRs) expression, at BBB level, during brain maturation has been described [70, 71]. This represents a great unmet need for suitable treatment options [72]. Such evidence, compounded with the elevated costs of ERT, has somehow put up for discussion the opportunity to treat Hunter patients presenting with a severe form [2, 11, 73], together with the criteria for initiating or ending the treatment with ERT [74].
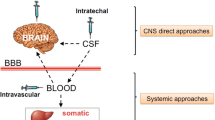
To tackle this issue, different approaches based on cell therapies, gene therapies, and genome-editing technologies, as well as on brain-targeting ERT approaches, have been studied in the last 10 years (Fig. 1). All of them are based on a biological process called cross-correction, by which the therapeutic enzyme is released into extracellular fluid and endocytosed by neighboring and distant cells through CI-M6PRs binding, thus correcting the enzyme deficit [64, 75, 76].

Major therapeutic approaches evaluated to target the CNS disease in lysosomal storage disorders. BBB: blood-brain barrier; CNS: central nervous system; ERT: enzyme replacement therapy; HSCT: hematopoietic stem cell transplantation; HSCGT: hematopoietic stem cell gene therapy. Created with BioRender.com
5 Enzyme Replacement Therapy: Advantages and Limitations
As cited above, ERT has long been the most promising and applied therapeutic strategy for Hunter disease, and still is. The enzyme substitution was developed and then approved by the US FDA in 2006 (FDA Biologic License Application: 125151), while in 2007 the European Medicines Agency (EMA) authorized its use in the European Union (EMA product number: EMEA/H/C/000700). Disease-specific treatment is available in the form of the recombinant enzyme idursulfase (Elaprase®, Shire Human Genetic Therapies, USA, now Takeda Pharmaceuticals USA, Inc., Lexington, MA, USA), administered intravenously weekly. Starting 2005, a registry called the Hunter Outcome Survey (HOS), a multinational long-term survey monitoring the natural history of the disease, as well as safety/efficacy of ERT with Elaprase® [3], was established, and it was completed at the end of 2022 (https://mpssociety.org.uk/files/hos-patient-report-2023.pdf). The study progressively enrolled 1405 patients from 34 countries and provided a huge amount of data, related to the disease and the outcomes of the treatment.
In addition, in 2013 a study described the Phase I/II clinical trial conducted with a second drug called idursulfase beta (Hunterase®, Green Cross Corp.,Yongin, Korea), a recombinant IDS that is produced using a genetic engineering approach in Chinese hamster ovary (CHO) cell line [77]. Hunterase® received its first approval by the Korean FDA in 2012 and afterwards from several other drug agencies, including the US FDA and EMA, and is presently marketed in 12 countries.
Analyses of ERT safety/efficacy in MPS II has been widely conducted. Both drugs are generally well tolerated and have shown an acceptable safety profile, although infusion-associated reactions have been quite often registered, most commonly controlled by an antihistaminic, antipyretic, or corticosteroids premedication [78,79,80]. Some cases of anaphylactoid reactions were also reported, representing a very serious complication of the treatment, and requiring an immediate stop of the infusion, and resuscitation [81]. Furthermore, the development of anti-idursulfase antibodies, likely due to the necessary long-term, continuous administration of the drug, is registered in about half of the treated patients [79, 80], and this may reduce ERT efficacy. Nevertheless, most of the infusion-associated reactions were mild to moderate, and in most cases, patients could receive subsequent infusions [11].
An efficacy evaluation evidenced the ability of the enzyme to improve somatic manifestations of Hunter disease, although the therapeutic benefit may vary in different patients, according to several variables such as clinical phenotype or age at start of treatment, and it also varies in the different regions of the body [11]. A reduction of urinary glycosaminoglycan levels together with a decrease of organomegaly (mainly liver and spleen) is commonly observed, from the start of treatment [41, 82]. Significant improvement of endurance (for example, 6 min walking test) was reported [77, 83], and in some cases an amelioration of joint stiffness [11, 84]. The frequency of respiratory infections was also significantly reduced [84]. However, heart hypertrophy and cardiac valves, heavily involved in the disease, did not benefit much from ERT [82, 84], and neither did adeno-tonsils hypertrophy, hearing disabilities, and corneal clouding [11].
Main limitations of the recombinant enzymes are represented by the high costs of ERT, which have caused difficulties in its application in developing countries [42, 85], and difficulties in compliance in patients and families due to the need for weekly hospitalization, since the enzyme is most commonly administered under medical control. Importantly, it was clear since the beginning that idursulfase administered via i.v. injection was not able to cross the blood–brain barrier and therefore could not have any effects on the cognitive and behavioral problems [3]. A paper published in 2020 analyzing fluid biomarkers in neuronopathic MPS II patients evidenced elevated levels of GAG (up to 30-fold) both in the CSF and in the serum of MPS II patients, compared with non-MPS patients, in whom it did not reduce following treatment with ERT [45]. In the same analysis, significantly elevated NfL levels were also detected in the CSF and serum, regardless of the treatment regimen to which patients were subjected.
From here, the idea to explore other delivery systems or strategies, to favor the enzyme crossing of the BBB, has long been considered.
6 Brain-Targeted ERT, an Update
For the brain-targeted therapeutic perspective by using ERT, there are two the main issues to take into consideration. The first one is the design of a delivery system able to transport high-molecular-weight proteins, such as enzymes, safely and efficiently across the barrier, no harming the BBB and the brain. The second issue to consider is the ability to maintain the enzyme activity all along the way, from the bloodstream to the correct target site within the cell; in this specific case the enzyme needs to maintain its degradative ability right inside the cell lysosomes. In addition, a positive effect on the improvement of the cognitive abilities of the patients, or at least in the delay of their progression, needs to be demonstrated. In fact, since many of the strategies evaluated are somehow risky or require an invasive procedure, the achievement of an optimal risk/benefit ratio is highly recommended and ethically required.
6.1 Intrathecal ERT (IT-ERT) and Intracerebroventricular ERT (ICV-ERT)
Despite the fact that ERT cannot efficiently cross BBB following intravenous administration [86], it has been known for many years that, if administered directly into the brain, it can readily enter neural cells through their CI-M6PRs [62, 87]. Indeed, these receptors are expressed in all areas of the brain [88], in most neuronal cells, including neurons, astrocytes, or still immature oligodendrocytes [89].
There are two main routes for a direct administration of ERT to the CNS, intrathecal (IT) and the intracerebroventricular (ICV). Both of these ways present advantages and limitations; they both deliver the enzyme into the CSF—in ICV into the lateral ventricle, while in the IT procedure, the enzyme is injected into the lumbar spine or subarachnoid space at the cisterna magna [90].
For IT administration, an intrathecal drug delivery device (IDDD) is commonly used, while for ICV injections, patients need to be implanted with a reservoir as a prerequisite to allow periodical ICV administrations [91]. Both devices require a surgical procedure, often posing technical difficulties especially in very young children and especially for ICV [91], to avoid risks of hemorrhagic complications. Despite being potentially promising in treating CNS problems in neuronopathic MPS II patients, these technical difficulties had already been reported a few years ago by Okuyama and colleagues, who described them as “significant hurdles to overcome” [92].
Both techniques were known since they had been previously used to treat other conditions or diseases, such as for anti-pain drugs in cancer [93] or in Parkinson’s disease [94].
Preclinical studies on IT and ICV administrations of ERT were widely conducted in different animal models of MPS II. Calias et al. in 2012 evaluated the IT and ICV injection of Elaprase® in monkey, dog, and mouse models [95]. Both ICV and lumbar IT administration to the monkeys and dogs determined a significant enzyme distribution in the brain parenchyma, together with an important cellular uptake; the spinal cord could also benefit from the lumbar IT injection, while a very little amount of idursulfase was seen following ICV administration. In the mouse model, injections of the enzyme through lumbar IT could reduce the brain cellular vacuolation, commonly seen in the histopathological analysis of the diseased samples.
ICV-ERT has recently received drug approval in Japan [96], where it is administered together with intravenous ERT. A recent study [97] reported the final results of a long-term Phase I/II clinical trial in six patients administered ICV idursulfase for 5 years, at progressive doses from 1 to 30 mg/ml every 4 weeks, together with intravenous weekly injections at the usual dosage of 0.5 mg/ml. The study evidenced good tolerability with no higher safety concerns with respect to the common i.v. treatment, and some positive results with a significant progressive decrease of CSF HS concentrations, which presenting a mean baseline value of 7.75 μg/ml, after 5 years of treatment were reduced to a mean value of 2.15 μg/ml (72.3% reduction). They also registered an increase in age in the developmental decline or at least a stabilization of the decline. As in most clinical studies, best results were obtained in patients where treatment was started before 3 years of age.
As for IT-ERT, starting in the Phase I/II clinical trial, the IDDD created numerous complications [98]; that is why, in Phase II/III of the trial, a smaller device was adopted [99]. Idursulfase-IT formulation was supplied as an isotonic solution at 10 mg/ml [99] every 4 weeks for overall 52 weeks, together with i.v. weekly injections of 0.5 mg/kg in 34 subjects (32 of whom were able to complete the study), while in 15 control patients no IT administration was performed. IT-ERT was generally well tolerated by the patients. Best results were obtained in patients younger than 6 years of age and carrying missense variants, in whom a significant GAG decrease was measured in CSF. However, analysis of the primary and secondary endpoints related to an improvement in overall intellectual ability and adaptive behavior ability, respectively, could not show a statistically significant result in the patients treated with IT-ERT versus non-treated patients [99]. Due to this insufficient efficacy outcome, authors concluded that the data obtained in the clinical study did not meet the “evidentiary standard to support regulatory filings,” although a trend toward a possible positive effect was observed.
All studies conducted so far underlined the need to initiate the treatment in very young patients, where the best efficacy was registered [97, 99]. Some of the borderline effects measured in these studies may in fact be due to the elevated age of the children at start of treatment. This is particularly important for brain-targeted therapies, due to the progressive and irreversible damage affecting CNS.
6.2 Nanoparticles as Delivery Vehicles
Nanoparticles are very small delivery vehicles, 200–300 nm in diameter [63], able to safely transport therapeutic molecules to specific target sites as well as to protect the delivered molecule. Polymeric nanoparticles can offer chemical-physical and biological stability, a long circulation timing, and a great bioavailability [100]. In addition, they can target specific organs or body districts, as well as cellular organelles, while still keeping the delivered compound active [100].
Their development has increased in the last few years, and their ability to deliver difficult macromolecules as proteins and enzymes across the blood–brain barrier has been demonstrated [63]. Specifically, the role of nanotechnology for the delivery of the enzymes needed for the treatment of lysosomal storage disorders has been highlighted in the last decade [61, 100]. In fact, nanoparticles not only can provide a delivery system but can also solve some enzyme difficulties related to their size, risk of degradability, and solubility requirements [63].
Therapeutic enzymes have been delivered to the CNS system in Gaucher disease [101] and Krabbe disease [102]. As for Hunter disease, the only preclinical in vivo study published so far was from our team in 2019 [103]. We used polylactide-co-glycolide (PLGA) biocompatible and biodegradable nanoparticles, approved by the FDA for human use, functionalized with a glycopeptide of seven amino acids (g7-NPs) [104, 105] for CNS targeting [103]. g7-NPs were loaded with the therapeutic IDS and, following their biochemical analysis and in vitro evaluation, they were administered via weekly i.v. injections for short-term evaluation in a mouse model of the disease. Mice were treated for 6 weeks, after which we observed a significant decrease of glycosaminoglycan deposits, both in the liver and in the brain parenchyma, together with a reduction of some neuro-inflammatory biomarkers such as GFAP, CD68, and LAMP-2 [103].
6.3 Brain-Targeted Enzyme Fusion Proteins
Several approaches based on receptor-mediated transcytosis such as BBB crossing strategy have been implemented in the last years and have revealed to be successful after more than 25 years of failed attempts [106]. To this aim, fusion proteins consisting of the functional enzyme fused with an antibody that binds endogenous protein receptors on the luminal side of the brain capillary endothelial cell are designed and delivered i.v.. In this way, the fusion proteins are endocytosed into the endothelial cell and afterwards released into brain parenchyma [107]. Pabinafusp alfa (JR141; JCR Pharmaceuticals) is a recombinant form of idursulfase where the IDS enzyme is fused with an anti-human transferrin receptor antibody. The fusion drug, administered through i.v. injection, binds to the transferrin receptor and, through receptor-mediated transcytosis, reaches the brain parenchyma [106]. At the moment, pabinafusp alfa is the only drug of this type that has completed the Phase III clinical trial, and in 2021 was authorized in Japan for use in mucopolysaccharidosis type II.
Preclinical experiments were conducted on the drug pabinafusp alfa in mice and monkeys, where a reduction of glycosaminoglycan levels both in the peripheral tissues and in the brain of a mouse model of MPS II were recorded [108], with a significant improvement of neurocognitive deficit [109]. The first Phase I/II open-label, multicenter, randomized clinical trial was published in February 2019 [92]. In the study, 14 patients were enrolled, and the effect of the drug on neurodegeneration was assessed with positive outcome by measuring heparan sulfate and dermatan sulfate levels in cerebrospinal fluid as biomarkers. A Phase II/III clinical trial was afterwards conducted, whose results were published in 2021 [110], while in the same year, a Phase II trial was conducted in Brazil [111]. Overall, 62 patients were involved in these clinical trials; almost 90% of them experienced stabilization or improvement in their neurocognitive impairment, and no drug-related severe adverse events [111].
In Japan, where the drug was first developed, the use of JR141 was authorized in 2021 (international patent WO 2016/208695 A1) [112], where it is currently available to MPS II patients of all phenotypic forms [113]. Very recently a paper reported the caregivers’ observations recorded for seven MPS II patients, under JR141 treatment for 3.3–3.5 years [114]; five of them were affected by the neuronopathic form, and two were classified as being more similar to the non-neuronopathic form. Data collected in the interviews suggest a trend toward improvement of multiple clinical aspects, including some cognitive signs, such as language skills, self-control, abilities to follow instructions, and expressing personal needs, among others, together with some somatic signs, mainly related to organ involvement, musculoskeletal apparatus impairment, and joint stiffness, generally improving the quality of life of patients and families [114]. In January 2023 the drug was granted rare disease designation from the US FDA.
Another two similar fusion proteins have been developed and tested in clinical trial: AGT-182 (ArmaGen) where idursulfase is fused to an anti-insulin receptor antibody, and DNL-310 (Denali Therapeutics), an enzyme fusion protein that contains a low-affinity transferrin-binding peptide [106, 115]. As for AGT-182, according to ClinicalTrials.gov, a Phase I clinical trial was completed on March 17, 2017; however, no outcomes were reported. Concerning DNL-310, a potential association of the drug with anemia in two of the five patients enrolled in the clinical trial in relation to the transferrin receptors suggested the requirement of a further evaluation [106].
7 BBB Disruption Strategies
Some strategies have been proposed to increase BBB permeability, thus allowing a temporary passage of drugs from the bloodstream to the CNS. Such strategies, generally applying invasive techniques, were based on neurosurgery or chemical/physical disruption of the barrier or via osmotic shift [116]. These techniques present, however, numerous drawbacks, mainly the invasiveness, the elevated costs of neurosurgery, and mainly high risks of infection and damages from toxins, because of the temporary opening of the BBB [117]. They were tested in some LSD, such as in MPS I, where in the mouse model of the disease, the application of the focus ultrasound, using magnetic resonance thermometry, allowed the delivery of the enzyme α-iduronidase across the BBB [118]. Osmotic shift using a co-injection of mannitol and adenovirus or adeno-associated virus were evaluated in Sandhoff disease [119], MPS IIIB [120], and CLN2 deficiency [121]. To the best of our knowledge, these strategies have not been evaluated for the treatment of mucopolysaccharidosis type II brain disease.
8 Brain-Targeted Cell and Gene Therapies
Cell and gene therapy approaches for MPS treatment involve delivering an exogenous gene or cells with the aim of expressing and secreting supraphysiologic levels of the functional enzyme able to elicit a therapeutic effect. To target and cure the brain disease in MPS II, several strategies have been implemented via both cell- and gene-based approaches or via combined ones.
8.1 Allogeneic HSCT
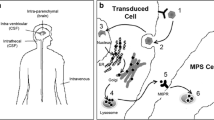
Allogeneic HSCT consists of transplantation of HLA-compatible bone marrow or umbilical cord blood stem cells, which can become long-term sources of functional therapeutic enzymes. Moreover, monocytic/phagocytic cells can reach the brain, by crossing the BBB, and being hosted as microglia, which in turn would be able to secrete the functional enzyme, thus correcting neuronal enzyme deficiency (Fig. 2). This was first demonstrated in 2009 by Araya, who, in a 6-year-old patient with severe MPS II, documented the distribution of donor-derived cells in the brain 10 months after cord blood stem cell transplantation [122]. To date, several authors have reported slight neurological improvements after HSCT. Ameliorations of brain magnetic resonance imaging (MRI) atrophy and category I and II brain lesions as well as of activity of daily living (ADL) were reported by Tanaka and colleagues in a nationwide follow-up in Japan [123]. Analogously stable or improved MRI brain lesions were observed by Kubaski and colleagues [124]. Improvements in cognitive, language, and motor skills were observed in a severe patient submitted to HSCT at 70 days old and followed for 7 years [9].

BBB crossing by monocytic/phagocytic cells and cross-correction mechanism. Healthy donor or genetically modified hematopoietic stem cells can reach the brain by crossing the BBB; then, cells can be hosted as microglia-like cells, which in turn will secrete the functional enzyme that will correct the neuronal enzyme deficit through the cross-correction mechanism. BBB: blood–brain barrier; CNS: central nervous system. Created with BioRender.com
A recent retrospective study in 109 Japanese Hunter patients has shown that HSCT might improve the ADL scores in both mild and severe patients [125]. However, the efficacy of HSCT on the neurological system for MPS II remains controversial, due to the limited information regarding the long-term outcomes. Moreover, HSCT remains a risky procedure, although with protocol refinement, a progressive reduction of risks of life-threatening graft-versus-host reactions and/or infections have been obtained [126, 127].
8.2 Gene Therapy
As MPS II is a monogenic disease with a well-known pathophysiology, it is a good candidate for gene therapy. Indeed, through the mechanism of cross-correction, the therapeutic enzyme produced after genetic modification by a depot organ can be captured by other cells and organs, and a healthy status can be obtained with at least 5–15% of the normal enzyme level [128]. Moreover, gene therapy might overcome many ERT limitations, such as the need for repeated administrations, the inability to cross the BBB, and the risk of developing neutralizing antibodies against the therapeutic enzyme [129]. In recent years, several gene therapy approaches have been attempted for MPS II to specifically target the brain; to this aim different vectors and administration routes, as well as in vivo and ex vivo procedures, were studied [130].
8.2.1 Hematopoietic Stem Cell Gene Therapy (HSCGT)
HSCGT has several advantages over allogeneic HSCT. It relies on the patient’s own cells, thus bypassing the risk of graft-versus-host disease (GVHD); moreover, as with other gene therapy approaches, it has the potential to achieve supra-physiological levels of the therapeutic enzyme. Finally, HSCGT might provide a sustained life-long source of the enzyme, without the need for frequent enzyme infusions, thus potentially improving patients’ quality of life [72].
Hematopoietic stem cell gene therapy using lentiviral vectors was successfully applied in the MPS II mouse model by Wakabayashi et al. [131]. A correction of neuronal manifestations via reduction of lysosomal storage and autophagic dysfunction in brain was observed together with an overall stabilization of neuronal functions. A recent study from the same group confirmed previously reported results and evidenced that strong preconditioning is required to obtain an efficient engraftment of the genetically modified stem cells and thus an amelioration of CNS involvement in MPS II mice [132]. However, lentiviruses’ random integration into the genome poses potential risks such as mutagenesis and neoplastic transformation.
Recently, Das and colleagues employed lentiviral vectors encoding an optimized human IDS cDNA in an ex vivo hematopoietic stem cell gene therapy approach, in both young pre-symptomatic and symptomatic MPS II mice, to assess the efficacy, feasibility, and safety of the protocol [127]. The study evidenced a robust and sustained engraftment of the infused cells 17–18 months post-transplantation, leading to long-term supraphysiological IDS enzymatic activity in bone marrow (BM) mononuclear cells, in serum as well as in brain. Normalization of the behavioral and neurocognitive disease manifestations of both pre-symptomatic and symptomatic treated MPS II mice was reported; benefit for the skeletal phenotype was also evidenced [127].
To date, only a clinical trial using lentiviral vectors is active. The study aims to evaluate safety and efficacy of ex vivo gene therapy, via autologous CD34+ hematopoietic stem cells transduced with a lentiviral vector containing the human IDS gene tagged with ApoEII (NCT05665166; https://clinicaltrials.gov/).
8.2.2 Adeno-Associated Viral Vectors
Adeno-associated viral vectors (AAVs) have emerged as a leading choice for gene therapy due to their ability to provide stable gene expression and deliver genes to both dividing and non-dividing cells. Major limitations of these viral vectors are the risk of immunogenicity and the potential for generating neutralizing antibodies [1]. Initial gene therapy attempts based on AAV used type 2/8 vectors administered intravenously in MPS II adult mice; both studies evidenced IDS activity restoration and full clearance of GAG storage in plasma and in several tissues including the brain [133, 134]. In 2016, Motas and colleagues administered AAV-9 vectors encoding IDS to the cerebrospinal fluid of MPS II mice through intracisternal injections and after 4 months evidenced a significant increase in IDS activity throughout the encephalon and full resolution of lysosomal storage lesions, and disappearance of neuroinflammation [135]. Hinderer and Laoharawee obtained the same results in two subsequent studies, using a similar strategy, but administering AAV-9 vectors via intracerebroventricular injections [136, 137].
The use of a less invasive route of administration to address CNS disease was tested by the Laoharawee’s group, who evidenced that a single dose of intravenously administered AAV9-hIDS may determine a global normalization of GAG on both sides of the BBB and prevention of neurocognitive decline in the treated MPS II mice [138].
Currently, two clinical trials (NCT03566043, NCT04571970; https://clinicaltrials.gov/) are ongoing using the AAV9.CB7.hIDS (RGX-121) produced by Regenxbio Inc., which is administered intracisternally or intracerebroventricularly with the aim of evaluating both safety and efficacy in improving CNS disease manifestations.
Recently, Smith and colleagues compared different doses of RGX-121 administered intravenously and intrathecally to determine whether intrathecal delivery alone can ameliorate both CNS and systemic manifestations. The relevant release of the vector into the periphery, after CSF-mediated delivery of AAV9 at a specific dose, suggests that this approach could be able to address both neurological and systemic disease manifestations in a murine model of MPS II and thus would be promising also for treating the human disease [139].
8.3 Brain-Targeted Genome Editing
Genome editing is a promising method whose aim is to repair the damage in a specific site of the genome through either non-homologous end joining or homologous recombination. To date, different editing platforms are available: zinc finger nucleases (ZNFs), transcription activator-like effector nucleases (TALENs), and, based on the CRISPR/Cas9 system, the CRISPR/Cas9- based editors and CRISPR/Cas9-prime editing tools [129].
So far, only the ZFN platforms have been studied for the treatment of MPS II, using the same strategy previously studied for the insertion in the albumin locus of human coagulation factor VIII (hF8) and factor IX (hF9) transgenes and subsequent correction of clotting defects in mouse models of hemophilia A and B, respectively [140,141,142]. Analogously, AAV8 vectors with albumin locus-targeting ZFN in hepatocytes were injected intravenously in MPS II mice at three different doses; treatment caused an increase of the IDS enzyme in blood and other peripheral tissues, as well as reduction of GAG in visceral organs and brain. Moreover, the high vector dose prevented the development of neurocognitive deficit in young MPS II mice, thus resulting in a promising treatment also for the CNS [143]. These results lead to the development of a Phase I/II clinical study (NCT03041324; https://clinicaltrials.gov/) with the first patients ever being treated with in vivo gene editing therapy. The trial results evidenced a favorable safety profile of the ZNF-based platform used; however, in vivo genome editing occurred at a very low level in the liver of the evaluated subjects with no long-term sustained enzyme expression in blood [144].
9 Small Molecules
Pharmacological chaperones (PCs) are low-molecular-weight molecules able to reach the endoplasmic reticulum (ER) and modify misfolded proteins to correct them, at least partially, and render them more active in their specific function. As for lysosomal storage disorders, PCs are mainly used to increase the enzyme activity of a mutated protein. It has, in fact, been suggested that aminoacidic changes, caused by sequence mutations, may render proteins less stable due to alterations of one or more of the several steps contributing to a correctly folded or glycosylated protein. This may lead to ER or Golgi apparatus retention or degradation of the protein, which renders the protein unsuitable for transport to the lysosomes, where it exerts its hydrolytic function [145]. PCs present some important advantages since they can be orally administered; they are able to reach most body regions, and they are less immunogenic than human recombinant proteins. In addition, if administered in combined protocols with ERT, they apparently can also stabilize the co-administered recombinant protein [146], which possibly suggests that they could enable a reduction of the ERT dosage.
Chaperones have been used for almost 25 years for the treatment of other lysosomal disorders such as Gaucher and Fabry diseases [145].
Recently, D2S0, a sulfated disaccharide derived from heparin with a similar structure to the natural substrate of IDS was evaluated in vitro as a PC-based potential treatment for MPS II patients [145]. In this study, MPS II fibroblasts, carrying the p.P231L mutation, treated with 10 M D2S0 for 8 days showed a 1.97-fold increase in IDS activity and a significant reduction in Alcian blue-positive granules. The same evaluation, conducted on HEK293T cells expressing p.N63D, p.L67P, p.A85T, p.R88H, p.Y108S, and p.P231L, showed an increased IDS activity between 1.6- and 39.6-fold but no effects in cells expressing p.L314P. These promising results showed that D2S0 may function in a mutation-dependent manner, but further optimization will be necessary to increase the efficacy of the molecule. This strategy could represent an interesting therapeutic approach since about 41% of mutations affecting the IDS gene are missense mutations and involve buried amino acids that can likely induce protein destabilization and misfolding [147].
The substrate reduction therapy (SRT) approach aims to reduce the bioavailability of the compound that cannot be fully metabolized by the defective enzyme (“substrate reduction”), thereby reducing the burden of the accumulating substrate [148]. This treatment can be administered orally, and it is potentially able to cross the BBB due to the size and charge of the molecules used. The evaluated strategies for MPS focus on indirect inhibition of GAG synthesis, as a direct approach would imply the use of toxic inhibitors of the enzymes related to the anabolic GAG synthetic pathway [128]. In 2008, an open-label pilot study evaluating the effectiveness of genistein, an isoflavone that reduces the synthesis of GAG, was completed on 10 patients with MPS IIIA or MPS IIIB. Patients evidenced improvements in urinary GAG and cognitive function and no adverse effects after 12 months of treatment [149]. However, these results were not replicated by the following studies [150]. Thus, to date, no effective SRT drugs for MPS II as well as for the other MPSs are available. Therefore, this approach still remains a potential therapeutic approach.
Another strategy could be the development of stop codon readthrough-inducing drugs. This approach could be applicable when the disease-causing mutation is a nonsense mutation and the patient could potentially benefit from molecules that promote the ribosome to read through the premature stop codon; this would allow protein translation to continue and, as a result, the restoration of the biosynthesis of the full-length protein [151]. To date, no studies are ongoing on candidate stop codon readthrough-inducing drugs for MPS II treatment.
10 Conclusions, Open Issues, and Future Perspectives
During the last 2 decades, several strategies have been evaluated to allow the crossing of therapeutics through the BBB, thus targeting them to the brain. In fact, the BBB, which positively protects the brain from most infective agents and circulating toxins, represents a difficult shield to cross or bypass for most therapeutic molecules. An important exception is via approaches providing healthy or genetically corrected cells, such as transplant or gene therapy approaches, or viral vectors, such as AAV, with both of these systems being able to cross the barrier. BBB crossing is particularly hard for high-molecular-weight drugs, such as recombinant proteins or enzymes such as those needed for the treatment of most lysosomal storage disorders affected by neurological impairment. Therefore, treatment of these diseases, including MPS II, remains challenging, given the fact that the mechanisms of the neuropathology are also not yet fully elucidated, thus making the identification of new therapeutic targets and biomarkers of diagnosis/efficacy difficult.
All approaches so far evaluated for MPS II present both advantages and limitations, and to date, no ideal therapeutic solution is available for the neurological disease. All of them are quite expensive, and this represents a significant problem for developing countries, for example, with i.v. ERT.
The timing of treatment is also a critical point: treatment of patients in their early stage of life could be more effective on the neurological disorder for most approaches—“the earlier the better.” However, this requires early clinical observation and suspicion, and good tools for differential diagnosis, as well as clear disease biomarkers, rapidly measurable through non-invasive procedures. The low number of patients who can be enrolled in the clinical trials, as MPS II is a rare disease, represents a further critical point. This entails rather a long time for an adequate safety/efficacy evaluation of each new therapeutic strategy, delaying its possible authorized release on the market.
Viral vectors as well as ERT approaches may lead to immunogenic responses that may subsequently reduce the treatment efficacy. Moreover, ERT-based approaches require repeated intravenous administration, and for intrathecal and intracerebroventricular routes, a continuous administration can be obtained via specific devices or reservoirs, which require a surgical procedure, which is often technically difficult. In contrast, cell and gene therapy approaches, which present the advantage of requiring a one-time administration and also look promising, are mostly still in the preclinical phase. Regarding costs, cell and gene therapies have relatively high costs related to technology development, which however could be counterbalanced by the single-administration needed and by the fact that the same technology could be applied to other disorders.
So far, the most promising approaches for MPS II appear to be fusion proteins, such as pabinafusp alfa, HSCGT approaches, and AAV vectors, all administered i.v., thus through the least invasive procedure. Nowadays, some of these approaches are available or will be available soon; therefore, the possibility of using only one therapy able to target both the somatic and the CNS disease is closer to being real or real in some cases. Specifically for the fusion proteins, in the future continuous i.v. administration might be proposed, at lower doses, mimicking the physiological production of the enzyme by the cells.
In recent years, the evaluation of several therapeutic strategies approaching the brain disease in MPS II from different sides has been revealed to be extremely positive, allowing dissection of their potential efficacy together with their limitations, thus suggesting possible improvements. These studies will also be useful in a wider scenario, developing similar approaches for other lysosomal storage disorders, with patients still suffering for the lack of therapy.
References
D’Avanzo F, Rigon L, Zanetti A, Tomanin R. Mucopolysaccharidosis type II: one hundred years of research, diagnosis, and treatment. Int J Mol Sci. 2020;21:E1258.
Wraith JE, Scarpa M, Beck M, Bodamer OA, De Meirleir L, Guffon N, et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167:267–77.
Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J, et al. Initial report from the Hunter Outcome Survey. Genet Med Off J Am Coll Med Genet. 2008;10:508–16.
Parini R, Jones SA, Harmatz PR, Giugliani R, Mendelsohn NJ. The natural history of growth in patients with Hunter syndrome: data from the Hunter Outcome Survey (HOS). Mol Genet Metab. 2016;117:438–46.
Wiśniewska K, Wolski J, Gaffke L, Cyske Z, Pierzynowska K, Węgrzyn G. Misdiagnosis in mucopolysaccharidoses. J Appl Genet. 2022;63:475–95.
Vellodi A, Young E, Cooper A, Lidchi V, Winchester B, Wraith JE. Long-term follow-up following bone marrow transplantation for Hunter disease. J Inherit Metab Dis. 1999;22:638–48.
Guffon N, Bertrand Y, Forest I, Fouilhoux A, Froissart R. Bone marrow transplantation in children with Hunter syndrome: outcome after 7 to 17 years. J Pediatr. 2009;154:733–7.
Scarpa M. Mucopolysaccharidosis type II. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993 [cited 2024 Jul 2]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1274/.
Barth AL, de Magalhães TSPC, Reis ABR, de Oliveira ML, Scalco FB, Cavalcanti NC, et al. Early hematopoietic stem cell transplantation in a patient with severe mucopolysaccharidosis II: a 7 years follow-up. Mol Genet Metab Rep. 2017;12:62–8.
Sreekantam S, Smith L, Stewart C, Kearney S, Lawson S, Raiman J, et al. Efficacy of early haematopoietic stem cell transplantation versus enzyme replacement therapy on neurological progression in severe Hunter syndrome: case report of siblings and literature review. Mol Genet Metab Rep. 2022;32: 100881.
Parini R, Deodato F. Intravenous enzyme replacement therapy in mucopolysaccharidoses: clinical effectiveness and limitations. Int J Mol Sci. 2020;21:2975.
Tuschl K, Gal A, Paschke E, Kircher S, Bodamer OA. Mucopolysaccharidosis type II in females: case report and review of literature. Pediatr Neurol. 2005;32:270–2.
Manara R, Rampazzo A, Cananzi M, Salviati L, Mardari R, Drigo P, et al. Hunter syndrome in an 11-year old girl on enzyme replacement therapy with idursulfase: brain magnetic resonance imaging features and evolution. J Inherit Metab Dis. 2010;33(Suppl 3):S67-72.
Jurecka A, Krumina Z, Żuber Z, Różdżyńska-Świątkowska A, Kłoska A, Czartoryska B, et al. Mucopolysaccharidosis type II in females and response to enzyme replacement therapy. Am J Med Genet A. 2012;158A:450–4.
Ago Y, Rintz E, Musini KS, Ma Z, Tomatsu S. Molecular mechanisms in pathophysiology of mucopolysaccharidosis and prospects for innovative therapy. Int J Mol Sci. 2024;25:1113.
Kowalewski B, Lamanna WC, Lawrence R, Damme M, Stroobants S, Padva M, et al. Arylsulfatase G inactivation causes loss of heparan sulfate 3-O-sulfatase activity and mucopolysaccharidosis in mice. Proc Natl Acad Sci. 2012;109:10310–5.
Velde HM, Reurink J, Held S, Li CHZ, Yzer S, Oostrik J, et al. Usher syndrome type IV: clinically and molecularly confirmed by novel ARSG variants. Hum Genet. 2022;141:1723–38.
Rasheeedah I, Patrick O, Abdullateef A, Mohammed A, Sherifat K, Gbadebo I. Challenges in the management of mucopolysaccharidosis type II (Hunter’s syndrome) in a developing country: a case report. Ethiop J Health Sci. 2015;25:279–82.
Chan M-J, Liao H-C, Gelb MH, Chuang C-K, Liu M-Y, Chen H-J, et al. Taiwan National Newborn Screening Program by tandem mass spectrometry for mucopolysaccharidoses types I, II, and VI. J Pediatr. 2019;205:176–82.
Chien Y-H, Lee N-C, Chen P-W, Yeh H-Y, Gelb MH, Chiu P-C, et al. Newborn screening for Morquio disease and other lysosomal storage diseases: results from the 8-plex assay for 70,000 newborns. Orphanet J Rare Dis. 2020;15:38.
Chuang C-K, Tu Y-R, Lee C-L, Lo Y-T, Chang Y-H, Liu M-Y, et al. Updated confirmatory diagnosis for mucopolysaccharidoses in Taiwanese infants and the application of gene variants. Int J Mol Sci. 2022;23:9979.
Lin H-Y, Chang Y-H, Lee C-L, Tu Y-R, Lo Y-T, Hung P-W, et al. Newborn screening program for mucopolysaccharidosis type II and long-term follow-up of the screen-positive subjects in Taiwan. J Pers Med. 2022;12:1023.
Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144:S27-34.
Filocamo M, Tomanin R, Bertola F, Morrone A. Biochemical and molecular analysis in mucopolysaccharidoses: what a paediatrician must know. Ital J Pediatr. 2018;44:129.
Zanetti A, D’Avanzo F, Bertoldi L, Zampieri G, Feltrin E, De Pascale F, et al. Setup and validation of a targeted next-generation sequencing approach for the diagnosis of lysosomal storage disorders. J Mol Diagn JMD. 2020;22:488–502.
Ghaffari SR, Rafati M, Shadnoush M, Pourbabaee S, Aghighi M, Mirab Samiee S, et al. Molecular characterization of a large cohort of mucopolysaccharidosis patients: Iran mucopolysaccharidosis RE-diagnosis study (IMPRESsion). Hum Mutat. 2022;43:e1-23.
Gul R, Firasat S, Schubert M, Ullah A, Peña E, Thuesen ACB, et al. Identifying the genetic causes of phenotypically diagnosed Pakistani mucopolysaccharidoses patients by whole genome sequencing. Front Genet. 2023;14:1128850.
Lau H, Harmatz P, Botha J, Audi J, Link B. Clinical characteristics and somatic burden of patients with mucopolysaccharidosis II with or without neurological involvement: an analysis from the Hunter Outcome Survey. Mol Genet Metab Rep. 2023;37: 101005.
Żuber Z, Kieć-Wilk B, Kałużny Ł, Wierzba J, Tylki-Szymańska A. Diagnosis and management of mucopolysaccharidosis type II (Hunter syndrome) in Poland. Biomedicines. 2023;11:1668.
Young ID, Harper PS. Mild form of Hunter’s syndrome: clinical delineation based on 31 cases. Arch Dis Child. 1982;57:828–36.
Jones SA, Almássy Z, Beck M, Burt K, Clarke JT, Giugliani R, et al. Mortality and cause of death in mucopolysaccharidosis type II-a historical review based on data from the Hunter Outcome Survey (HOS). J Inherit Metab Dis. 2009;32:534–43.
Burton BK, Jego V, Mikl J, Jones SA. Survival in idursulfase-treated and untreated patients with mucopolysaccharidosis type II: data from the Hunter Outcome Survey (HOS). J Inherit Metab Dis. 2017;40:867–74.
Martin R, Beck M, Eng C, Giugliani R, Harmatz P, Muñoz V, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics. 2008;121:e377-386.
Shah GS, Mahal T, Sharma S. Atypical clinical presentation of mucopolysaccharidosis type II (Hunter syndrome): a case report. J Med Case Rep. 2010;4:154.
Gupta A, Uttarilli A, Dalal A, Girisha KM. Hunter syndrome with late age of presentation: clinical description of a case and review of the literature. BMJ Case Rep. 2015;2015:bcr2015209305.
Kamin W. Diagnosis and management of respiratory involvement in Hunter syndrome. Acta Paediatr Oslo Nor. 1992;2008(97):57–60.
Fesslová V, Corti P, Sersale G, Rovelli A, Russo P, Mannarino S, et al. The natural course and the impact of therapies of cardiac involvement in the mucopolysaccharidoses. Cardiol Young. 2009;19:170–8.
Dehghan B, Rostampour N, Sedighi M, Saryazdi MH, Rizi MJ, Mostofizadeh N, et al. Evaluation of cardiac findings in mucopolysaccharidosis. Int J Cardiovasc Imaging. 2024;40:73–8.
Link B, Botha J, Giugliani R. Characterization of orthopedic manifestations in patients with mucopolysaccharidosis II using data from 15 years of the Hunter Outcome Survey. JIMD Rep. 2024;65:17–24.
Jones SA, Parini R, Harmatz P, Giugliani R, Fang J, Mendelsohn NJ, et al. The effect of idursulfase on growth in patients with Hunter syndrome: data from the Hunter Outcome Survey (HOS). Mol Genet Metab. 2013;109:41–8.
Tomanin R, Zanetti A, D’Avanzo F, Rampazzo A, Gasparotto N, Parini R, et al. Clinical efficacy of enzyme replacement therapy in paediatric Hunter patients, an independent study of 3.5 years. Orphanet J Rare Dis. 2014;9:129.
Racoma MJC, Calibag MKKB, Cordero CP, Abacan MAR, Chiong MAD. A review of the clinical outcomes in idursulfase-treated and untreated Filipino patients with mucopolysaccharidosis type II: data from the local lysosomal storage disease registry. Orphanet J Rare Dis. 2021;16:323.
Fiorenza MT, Moro E, Erickson RP. The pathogenesis of lysosomal storage disorders: beyond the engorgement of lysosomes to abnormal development and neuroinflammation. Hum Mol Genet. 2018;27:R119–29.
Bigger BW, Begley DJ, Virgintino D, Pshezhetsky AV. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol Genet Metab. 2018;125:322–31.
Bhalla A, Ravi R, Fang M, Arguello A, Davis SS, Chiu C-L, et al. Characterization of fluid biomarkers reveals lysosome dysfunction and neurodegeneration in neuronopathic MPS II patients. Int J Mol Sci. 2020;21:5188.
Salvalaio M, D’Avanzo F, Rigon L, Zanetti A, D’Angelo M, Valle G, et al. Brain RNA-Seq profiling of the mucopolysaccharidosis type II mouse model. Int J Mol Sci. 2017;18:E1072.
Varga E, Nemes C, Bock I, Varga N, Fehér A, Dinnyés A, et al. Generation of mucopolysaccharidosis type II (MPS II) human induced pluripotent stem cell (iPSC) line from a 1-year-old male with pathogenic IDS mutation. Stem Cell Res. 2016;17:482–4.
Casamassa A, Zanetti A, Ferrari D, Lombardi I, Galluzzi G, D’Avanzo F, et al. Generation of an induced pluripotent stem cells line, CSSi014-A 9407, carrying the variant c.479C>T in the human iduronate 2-sulfatase (hIDS) gene. Stem Cell Res. 2022;63:102846.
Rybová J, Ledvinová J, Sikora J, Kuchař L, Dobrovolný R. Neural cells generated from human induced pluripotent stem cells as a model of CNS involvement in mucopolysaccharidosis type II. J Inherit Metab Dis. 2018;41:221–9.
Kobolák J, Molnár K, Varga E, Bock I, Jezsó B, Téglási A, et al. Modelling the neuropathology of lysosomal storage disorders through disease-specific human induced pluripotent stem cells. Exp Cell Res. 2019;380:216–33.
Hong J, Cheng Y-S, Yang S, Swaroop M, Xu M, Beers J, et al. iPS-derived neural stem cells for disease modeling and evaluation of therapeutics for mucopolysaccharidosis type II. Exp Cell Res. 2022;412: 113007.
Hendriksz CJ, Muenzer J, Vanderver A, Davis JM, Burton BK, Mendelsohn NJ, et al. Levels of glycosaminoglycans in the cerebrospinal fluid of healthy young adults, surrogate-normal children, and Hunter syndrome patients with and without cognitive impairment. Mol Genet Metab Rep. 2015;5:103–6.
Shapiro EG, Eisengart JB. The natural history of neurocognition in MPS disorders: a review. Mol Genet Metab. 2021;133:8–34.
Yund B, Rudser K, Ahmed A, Kovac V, Nestrasil I, Raiman J, et al. Cognitive, medical, and neuroimaging characteristics of attenuated mucopolysaccharidosis type II. Mol Genet Metab. 2015;114:170–7.
Manara R, Priante E, Grimaldi M, Santoro L, Polonara G, Parini R, et al. Closed Meningo(encephalo)cele: a new feature in Hunter syndrome. AJNR Am J Neuroradiol. 2012;33:873–7.
Nan H, Park C, Maeng S. Mucopolysaccharidoses I and II: brief review of therapeutic options and supportive/palliative therapies. BioMed Res Int. 2020;2020:2408402.
Scarpa M, Bellettato CM, Lampe C, Begley DJ. Neuronopathic lysosomal storage disorders: approaches to treat the central nervous system. Best Pract Res Clin Endocrinol Metab. 2015;29:159–71.
Hampe CS, Yund BD, Orchard PJ, Lund TC, Wesley J, McIvor RS. Differences in MPS I and MPS II disease manifestations. Int J Mol Sci. 2021;22:7888.
Muenzer J, Beck M, Eng CM, Escolar ML, Giugliani R, Guffon NH, et al. Multidisciplinary management of Hunter syndrome. Pediatrics. 2009;124:e1228-1239.
Eisengart JB, King KE, Shapiro EG, Whitley CB, Muenzer J. The nature and impact of neurobehavioral symptoms in neuronopathic Hunter syndrome. Mol Genet Metab Rep. 2020;22: 100549.
Critchley BJ, Gaspar HB, Benedetti S. Targeting the central nervous system in lysosomal storage diseases: strategies to deliver therapeutics across the blood–brain barrier. Mol Ther J Am Soc Gene Ther. 2023;31:657–75.
Calias P. Drug delivery to the CNS. Drug Deliv Transl Res. 2012;2:143–4.
Tosi G, Duskey JT, Kreuter J. Nanoparticles as carriers for drug delivery of macromolecules across the blood–brain barrier. Expert Opin Drug Deliv. 2020;17:23–32.
Fratantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science. 1968;162:570–2.
Kim C, Seo J, Chung Y, Ji H-J, Lee J, Sohn J, et al. Comparative study of idursulfase beta and idursulfase in vitro and in vivo. J Hum Genet. 2017;62:167–74.
Brady RO, Tallman JF, Johnson WG, Gal AE, Leahy WR, Quirk JM, et al. Replacement therapy for inherited enzyme deficiency. N Engl J Med. 1973;289:9–14.
Brady RO, Pentchev PG, Gal AE, Hibbert SR, Dekaban AS. Replacement therapy for inherited enzyme deficiency. N Engl J Med. 1974;291:989–93.
Bradley LA, Haddow HRM, Palomaki GE. Treatment of mucopolysaccharidosis type II (Hunter syndrome): results from a systematic evidence review. Genet Med. 2017;19:1187–201.
Rabiee N, Ahmadi S, Afshari R, Khalaji S, Rabiee M, Bagherzadeh M, et al. Polymeric nanoparticles for nasal drug delivery to the brain: relevance to Alzheimer’s disease. Adv Ther. 2021;4:2000076.
Urayama A, Grubb JH, Sly WS, Banks WA. Developmentally regulated mannose 6-phosphate receptor-mediated transport of a lysosomal enzyme across the blood–brain barrier. Proc Natl Acad Sci USA. 2004;101:12658–63.
Gauthier C, El Cheikh K, Basile I, Daurat M, Morère E, Garcia M, et al. Cation-independent mannose 6-phosphate receptor: from roles and functions to targeted therapies. J Control Release Off J Control Release Soc. 2024;365:759–72.
Horgan C, Jones SA, Bigger BW, Wynn R. Current and future treatment of mucopolysaccharidosis (MPS) type II: is brain-targeted stem cell gene therapy the solution for this devastating disorder? Int J Mol Sci. 2022;23:4854.
Muenzer J, Bodamer O, Burton B, Clarke L, Frenking GS, Giugliani R, et al. The role of enzyme replacement therapy in severe Hunter syndrome-an expert panel consensus. Eur J Pediatr. 2012;171:181–8.
Scarpa M, Almássy Z, Beck M, Bodamer O, Bruce IA, De Meirleir L, et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72.
Sands MS, Davidson BL. Gene therapy for lysosomal storage diseases. Mol Ther. 2006;13:839–49.
Christensen CL, Ashmead RE, Choy FYM. Cell and gene therapies for mucopolysaccharidoses: base editing and therapeutic delivery to the CNS. Dis Basel Switz. 2019;7:47.
Sohn YB, Lee J, Cho SY, Kim SJ, Ko A-R, Nam MH, et al. Improvement of CNS defects via continuous intrathecal enzyme replacement by osmotic pump in mucopolysaccharidosis type II mice. Am J Med Genet A. 2013;161A:1036–43.
Miebach E. Management of infusion-related reactions to enzyme replacement therapy in a cohort of patients with mucopolysaccharidosis disorders. Int J Clin Pharmacol Ther. 2009;47(Suppl 1):S100-106.
Burton BK, Whiteman DAH, HOS Investigators. Incidence and timing of infusion-related reactions in patients with mucopolysaccharidosis type II (Hunter syndrome) on idursulfase therapy in the real-world setting: a perspective from the Hunter Outcome Survey (HOS). Mol Genet Metab. 2011;103:113–20.
Chan M-Y, Nelson AJ, Ngu L-H. Long-term experience with idursulfase beta (Hunterase) in two adolescent patients with MPS II: a case series. Mol Genet Metab Rep. 2023;36: 100991.
Ngu L-H, Ong Peitee W, Leong HY, Chew HB. Case report of treatment experience with idursulfase beta (Hunterase) in an adolescent patient with MPS II. Mol Genet Metab Rep. 2017;12:28–32.
Parini R, Rigoldi M, Tedesco L, Boffi L, Brambilla A, Bertoletti S, et al. Enzymatic replacement therapy for Hunter disease: up to 9 years experience with 17 patients. Mol Genet Metab Rep. 2015;3:65–74.
Muenzer J, Beck M, Eng CM, Giugliani R, Harmatz P, Martin R, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med Off J Am Coll Med Genet. 2011;13:95–101.
Lampe C, Bosserhoff A-K, Burton BK, Giugliani R, de Souza CF, Bittar C, et al. Long-term experience with enzyme replacement therapy (ERT) in MPS II patients with a severe phenotype: an international case series. J Inherit Metab Dis. 2014;37:823–9.
Afroze B, Brown N. Ethical issues in managing lysosomal storage disorders in children in low and middle income countries. Pak J Med Sci. 2017;33:1036–41.
Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37:13–25.
Begley DJ, Pontikis CC, Scarpa M. Lysosomal storage diseases and the blood–brain barrier. Curr Pharm Des. 2008;14:1566–80.
Wang Y, MacDonald RG, Thinakaran G, Kar S. Insulin-like growth factor-II/cation-independent mannose 6-phosphate receptor in neurodegenerative diseases. Mol Neurobiol. 2017;54:2636–58.
Kaminski D, Yaghootfam C, Matthes F, Reßing A, Gieselmann V, Matzner U. Brain cell type-specific endocytosis of arylsulfatase A identifies limitations of enzyme-based therapies for metachromatic leukodystrophy. Hum Mol Genet. 2021;29:3807–17.
Scarpa M, Orchard PJ, Schulz A, Dickson PI, Haskins ME, Escolar ML, et al. Treatment of brain disease in the mucopolysaccharidoses. Mol Genet Metab. 2017;122S:25–34.
Mizushima M, Kawabori M, Yamazaki K, Egawa K, Fujimura M. Ommaya reservoir placement using ultrasound guidance via anterior fontanelle combined with frameless electromagnetic neuronavigation in patients with mucopolysaccharidosis type 2: case reports and review of the literature. Childs Nerv Syst ChNS Off J Int Soc Pediatr Neurosurg. 2024;40(5):1603–1607.
Okuyama T, Eto Y, Sakai N, Minami K, Yamamoto T, Sonoda H, et al. Iduronate-2-sulfatase with anti-human transferrin receptor antibody for neuropathic mucopolysaccharidosis II: a phase 1/2 trial. Mol Ther J Am Soc Gene Ther. 2019;27:456–64.
Ghafoor VL, Epshteyn M, Carlson GH, Terhaar DM, Charry O, Phelps PK. Intrathecal drug therapy for long-term pain management. Am J Health-Syst Pharm AJHP Off J Am Soc Health-Syst Pharm. 2007;64:2447–61.
Patel NK, Gill SS. GDNF delivery for Parkinson’s disease. Acta Neurochir Suppl. 2007;97:135–54.
Calias P, Papisov M, Pan J, Savioli N, Belov V, Huang Y, et al. CNS penetration of intrathecal-lumbar idursulfase in the monkey, dog and mouse: implications for neurological outcomes of lysosomal storage disorder. PLoS ONE. 2012;7: e30341.
Seo J-H, Kosuga M, Hamazaki T, Shintaku H, Okuyama T. Impact of intracerebroventricular enzyme replacement therapy in patients with neuronopathic mucopolysaccharidosis type II. Mol Ther Methods Clin Dev. 2021;21:67–75.
Seo J-H, Kosuga M, Hamazaki T, Shintaku H, Okuyama T. Intracerebroventricular enzyme replacement therapy in patients with neuronopathic mucopolysaccharidosis type II: final report of 5-year results from a Japanese open-label phase 1/2 study. Mol Genet Metab. 2023;140: 107709.
Muenzer J, Hendriksz CJ, Fan Z, Vijayaraghavan S, Perry V, Santra S, et al. A phase I/II study of intrathecal idursulfase-IT in children with severe mucopolysaccharidosis II. Genet Med Off J Am Coll Med Genet. 2016;18:73–81.
Muenzer J, Burton BK, Harmatz P, Gutiérrez-Solana LG, Ruiz-Garcia M, Jones SA, et al. Intrathecal idursulfase-IT in patients with neuronopathic mucopolysaccharidosis II: results from a phase 2/3 randomized study. Mol Genet Metab. 2022;137:127–39.
Martín-Banderas L, Holgado MA, Durán-Lobato M, Infante JJ, Álvarez-Fuentes J, Fernández-Arévalo M. Role of nanotechnology for enzyme replacement therapy in lysosomal diseases. A focus on Gaucher’s disease. Curr Med Chem. 2016;23:929–52.
Goldsmith M, Abramovitz L, Braunstein H, Horowitz M, Peer D. Quantitative analysis of recombinant glucocerebrosidase brain delivery via lipid nanoparticles. Nano Futur. 2018;2: 045003.
Del Grosso A, Galliani M, Angella L, Santi M, Tonazzini I, Parlanti G, et al. Brain-targeted enzyme-loaded nanoparticles: a breach through the blood–brain barrier for enzyme replacement therapy in Krabbe disease. Sci Adv. 2019;5:eaax7462.
Rigon L, Salvalaio M, Pederzoli F, Legnini E, Duskey JT, D’Avanzo F, et al. Targeting brain disease in MPSII: preclinical evaluation of IDS-loaded PLGA nanoparticles. Int J Mol Sci. 2019;20:2014.
Tosi G, Costantino L, Rivasi F, Ruozi B, Leo E, Vergoni AV, et al. Targeting the central nervous system: in vivo experiments with peptide-derivatized nanoparticles loaded with loperamide and rhodamine-123. J Control Release Off J Control Release Soc. 2007;122:1–9.
Tosi G, Fano RA, Bondioli L, Badiali L, Benassi R, Rivasi F, et al. Investigation on mechanisms of glycopeptide nanoparticles for drug delivery across the blood–brain barrier. Nanomedicine. 2011;6:423–36.
Sato Y, Minami K, Hirato T, Tanizawa K, Sonoda H, Schmidt M. Drug delivery for neuronopathic lysosomal storage diseases: evolving roles of the blood brain barrier and cerebrospinal fluid. Metab Brain Dis. 2022;37:1745–56.
Sato Y, Okuyama T. Novel enzyme replacement therapies for neuropathic mucopolysaccharidoses. Int J Mol Sci. 2020;21:400.
Sonoda H, Morimoto H, Yoden E, Koshimura Y, Kinoshita M, Golovina G, et al. A blood–brain-barrier-penetrating anti-human transferrin receptor antibody fusion protein for neuronopathic mucopolysaccharidosis II. Mol Ther J Am Soc Gene Ther. 2018;26:1366–74.
Sonoda H, Morimoto H, Koshimura Y, Kinoshita M. Correction of central nervous system deficits in the mouse model of Hunter syndrome by recombinant iduronate 2-sulfatase crossing the blood–brain barrier. Mol Genet Metab. 2017;120:S125–6.
Okuyama T, Eto Y, Sakai N, Nakamura K, Yamamoto T, Yamaoka M, et al. A phase 2/3 trial of pabinafusp alfa, IDS fused with anti-human transferrin receptor antibody, targeting neurodegeneration in MPS-II. Mol Ther J Am Soc Gene Ther. 2021;29:671–9.
Giugliani R, Martins AM, So S, Yamamoto T, Yamaoka M, Ikeda T, et al. Iduronate-2-sulfatase fused with anti-hTfR antibody, pabinafusp alfa, for MPS-II: a phase 2 trial in Brazil. Mol Ther J Am Soc Gene Ther. 2021;29:2378–86.
Yamamoto R, Kawashima S. Pharmacological property, mechanism of action and clinical study results of pabinafusp alfa (genetical recombination) (IZCARGO® I.V. Infusion 10 mg) as the therapeutic for mucopolysaccharidosis type-II (Hunter syndrome). Nihon Yakurigaku Zasshi Folia Pharmacol Jpn. 2022;157:62–75.
Imakiire A, Morimoto H, Suzuki H, Masuda T, Yoden E, Inoue A, et al. Transferrin receptor-targeted iduronate-2-sulfatase penetrates the blood-retinal barrier and improves retinopathy in mucopolysaccharidosis II mice. Mol Pharm. 2023;20:5901–9.
Nakamura K, Sakai N, Hossain MA, Eisengart JB, Yamamoto T, Tanizawa K, et al. Analysis of caregiver perspectives on patients with mucopolysaccharidosis II treated with pabinafusp alfa: results of qualitative interviews in Japan. Orphanet J Rare Dis. 2024;19:104.
Arguello A, Meisner R, Thomsen ER, Nguyen HN, Ravi R, Simms J, et al. Iduronate-2-sulfatase transport vehicle rescues behavioral and skeletal phenotypes in a mouse model of Hunter syndrome. JCI Insight. 2021;6: e145445.
Siegal T, Rubinstein R, Bokstein F, Schwartz A, Lossos A, Shalom E, et al. In vivo assessment of the window of barrier opening after osmotic blood–brain barrier disruption in humans. J Neurosurg. 2000;92:599–605.
Salvalaio M, Rigon L, Belletti D, D’Avanzo F, Pederzoli F, Ruozi B, et al. Targeted polymeric nanoparticles for brain delivery of high molecular weight molecules in lysosomal storage disorders. PLoS ONE. 2016;11: e0156452.
Hsu Y-H, Liu R-S, Lin W-L, Yuh Y-S, Lin S-P, Wong T-T. Transcranial pulsed ultrasound facilitates brain uptake of laronidase in enzyme replacement therapy for mucopolysaccharidosis type I disease. Orphanet J Rare Dis. 2017;12:109.
Bourgoin C, Emiliani C, Kremer EJ, Gelot A, Tancini B, Gravel RA, et al. Widespread distribution of beta-hexosaminidase activity in the brain of a Sandhoff mouse model after coinjection of adenoviral vector and mannitol. Gene Ther. 2003;10:1841–9.
McCarty DM, DiRosario J, Gulaid K, Muenzer J, Fu H. Mannitol-facilitated CNS entry of rAAV2 vector significantly delayed the neurological disease progression in MPS IIIB mice. Gene Ther. 2009;16:1340–52.
Foley CP, Rubin DG, Santillan A, Sondhi D, Dyke JP, Crystal RG, et al. Intra-arterial delivery of AAV vectors to the mouse brain after mannitol mediated blood brain barrier disruption. J Control Release Off J Control Release Soc. 2014;196:71–8.
Araya K, Sakai N, Mohri I, Kagitani-Shimono K, Okinaga T, Hashii Y, et al. Localized donor cells in brain of a Hunter disease patient after cord blood stem cell transplantation. Mol Genet Metab. 2009;98:255–63.
Tanaka A, Okuyama T, Suzuki Y, Sakai N, Takakura H, Sawada T, et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: a nationwide survey in Japan. Mol Genet Metab. 2012;107:513–20.
Kubaski F, Yabe H, Suzuki Y, Seto T, Hamazaki T, Mason RW, et al. Hematopoietic stem cell transplantation for patients with mucopolysaccharidosis II. Biol blood marrow transplant. J Am Soc Blood Marrow Transplant. 2017;23:1795–803.
Suzuki Y, Taylor M, Orii K, Fukao T, Orii T, Tomatsu S. Assessment of activity of daily life in mucopolysaccharidosis type II patients with hematopoietic stem cell transplantation. Diagn Basel Switz. 2020;10:46.
Selvanathan A, Ellaway C, Wilson C, Owens P, Shaw PJ, Bhattacharya K. Effectiveness of early hematopoietic stem cell transplantation in preventing neurocognitive decline in mucopolysaccharidosis type II: a case series. In: Morava E, Baumgartner M, Patterson M, Rahman S, Zschocke J, Peters V, editors. JIMD Rep Vol 41 focus issue adults metab [Internet]. Berlin: Springer; 2018 [cited 2024 Mar 20]. p. 81–9. https://doi.org/10.1007/8904_2018_104.
Das S, Rruga F, Montepeloso A, Dimartino A, Spadini S, Corre G, et al. An empowered, clinically viable hematopoietic stem cell gene therapy for the treatment of multisystemic mucopolysaccharidosis type II. Mol Ther J Am Soc Gene Ther. 2024;S1525–0016(24):00034.
Sawamoto K, Stapleton M, Alméciga-Díaz CJ, Espejo-Mojica AJ, Losada JC, Suarez DA, et al. Therapeutic options for mucopolysaccharidoses: current and emerging treatments. Drugs. 2019;79:1103–34.
Zapolnik P, Pyrkosz A. Gene therapy for mucopolysaccharidosis type II—a review of the current possibilities. Int J Mol Sci. 2021;22:5490.
Rossi A, Brunetti-Pierri N. Gene therapies for mucopolysaccharidoses. J Inherit Metab Dis. 2024;47:135–44.
Wakabayashi T, Shimada Y, Akiyama K, Higuchi T, Fukuda T, Kobayashi H, et al. Hematopoietic stem cell gene therapy corrects neuropathic phenotype in murine model of mucopolysaccharidosis type II. Hum Gene Ther. 2015;26:357–66.
Miwa S, Watabe AM, Shimada Y, Higuchi T, Kobayashi H, Fukuda T, et al. Efficient engraftment of genetically modified cells is necessary to ameliorate central nervous system involvement of murine model of mucopolysaccharidosis type II by hematopoietic stem cell targeted gene therapy. Mol Genet Metab. 2020;130:262–73.
Cardone M, Polito VA, Pepe S, Mann L, D’Azzo A, Auricchio A, et al. Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene delivery. Hum Mol Genet. 2006;15:1225–36.
Jung S-C, Park E-S, Choi EN, Kim CH, Kim SJ, Jin D-K. Characterization of a novel mucopolysaccharidosis type II mouse model and recombinant AAV2/8 vector-mediated gene therapy. Mol Cells. 2010;30:13–8.
Motas S, Haurigot V, Garcia M, Marcó S, Ribera A, Roca C, et al. CNS-directed gene therapy for the treatment of neurologic and somatic mucopolysaccharidosis type II (Hunter syndrome). JCI Insight. 2016;1: e86696.
Hinderer C, Katz N, Louboutin J-P, Bell P, Yu H, Nayal M, et al. Delivery of an adeno-associated virus vector into cerebrospinal fluid attenuates central nervous system disease in mucopolysaccharidosis type II mice. Hum Gene Ther. 2016;27:906–15.
Laoharawee K, Podetz-Pedersen KM, Nguyen TT, Evenstar LB, Kitto KF, Nan Z, et al. Prevention of neurocognitive deficiency in mucopolysaccharidosis type II mice by central nervous system-directed, AAV9-mediated iduronate sulfatase gene transfer. Hum Gene Ther. 2017;28:626–38.
Laoharawee K, Podetz-Pedersen KM, Nguyen TT, Singh SM, Smith MC, Belur LR, et al. Non-invasive intravenous administration of AAV9 transducing iduronate sulfatase leads to global metabolic correction and prevention of neurologic deficits in a mouse model of Hunter syndrome. Mol Genet Metab Rep. 2023;34: 100956.
Smith MC, Belur LR, Karlen AD, Erlanson O, Furcich J, Lund TC, et al. Comparative dose effectiveness of intravenous and intrathecal AAV9.CB7.hIDS, RGX-121, in mucopolysaccharidosis type II mice. Mol Ther Methods Clin Dev. 2024;32:101201.
Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–21.
Anguela XM, Sharma R, Doyon Y, Miller JC, Li H, Haurigot V, et al. Robust ZFN-mediated genome editing in adult hemophilic mice. Blood. 2013;122:3283–7.
Sharma R, Anguela XM, Doyon Y, Wechsler T, DeKelver RC, Sproul S, et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood. 2015;126:1777–84.
Laoharawee K, DeKelver RC, Podetz-Pedersen KM, Rohde M, Sproul S, Nguyen H-O, et al. Dose-dependent prevention of metabolic and neurologic disease in murine MPS II by ZFN-mediated in vivo genome editing. Mol Ther J Am Soc Gene Ther. 2018;26:1127–36.
Harmatz P, Prada CE, Burton BK, Lau H, Kessler CM, Cao L, et al. First-in-human in vivo genome editing via AAV-zinc-finger nucleases for mucopolysaccharidosis I/II and hemophilia B. Mol Ther J Am Soc Gene Ther. 2022;30:3587–600.
Losada Díaz JC, Cepeda Del Castillo J, Rodriguez-López EA, Alméciga-Díaz CJ. Advances in the development of pharmacological chaperones for the mucopolysaccharidoses. Int J Mol Sci. 2019;21:232.
Shen J-S, Edwards NJ, Hong YB, Murray GJ. Isofagomine increases lysosomal delivery of exogenous glucocerebrosidase. Biochem Biophys Res Commun. 2008;369:1071–5.
Demydchuk M, Hill CH, Zhou A, Bunkóczi G, Stein PE, Marchesan D, et al. Insights into Hunter syndrome from the structure of iduronate-2-sulfatase. Nat Commun. 2017;8:15786.
Yue WW, Mackinnon S, Bezerra GA. Substrate reduction therapy for inborn errors of metabolism. Emerg Top Life Sci. 2019;3:63–73.
Piotrowska E, Jakóbkiewicz-Banecka J, Tylki-Szymanska A, Liberek A, Maryniak A, Malinowska M, et al. Genistin-rich soy isoflavone extract in substrate reduction therapy for Sanfilippo syndrome: an open-label, pilot study in 10 pediatric patients. Curr Ther Res. 2008;69:166–79.
de Ruijter J, Valstar MJ, Narajczyk M, Wegrzyn G, Kulik W, IJlst L, et al. Genistein in Sanfilippo disease: a randomized controlled crossover trial. Ann Neurol. 2012;71:110–20.
Giugliani R, Vairo F, Kubaski F, Poswar F, Riegel M, Baldo G, et al. Neurological manifestations of lysosomal disorders and emerging therapies targeting the CNS. Lancet Child Adolesc Health. 2018;2:56–68.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open access funding was provided by Università degli Studi di Padova within the CRUI-CARE Agreement.
Conflict of interest
Authors declare no conflicts of interest directly or indirectly related to the work presented here.
Availability of data and material
Not applicable.
Ethics approval
Not applicable.
Patient consent to participate/publish
Not applicable.
Code availability
Not applicable.
Author contributions
Both authors contributed equally to this work. Both authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Zanetti, A., Tomanin, R. Targeting Neurological Aspects of Mucopolysaccharidosis Type II: Enzyme Replacement Therapy and Beyond. BioDrugs 38, 639–655 (2024). https://doi.org/10.1007/s40259-024-00675-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-024-00675-0