
Abstract
Objectives
The aim of this review is to assess the current state of empirical research regarding the effectiveness of national pricing regulations of the patent-protected market for prescription pharmaceuticals. Effectiveness is understood to be the capacity of policies to have a desired impact on outcomes, such as health status, patient access, healthcare expenditure, and research investments, among others.
Methods
A systematic review of the published literature on pricing regulations in OECD countries was performed. The PubMed, MEDLINE, Scopus, Web of Science, Cochrane Library and the OECD iLibrary databases were searched in September 2016 and December 2017, with an update in August 2018. Interrupted time series studies and additional empirical studies were included, as well as systematic reviews if appropriate methods were applied. The risk of bias was assessed based on the recommendations of the BMJ guidelines, Cochrane EPOC criteria, QHES instrument, HTA good practice guidelines, CRD’s guidance and the CHEC criteria. The quality of evidence was evaluated using the suggestions from EPOC and GRADE.
Results
Thirty-one publications met the inclusion criteria. Most of the assessed empirical research included therapeutic (TRP) and/or external reference pricing (ERP), with a clear majority focusing on TRP. The main outcomes that were analysed were drug prices, expenditures and drug use. For value-based pricing (VBP), only limited empirical data were found.
Conclusions
We found evidence that TRP may reduce pharmaceutical prices and expenditures in the short term. Furthermore, TRP may lead to substitution effects towards lower-priced pharmaceuticals. The effects of TRP on patient access, healthcare utilisation and R&D investments were found to be uncertain. No conclusions were drawn for ERP and VBP. No evidence was found for the effects on health outcomes for any of the analysed policies. There is a strong need for evidence generation regarding effective pricing policies, particularly for VBP, managed entry agreements and non-financial outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
We found evidence that therapeutic reference pricing may reduce pharmaceutical prices and expenditures in the short term and may lead to substitution effects towards lower-priced drugs in the short term. |
Apart from the above, the existing research does not provide credible evidence that the current policies have an impact on healthcare utilisation, patient access, health outcomes or R&D investments. |
There is a strong need for evidence generation regarding effective pricing policies, particularly for value-based pricing policies and for non-financial outcomes. |
Policy makers should systematically define the assessment of policy effectiveness during the policy formulation process. In addition, experimental research on the topic could augment the scarce amount of available evidence, especially for value-based pricing policies. |
1 Introduction and Background
1.1 Description of the Policy Issue
In most OECD (Organisation for Economic Co-operation and Development) countries, laws regulate the pricing of prescription pharmaceuticals. Parliaments and governments aim to control public spending on pharmaceuticals, while securing access to innovative treatments for their citizens.
Desired policy outcomes of pricing and reimbursement regulations are focused on short-term reduction and mid-term stabilisation of health expenditures [1]. In light of the financial and economic crisis, a majority of the countries in Western Europe in recent years decreed one-off as well as permanent, cost-containment policy measures to increase the control of spending on pharmaceuticals [1, 2]. However, these policies face increasing criticism that they are not sufficiently effective, not only regarding impacts on public finances, but even more due to their potential consequences on access for patients to new treatment options [3, 4], although new procedures for early access (conditional approval) and managed entry (coverage with evidence) have been introduced in Europe to make new therapies with “unsettled benefit–risk profiles” available [5].
Currently, the calls for reform are getting louder, in light of a growing number of new molecules and increasingly complex treatments, especially in oncology. According to IMS, the R&D pipeline for oncology “has expanded by more than 60% over the past decade”, amounting to 710 late-phase therapies in 2017 [6, 7]. Knowledge of the clinical benefits of the new therapies is often scarce at the time of marketing authorisation [8], and the promised survival benefits imply a longer therapy duration, and therefore, additional budget impacts. Additionally, an increasing number of molecules are used in combination with other medications [6, 7]. Most of the national health systems have struggled to adapt to this new challenge, resulting in the reduced availability and coverage of new treatments [6].
The current regulations might have not only adverse incentives for short-term availability, they are also claimed to have further undesirable effects on the long-term research decisions of industry, reducing related investments [9, 10].
Finally, the ongoing change in the pharmaceutical sector, shifting from a “traditional blockbuster model” to more segmented markets offering therapies for smaller patient populations, leads to the discussion among authorities of whether current national policy approaches set the right incentives for industry to optimise research towards “value for money” [1, 11, 12]. In general, new drugs entering an established market can address previously unmet needs or target new populations for existing therapies (e.g. paediatric) and, consequently, increase treatment options [1]. While new therapies with high cure rates (e.g. for hepatitis) may be considered as cost effective even at high nominal costs per patient and month, other innovations may seem hardly worth a premium compared to existing standards [13]. Most recently, OECD classified “many high-cost cancer drugs” as low-value (“type C”) technologies, if value is “based solely on the cost per life year or per quality-adjusted life year” [13]. From a health system perspective, there is a clear need for the evolvement of current reimbursement policies in industrialised countries to better reflect “value for money” of new treatments [14]. This calls for pricing regulations that promote fast access to essential innovations, while not incentivising investments in “me-too” compounds that offer no additional benefit. The challenging part of such policy reforms towards a more value-based assessment is not only a technical but also a normative one: how to find consensus on which technologies are “essential” (funded on a system level) and which are “nice to have” (individual funding). The discussion on how to measure value for money and whether the new pricing schemes are increasing is still ongoing [15,16,17]. Figure 1 provides an overview of our policy analysis, summarising the most common pricing policies and the most discussed policy outcomes. The latter were defined with consideration to recent policy papers from the WHO and the OECD, as well as existing reviews of the topic [1, 12, 18,19,20,21,22]. We refined the effect taxonomy after the first abstract screenings. The differentiation between intermediate (behaviour, result) and ultimate policy effects represents our own proposal, which reflects the screened study designs.

Layout of pharmaceutical pricing policy analysis. *Includes affordability and availability (supply). MAH marketing authorisation holder, P&R pricing and reimbursement
1.2 Description of Current Policy Designs to Address the Policy Issue
The most common pricing policies for pharmaceuticals in industrialised countries are therapeutic (internal) reference pricing, international (external) reference pricing and value-based assessments. These are generally combined with positive reimbursement lists (formularies), regular price reviews (individual or in groups) and tendering. These direct pricing policies are often supplemented with measures on the demand side to set desired incentives for prescribers, pharmacies and patients. Finally, authorities can link reimbursement authorisations to managed entry agreements, including coverage with evidence obligations and pay-for-performance schemes. The definitions below closely follow the suggestions of the “Glossary of Pharmaceutical Terms” issued by the WHO Collaborating Centre (CC) for Pharmaceutical Pricing and Reimbursement Policies.
1.2.1 Therapeutic (Internal) Reference Pricing (TRP)
TRP is defined as the governmental practice of using the prices of identical (ATC 5 level) or similar medicines (ATC 4) or therapeutically equivalent treatments (beyond medicines) in a country. One or a set of therapies are used as benchmark or reference to set or negotiate the reimbursable price of a product in a given country [20, 23]. The concrete mechanics can differ between the national systems (average or lowest of “equal” treatments, weighted for multiple indications, or not, measured per cycle, month or year). TRP can also be combined with other pricing instruments, such as external reference pricing (ERP), and may also allow for an “innovation premium” for added therapeutic value (both are currently the case in Switzerland, based on Art. 65b KVV [24]). TRP sets the reimbursable price and thus implies that a third party, normally the patient (or insurance), has to cover the costs beyond the fixed price.
1.2.2 International (External) Reference Pricing (ERP)
ERP is the practice of using the prices of a medicine in one or several countries to derive a benchmark or reference to set or negotiate the price of a product in a given country [20, 23]. All but two EU member countries (Sweden, UK) have implemented some type of ERP [15]. While the policy intends to affect the national market of the issuing state, its external effects are the subject of debate, since marketing authorisation holders are incentivised to launch new therapies in countries with higher prices first and delay in others [15].
1.2.3 Value-Based Pricing (VBP)
The value-based assessment for pharmaceutical pricing is also referred to as “cost effectiveness-based”, “outcome-based” or “pharma economic” pricing. There is still “no widely accepted definition” of the value part of this instrument [23]. Following the WHO glossary, when referring to VBP, we mean the practice of authorities of “setting prices for new medicines and/or deciding on reimbursement based on the therapeutic value which the medicine offers, usually assessed through a health technology assessment (HTA)”. VBP can be combined with cost-effectiveness thresholds, such as in Sweden or the UK, to define a range for the acceptable price of a treatment. VBP agreements are predestined for combination with managed entry schemes (see below), such as the Patient Access Scheme (PAS) in the UK, which allows the manufacturer to adjust the offer to the authorities (bringing the ICER down). However, most managed entry agreements (MEAs) and PASs are still volume based and not value based [1, 2, 23, 25, 26].
1.2.4 Managed Entry Agreements (MEAs)
A MEA is an “arrangement between a manufacturer and a payer/provider that enables access to (coverage/reimbursement of) a health technology subject to specified conditions” [27]. It has established itself as the overarching term for arrangements such as “performance-based schemes”, “risk-sharing agreements” (RSAs), “patient access schemes” (PASs) or “coverage with evidence” schemes. MEAs aim to address uncertainties of new health technologies regarding clinical performance, cost effectiveness, effective use or long-term budget impacts [23, 27].
While some MEAs focus on real-world evidence collection after the market authorisation, others are set up as profit controls, volume-based agreements or other forms of predefined (“capped”) quantities or annual expenses for a new treatment. According to the ISPOR taxonomy, “cost-sharing” arrangements fall outside the definition of performance-based risk-sharing arrangements (PBRSA), since they are not linked to underlying cost effectiveness [28]. While we do not limit our analysis to “financial schemes” [26], our focus lies on MEAs as part of or linked to pharmaceutical pricing regulations, following the WHO definition [23] (see inclusion criteria below under Sect. 2.1).
1.2.5 Other Pricing and Reimbursement Policies
In addition, the simple instrument of issuing a positive (or negative) reimbursement list is a widely used component of pharmaceutical pricing regulation. Further, in several countries, the official reimbursement label is subject to negotiations for official reimbursement authorisation. In addition to price, it often defines additional limitations of reimbursable use, e.g. the eligible population, sometimes including mandatory tests to prove eligibility, subject to approval by a medical examiner (insurance physician). This practice was recently applied to control costs of the new hepatitis C therapies, which led to a controversial discourse on politics and health sciences [1, 13, 29].
Finally, an interesting complement to TRP, VBP and other pricing policies under discussion is “indication-based pricing”, which aims to “capture the differential value of drugs across indications or patient groups” [25]. Thus far, this has been applied to only a very few special therapies across the European countries [30]. One example is bevacizumab, which in Switzerland has the same official list price for all of its seven reimbursable indications. However, for two of these, the official reimbursement label defines an indication-specific (renal cell carcinoma and breast cancer) payback that is reimbursable by the health insurance of the treated patient [31]. One common challenge for indication-based pricing is that existing administrative structures in many countries are not designed to “track patients’ indications” for reimbursement purposes [32].
1.3 Why This Review is Important
Since the 1990s, scientific interest in pharmaceutical pricing regulation has steadily increased, with a clear rise in publications in PubMed since 2013 [33]. In recent years, there have been a few systematic reviews published on existing evidence regarding pharmaceutical pricing regulation. Most of these reviews are limited to specific policies and have not been updated to reflect the most recent studies published on various health system and policy changes, especially regarding value-based pricing. The Cochrane Library has published seven systematic reviews in the research area in which we are interested. The review by Acosta et al. [20] entitled “Pharmaceutical policies: effects of reference pricing, other pricing, and purchasing policies” was found to be a very comprehensive, but was also a very restrictive, structured review.
Acosta et al. included only interrupted time series (ITS) and repeated measures (RM) studies. A more inclusive analysis of the existing body of research is necessary to provide a broader picture on current state of the discourse, which often takes place in the political arena. Otherwise, the analysis might reach an exclusive expert audience only. Finally, and most importantly, the previous reviews did not focus on the patent-protected market and did not simultaneously include all relevant policies. Most studies actually did the opposite—including or focusing on the off-patent market, while limiting the analysis to a single or a few selected policies. Against the background of the mentioned challenges for financing and incentivising new, innovative health technologies, we consider the inverse focus to be essential.
1.4 Objectives
The aim of this review is to assess and summarise the current state of empirical research on the effectiveness of national pricing regulations on the patent-protected market for prescription pharmaceuticals. Effectiveness is understood to be the capacity of policies to have a desired impact on outcomes such as health state, patient access, drug use, healthcare utilisation, drug prices, healthcare expenditure, research investment, individual or organisational benefit, overall welfare, behaviour other than drug use and equity.
The study aims to contribute to the discourse around reform options of pricing regulations for new (“innovative”) pharmaceuticals in developed health systems with established public policies. Therefore, the analysis focuses on high-income OECD countries. There is undoubtedly a high need for health systems and related pharmaceutical policies to evolve in developing countries. However, the majority of the respective policy settings and the data and evidence availability and quality were rated to not be supportive for the overarching research aims of this review.
2 Methods
2.1 Inclusion Criteria for Studies
2.1.1 Type of Studies
Controlled before-after studies (CBA), interrupted time series studies (ITS) and repeated measures studies (RM) were included. Inclusion was generally based on the recommendations from the Effective Practice and Organisation of Care (EPOC) for review authors [34]. However, to gain a broader picture of the current research, empirical studies not fulfilling the EPOC criteria were included if appropriate quantitative data and methods for inductive statistics were used. Additionally, existing systematic reviews were included if appropriate methodologies were applied.
2.1.2 Type of Policy Designs
National pricing regulations for patent-protected pharmaceuticals, including international (external) reference pricing, TRP, VBP (based on clinical and pharma economic data), MEA, positive reimbursement lists (formularies), and regular price reviews (individual or in groups) were included if they were linked to price decisions. Not included were studies focusing on demand-side policies only, such as co-payments (patients), prescription quotas or budgeting (physicians), and substitution and margins (pharmacists).
Our definition of “pricing policies” follows the Glossary of Pharmaceutical Terms, published by the WHO CC for Pharmaceutical Pricing and Reimbursement Policies: “Regulations and processes used by government authorities to set the price of medicine as part of exercising price control” [23]. Based on this definition, we excluded analyses of MEAs not linked to price setting.
In contrast to the definition of the WHO CC and the Cochrane review by Acosta et al. [20], our scope was limited to official pricing policies issued by public authorities (i.e. national parliaments or governments, but not regulations issued by single health plans). Furthermore, effects of national pricing policies were considered only if they were realised in the addressed (sovereign) territory. In consequence, impact assessments of local policies on other countries, especially of ERP, were excluded. We acknowledge, however, that international effects are indirectly affecting the performance of national pricing policies [35].
2.1.3 Type of Outcome Measures
Outcomes of interest were policy effects such as health state (individual/societal), patient access, drug use, healthcare utilisation, drug prices, healthcare expenditure, research and development (R&D) investments, welfare (economic benefit for an individual or organisation, along with overall social welfare), behaviour other than drug use and equity (see Fig. 1).
2.1.4 Scope of the Studies: Health Systems Considered
Only research focusing on “high-income” (World Bank classification) OECD countries was included, considering the historical changes of countries in their classification [36]. The reason for the defined scope is laid out above (Sect. 1.4).
2.2 Search Methods
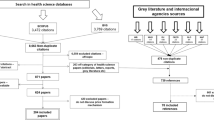
Published studies were identified by an electronic database search using the following sources: PubMed, MEDLINE, Scopus, Web of Science, Cochrane Library, and the OECD iLibrary. Searches with different word variations and combinations of “pharmaceutical” and “pricing” or “prices”, as well as “policies” and “regulation” were performed. Publications on “reference pricing”, “value-based pricing” and combinations with “patient access” and “innovative” were also searched. Further, a supplementary check on publications on “managed entry” or “risk sharing” agreements not flagged with “pricing” keywords was performed. The complete search strategy is listed in Supplementary Appendix 1, Table 3.
The searches were performed in September 2016, December 2017 and August 2018 (update of PubMed, Scopus and Web of Science search) and were restricted to records published in 2005 or later. No additional filters for language or quality of evidence (e.g. “peer-reviewed journal only”) were applied at this stage. Only duplicate records (394) were excluded for initial abstract screening. Four additional publications not identified through the keyword search were added manually for abstract screening, since they were cited by at least one of the included systematic reviews on the topic.
2.3 Data Extraction and Analysis
As revealed in the PRISMA study flow [37] in Fig. 2, the review was performed in five main phases: (a) identification (electronic database searches), (b) abstract screening, (c) research categorisation (full text), and (d) assessment of risk of bias with (e) subsequent qualitative synthesis. The first author (DJW) conducted the literature search and extracted the articles for initial abstract screening and research categorisation. When articles met any of the exclusion criteria, they were excluded from further assessment. If the assignment was ambiguous or unclear, the first author (DJW) consulted with the second author (SB) to reach an agreement.

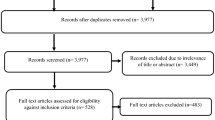
Study flow (PRISMA diagram). *Four studies were added manually because they were cited by at least one of the included systematic reviews. DB databases
In the abstract screening, the exclusion criteria (Fig. 2) were tested sequentially. If no exclusion criteria were met, then the publication was included in the research categorisation step (c).
In the research categorisation step (c), the full texts of the selected publications were analysed, and they were classified according to “research type” (16 types, see Supplementary Appendix 1, Table 4) and “research objective” (see Supplementary Appendix 1, Table 5). If the publication was not classified as one of the six predefined research types, it was excluded. At the same time, the research objective of the publication was assigned to one of 24 predefined research objective pairs (see Table 5 in Supplementary Appendix 1, based on the policy analysis layout in Fig. 1). Studies were excluded if pricing policy was not the independent variable. Further, if no policy change was analysed (no change over time or no comparison with another country or district with a different policy setting), then the study was categorised as “descriptive” and was excluded.
For poster abstracts included after screening, research categorisation was performed on the available abstract text. If, based on the available information, no exclusion criteria were applicable, then the abstract was included in the risk of bias assessment and qualitative synthesis.
2.4 Assessment of Risk of Bias
The assessment of the risk of bias was based on a synthesis of existing tools. The reason for this was that none of the commonly used tools were designed for an inclusive analysis of all of the types of research included in this analysis: empirical research, analytical microeconomic models and systematic literature reviews. The most common tools were designed for health interventions and had to be adjusted for health policy designs. The integrated questionnaire was based on a selection and synthesis of the recommendations from:
-
the BMJ guidelines [38] and a later, additional questionnaire by Drummond et al. [39],
-
the Cochrane EPOC criteria [40],
-
the Quality of Health Economic Studies (QHES) instrument [41],
-
the good practice guidelines for modelling in HTA by Philipps et al. [42],
-
the CRD’s guidance for reviews [43] and the criteria list from the Consensus on Health Economic Criteria (CHEC) project [44].
The questionnaire comprised a total of 18 questions, four on the study design (S1–S4), four on the data source and handling (D1–D4), three for the analytical modelling (M1–M3) and seven on the empirical analysis, synthesis and conclusions (E1–E7). The full questionnaire is given in Supplementary Appendix 1, Table 7.
For each publication and each question, the risk of bias was assessed as either low, unclear or high, following the EPOC suggestions [40]. Since the EPOC criteria were integrated with other assessment questionnaires, the scoring recommendation had to be adapted. In general, the score “unclear” was used restrictively. This was a necessary adaption for the rating of policy “interventions” compared to specific health interventions. If a question could not be answered since it was “not specified in paper” [40], the risk was rated high. The rating “unclear” was assigned only if credible arguments against relevant bias of the conclusion were indicated in the publication. For example, D4 (intervention dependent on other changes) was only rated low if it was plausible that the stated impact of the policy on the defined outcome was not influenced by other changes. It was rated unclear if efforts were displayed to control one or some of the most important confounders (e.g. other pharmaceutical policies implemented in parallel, other relevant healthcare policy measures in parallel, changes of the relevant market structure, relevant trends in technology, or relevant macroeconomic factors on the distribution or demand side).
Missing information from assessed poster abstracts was rated with a high risk of bias.
Low was scored with 2 points, unclear with 1 and high with 0. The final SCORE per record was the ratio of scored points to the total possible points, in percent. With the risk of bias defined as 1-SCORE, the grouping was set as: 0% no risk, 1%–19% low, 20%–39% moderate, 40%–59% medium, and 60%–100% high risk of bias.
2.5 Synthesis of Results
For each of the assessed publications all policy-outcome pairs analysed were listed and grouped into the four main policy designs (TRP, ERP, VBP, other), as described in Sect. 1.2. The results per pair and publication were assigned to one of the following six policy-outcome effects:
-
positive (↑),
-
negative (↓),
-
contradictory/inconclusive (↕),
-
effect exists (different directions possible, →),
-
no effect (X),
-
no conclusion (–).
The effects were contrasted with the risk of bias for the respective analysis for a qualitative synthesis per policy-outcome pair. We used the GRADE method [45] to rate the quality of evidence (high, moderate, low, very low), and based on that, followed the EPOC guidelines [46] to report the effect per pair.
3 Results
3.1 Included Studies
A total of 31 publications met the inclusion criteria, and none were classified as CBA or RM. Eight publications were based on interrupted time series studies (ITS), and seven were included because they met the requirements for quantitative data and methods. A further four publications presenting analytical models were included, since empirical validation was performed. Finally, 12 systematic reviews were also included.
Included ITS: Armeni et al. 2016, Augurzky et al. 2009, Barros et al. 2010, Grootendorst et al. 2005, Grootendorst et al. 2006, Morgan et al. 2008, Stargardt 2010, Yfantopoulos 2007 [47,48,49,50,51,52,53,54].
Additional empirical research included (inductive statistics but no qualified ITS): Golec et al. 2010, Kalo et al. 2012, Kanavos et al. 2011, Kyle 2007, Leopold et al. 2012, Stargardt 2011, von der Schulenburg et al. 2011 [10, 55,56,57,58,59,60].
Included analytical models with empirical validation: Atella et al. 2012, Comanor et al. 2018, Kaiser et al. 2014, Koenig et al. 2011 [61,62,63,64].
Included systematic reviews: Acosta et al. 2014, Espin et al. 2011, Galizzi et al. 2011, Green et al. 2010, Lee et al. 2012, Lee et al. 2015, Morgan et al. 2009, Puig-Junoy 2005, Relakis et al. 2013 (poster abstract), Rémuzat et al. 2015, van der Gronde et al. 2017, Zhou et al. 2014 (poster abstract) [19,20,21,22, 65,66,67,68,69,70,71,72].
Table 1 provides an overview of the included studies.
3.2 Excluded Studies
Of the 871 publications included for initial abstract screening, 666 did not meet the inclusion criteria. Most of them (281) did not focus on pharmaceutical pricing regulations according to the definition. A total of 169 did not focus on OECD and/or on high-income countries; 82 focused on the off-patent market. Of the 205 records included for full-text research categorisation, 174 were excluded because they did not analyse a policy change (134) and/or did not classify as an appropriate research type (147). Full details on every screened record and summary per criteria are given in Supplementary Appendix 2, Table 9.
The main reasons for the exclusion of empirical studies, which were included in other systematic reviews on the topic (included in this review as well), were that they were either published before the defined time frame (23), did not meet our definition of pricing policy (17), or focused on the off-patent (4) or free-pricing (2) market. Details are displayed in Supplementary Appendix 1, Table 6.
3.3 Summary of Risk of Bias
Study design: For all the non-ITS empirical studies and empirically verified theoretical studies, the risk of bias was rated high. The main reasons for this were the lack of measures to control relevant underlying trends and confounding variables and limited time points before and after the analysed policy change. Further, the policy intervention for some of the studies was not clearly defined [73,74,75,76]. For example, Kaiser et al. defined the policy intervention as a “change from external to internal reference pricing” in Denmark in 2005 [62], while Atella et al. bundled “regulatory regimes” with or without price controls for the USA and Italy [61]. In the latter, since effective (potentially interacting) policies remained unspecified, and no indication and justification for the data range was given, the risk of bias for the section study design was rated as high. Golec/Vernon investigated the impact of overall price controls on R&D spending and profitability of EU versus US firms [10]. However, the basic design lacks plausible methods to isolate the policy effects from confounding drivers for R&D spending or stock market returns.
Data: Most studies did not report techniques, evidence or plausible arguments to prevent outcomes from being influenced by other confounding variables during the analysed period [40]. Both Grootendorst/Steward and Morgan/Cunningham analysed the implementation of TRP policies in British Columbia [51, 52]. The first study reports a negative (decreasing) effect on drug expenditures, while the latter concluded that there was no impact on local R&D spending. While the risk of bias for the analysis of Grootendorst/Steward was rated low overall, they admit that the introduction of low-cost generics during the analysed period could not be adequately controlled for [51]. Morgan/Cunningham neither control for nor discuss changes in the market in their analysis. Differences between the two systems regarding market structure (e.g. R&D intensity, companies entering/leaving the market) were neither controlled for, nor disclosed or discussed [52].
Empirical analysis: Disaggregated results were missing for some of the studies and for some of the reviews. Additionally, some of the conclusions were enriched with opinion statements that were not founded on the analysed data and presented results. Yfantopoulos concludes that the introduction of positive list pricing in Greece was not effective in controlling pharmaceutical expenditures [54]. However, the underlying parameter is not significant, and many more details on the expenditure data that were used would be needed to reduce the risk of bias. The literature review from Espin/Rovira/Labry and that of Rémuzat et al. lacked an assessment of the evidence quality and risk of bias [19, 70]. For both, it remains unclear whether the summary and the reported conclusions were based on the performed systematic review; additionally, no potential limitations of the analysis were disclosed.
Overall risk: The average risk of bias for the included ITS studies was rated low. Only the analysis from Yfantopoulos [54] left concerns, since information was missing on the independence of the effects from other changes, as well as on the aggregation of data and basis for conclusions, resulting in a medium risk of bias. The included analytical models with empirical verification scored lower, with moderate risk, mainly due to leaving doubts regarding the independence of the analysed changes and due to the lack of data reported on a disaggregated level. The non-ITS empirical studies were assessed only partly, since their risk of bias was judged as high after the initial assessment of the study design and data selection (except Kyle and Stargardt 2011, which was medium [57, 59]). Most of the selected systematic reviews had a credible low risk of bias, with the exception of Rémuzat [70], Espin [19] and van der Gronde [72]. In Espin et al., more than half of the included studies were classified as opinions, while information on the included research designs was found to be incomplete. For the analysis of Relakis [69] and Zhou [71], no full results completing the published poster abstracts were found. Van der Gronde et al. [72] provided a comprehensive and up-to-date overview on the topic of pharmaceutical pricing; however, their paper did not disclose relevant information on the exclusion criteria (screening, eligibility), criteria for additional inclusions, the chosen time period, and selection criteria for newspapers. Furthermore, no assessment of the risk of bias or the method of synthesis was disclosed (see PRISMA recommendations [37]), and central statements were either based on a single publication or not referred at all. An overview of the assessment of the risk of bias is provided in Supplementary Appendix 1, Table 8.
3.4 Effects of the Policy Changes and Quality of Evidence
Table 2 provides an overview of the synthesised effects of the policy changes reported in the assessed empirical publications, as well as in the analysed systematic reviews. We found result statements for eight outcome categories. The only outcome category completely missing in the assessed publications was “equity”. No research intention was formulated for 7 potential policy-outcome pairs, leading to 17 potential effects for synthesis. A conclusive statement could be derived for only 11 pairs.
TRP on drug prices: Five of the empirical studies and six systematic reviews performed an analysis of the impact of TRP on drug prices. Three of the original studies found evidence for prices to decrease after the introduction of TRP, one study reported inconclusive results (increase as well as decrease) and one study did not find an impact. Four of the reviews concluded that the introduction of TRP was likely to have a negative (reducing) impact on prices, while one publication reported “minimal” impact, with no details. For a general statement of the respective correlations, the evidence quality was rated low. We conclude that TRP may reduce drug prices [22, 48, 56, 59, 60, 62, 65, 66, 68, 71, 72].
TRP on healthcare expenditures: Seven of the empirical studies and six systematic reviews performed a respective analysis. Four of the original studies reported reduced reimbursed expenditures. One study stated a transitory reduction with no impact on the trend. Furthermore, one study found that TRP did not reduce expenditures, and a recent (2016) analysis concluded that TRP leads to higher expenditures. Of the systematic reviews, all five reported that TRP reduced the amount of drug expenditures. However, the definition of savings and expenditures was heterogeneous, and uncertainties on sustainability of the effects were reported. Although the average risk of bias was moderate, the effect was ambiguous, and for half of the studies the evidence was limited to a specific therapy or substance. In consequence, the overall quality of evidence for a general statement was rated as low for short-term expenditures and very low for mid-term expenditures. We conclude that TRP may reduce drug expenditures in the short term, while the long-term effect is uncertain [20, 22, 47, 49,50,51, 53, 55, 62, 65,66,67, 71]. This finding is in line with that of Acosta et al. [20].
TRP on drug use: Four of the empirical studies and five systematic reviews reported a respective analysis. All of the publications reported, in some form, shifts, switches or substitutions due to TRP between reference and referenced drugs. Very limited information and evidence was reported on overall drug use for the affected therapies. The empirical studies were limited to four therapeutic/pharmacological subgroups: ACE inhibitors, calcium channel blockers, nonsteroidal anti-inflammatory drugs (ATC level 3) and statins (ATC level 4). Consequently, the overall quality of evidence for a general statement was found to be low, although the average risk of bias was low for the reported substitution effects. We found that for a “moderate” rating, broader empirical research is needed that includes different therapies [20, 22, 50, 51, 53, 62, 66, 67, 71].
For TRP and the outcomes healthcare utilisation, patient access and R&D investment, the quality of evidence was rated very low, and we conclude that it is uncertain whether a respective policy impact exists. No evidence was reported for effects on health outcome or overall welfare.
As displayed in Table 2, the body of research was markedly limited for ERP and very limited for VBP policies. We found the negative impact of both policies on prices and patient access to be uncertain, similar to the impact of ERP on research investments. For all other possible outcomes, no clear conclusion (or no conclusion) was reported in the analysed literature.
Further effects were reported for other policies such as administered price reductions (Spain) [49], positive list pricing (Greece) [54], price freeze (Germany) [59] or profit controls [60]. A Cochrane review from 2010 found that “Implementing restrictions to coverage and reimbursement of selected medications can decrease third-party drug spending without increasing the use of other health services” based on the results from six ITS studies [21]. We found no evidence for impacts from MEA in the context of pricing policies. There is a growing interest, with a corresponding increase of publications, on “use, advantages and disadvantages” [77], while evidence on these outcomes remains scarce [26, 78, 79].
4 Conclusions and Discussion
OECD countries have implemented numerous policy reforms on pharmaceutical pricing in the last decade [1]. The scientific evidence of whether these policy adjustments have the desired or unwanted effects on debated outcomes is, however, still scarce. In particular, for patent-protected pharmaceuticals (“new innovative technologies”), qualified evidence on policy effects remains very limited. The majority of empirical studies and reviews included in this review focused on therapeutic reference pricing. We found evidence that TRP may reduce pharmaceutical prices and expenditures in the short-term and may lead to substitution effects towards lower-priced drugs in the short term. Apart from that, the existing research does not provide certain evidence that the current policies have an impact on healthcare utilisation, patient access, health outcomes or R&D investments.
The aim of this paper was to analyse qualified empirical research on the effectiveness of existing policies. It was not the aim to perform an impact analysis of the existing research on current policy debates. To consider potential reasons for the scarce evidence that we found, we investigated the inclusion of the analysed research in selected topical policy publications from the OECD and the European Union. Of the 205 publications included in our initial research categorisation, only two (Paris et al. [12] and Vogler et al. [80], both qualified as “descriptive”) were referenced in the recent 231-page OECD paper on “New Health Technologies: Managing Access, Value and Sustainability” [13]. Additionally, the very recent report from the European Commission (Expert Panel on Effective ways of Investing in Health EXPH) on “Innovative payment models for high-cost innovative medicines” [14] refers only to one study (again, Paris et al. [12]). The cited OECD working paper No. 63 “Value in Pharmaceutical Pricing” [12] itself referrers to one of the categorised studies (Leopold [81], qualified as “descriptive”). The more recent OECD working paper No. 87 on “Pharmaceutical Expenditure and Policies” [1] makes reference to one of the included and assessed studies, the systematic review from Lee et al. [66], without placing it into context of other systematic reviews or original research.Footnote 1
For a general conclusion, a proper impact analysis would be needed that would consider the sequential influence of original research (e.g. using a “payback model”, see [82, 83]) in a broader selection of recent policy papers. Yet the conclusions might be noted for their discrepancy between imminent legislative discussions and available scientific evidence. While external and therapeutic reference pricing are still broadly applied and are not expected to be abandoned, new “innovative payment models for new medicines” are desired by authorities [14] and by the pharmaceutical industry [84]. There seems to be a high-level consensus that new models should reflect therapeutic value added for the society and focus on patient-relevant outcomes. Several OECD countries have already incorporated value-based elements into their pharmaceutical pricing policies; for example, Sweden has been doing this for more than 10 years [12, 18]. The trend in OECD countries is clear, with HTA being “increasingly used to inform coverage decisions” [1]. In this context, the lack of evidence on value-based pricing policies is consternating, if not alarming, especially given the current scepticism of the European Parliament, where value-based pricing “can be misused as a profit maximising economic strategy” [85]. As was very recently stated by Vogler (WHO) and Paris (OECD) et al. “regular reviews and evaluations of the impact of pricing and reimbursement policies” are critical to determine their effectiveness in “achieving the intended aims” [15]. There is a particular “need for impact assessments of managed entry agreements, value-based pricing and HTA” [15, 77].
Why is the value-based pricing policy debate not informed by a more solid body of empirical evidence? There are admittedly methodological challenges with the empirical analysis of such policy changes in general. The different settings of the included studies in this paper reveal an unsurprising picture. The reported results (regarding TRP) with a lower risk of bias focused on only a few, established therapies or substances with a certain technological “stability” over a few years. Some of those studies could hardly be repeated for current oncologic treatments, where an increasing number of completely new therapies (e.g. targeted immune oncology) are coming to the market. This technological trend is “multiplied” by the increasing number of interdependent policy changes in recent years in most OECD healthcare systems.
These scientific challenges are confronted by the changing needs of the policy makers—the emerging legislative necessity to move from generally valid, static regulations to adaptive policies based on a mix of models [14]. In addition, the effort of designing and evaluating policy adaptions is a disproportionately higher burden for smaller countries [86].
Finally, value-based, performance-oriented agreements are often linked to MEAs and RSAs [78]. The resulting prices and conditions are, in most countries, confidential and are therefore difficult to access for scientific evaluation [19, 26, 28, 78, 87].
It has never been more obvious that the interaction of health policy formulation and academic policy research needs to move beyond an “iterative” process (Habermas [88]) to a more “co-creative”, “co-evolutionary” process (Jasanoff [89]). This could also increase legitimacy. In the concrete example of value-based pricing of new pharmaceutical therapies, this should mean the following:
-
Need for the early and regular involvement of policy makers in the design of new policy research. Define the concept of qualified policy effectiveness analysis in the policy formulation phase, and roll it out jointly with the regulation change in the policy implementation phase.
-
Need for research institutions to proactively approach the changing needs of the policy formulation process with appropriate research tools. Promote applicable empirical tools to analyse smaller, faster and interdependent policy changes. Additionally, complement the classical empirical tools, such as interrupted time series studies, with economic (field or laboratory) experiments to test policy hypotheses at an early stage.
-
Need for the pharmaceutical industry, healthcare providers and insurance companies to set up and cooperatively participate in health economic studies beyond established clinical trials (post-marketing authorisation), using real-world data on clinical and non-clinical outcomes.
Main limitation: While our restrictive focus on qualified study designs and the chosen methodology to adjust for the risk of bias follows the current practices [34, 40], it comes at a cost. The current state of the underlying policy reform might call for a broader analysis of survey data, discussion papers and policy statements—from scientific and from political stakeholders. These data were not included in the findings of our study. On the other hand, the broader selection of designs, compared to the recommendations from EPOC [34, 40], and the inclusion of meta-literature might also have skewed our findings.
Nevertheless, we conclude that there is a high need for evidence generation on effective pricing policies. In particular, the emerging value-based pricing policies and managed entry agreements should be assessed continuously on a scientific basis to inform policy formulation processes in OECD countries [15]. In general, it is recommended to define policy effectiveness analysis in the policy formulation and implementation process.
Data Availability Statement
The datasets generated and/or analysed during the course of the current study are available in the electronic supplementary material (online version).
Notes
The three papers from the OECD and EXPH refer to some of the recent literature on MEAs, which we excluded since none of them provides a policy effect analysis (see above).
References
Belloni A, Morgan D, Paris V. Pharmaceutical expenditure and policies: past trends and future challenges. OECD Health Working Papers. 2016;(87):0_1–4,8–73.
Garattini L, Curto A, Freemantle N. Pharmaceutical price schemes in Europe: time for a ‘continental’ one? PharmacoEconomics. 2016;34(5):423–6.
Kheirandish M, Rashidian A, Kebriaeezade A, Cheraghali AM, Soleymani F. A review of pharmaceutical policies in response to economic crises and sanctions. J Res Pharm Pract. 2015;4(3):115–22.
Vogler S, Zimmermann N, Ferrario A, et al. Pharmaceutical policies in a crisis? Challenges and solutions identified at the PPRI conference. J Pharm Policy Pract. 2016;9:9.
Banzi R, Gerardi C, Bertele V, Garattini S. Approvals of drugs with uncertain benefit-risk profiles in Europe. Eur J Intern Med. 2015;26(8):572–84.
Aitken M, Kleinrock M. Global oncology trend report: a review of 2015 and outlook to 2020. IMS Institute for Healthcare Informatics; 2016.
Aitken M, Kleinrock M, Simorellis A, Nass D. Global oncology trends 2018, innovation, expansion and disruption. IQVIA Institute for Human Data Science; 2018. https://www.iqvia.com/institute/reports/global-oncology-trends-2018.
Grossmann N, Wild C. Between January 2009 and April 2016, 134 novel anticancer therapies were approved: what is the level of knowledge concerning the clinical benefit at the time of approval? ESMO Open. 2016;1(6):e000125.
Vernon JA. Examining the link between price regulation and pharmaceutical R&D investment. Health Econ. 2005;14(1):1–16.
Golec J, Vernon JA. Financial effects of pharmaceutical price regulation on R&D spending by EU versus US firms. PharmacoEconomics. 2010;28(8):615–28.
Pani L, Montilla S, Nemeth G, Russo P, Viceconte G, Vogler S. Balancing access to medicines and sustainability in Europe: an analysis from the network of competent authorities on pricing and reimbursement (CAPR). Pharmacol Res. 2016;111:247–50.
Paris V, Belloni A. Value in pharmaceutical pricing. OECD Health Working Papers, No. 63. Paris: OECD Publishing; 2013. https://doi.org/10.1787/5k43jc9v6knx-en.
OECD. New health technologies: managing access, value and sustainability. Paris: OECD Publishing; 2017. https://doi.org/10.1787/9789264266438-en.
Expert Panel on effective ways of investing in Health (EXPH). Innovative payment models for high-cost innovative medicines. European Commission; 2018. https://ec.europa.eu/health/expert_panel/sites/expertpanel/files/docsdir/opinion_innovative_medicines_en.pdf.
Vogler S, Paris V, Ferrario A, et al. How can pricing and reimbursement policies improve affordable access to medicines? Lessons learned from European countries. Appl Health Econ Health Policy. 2017;15(3):307–21.
Lorenzoni L, Murtin F, Springare L-S, Auraaen A, Daniel F. Which policies increase value for money in health care? OECD Health Working Papers, No. 104. Paris: OECD Publishing. 2018. https://doi.org/10.1787/a46c5b1f-en.
Cole A, Towse A, Lorgelly P, Sullivan R. Economics of innovative payment models compared with single pricing of pharmaceuticals. OHE Research Paper 18/04, London: Office of Health Economics; 2018. https://www.ohe.org/publications/economics-innovative-payment-models-compared-single-pricing-pharmaceuticals#overlay-context=publications.
Panteli D, Arickx F, Cleemput I, et al. Pharmaceutical regulation in 15 European countries review. Health Syst Transit. 2016;18(5):1–122.
Espin J, Rovera J, de Labry AO. Working paper 1: external price referencing—review series on pharmaceutical pricing policies and interventions. Geneva: World Health Organization and Health Action International; 2011.
Acosta A, Ciapponi A, Aaserud M, et al. Pharmaceutical policies: effects of reference pricing, other pricing, and purchasing policies. Cochrane Database Syst Rev. 2014(10):CD005979.
Green CJ, Maclure M, Fortin PM, Ramsay CR, Aaserud M, Bardal S. Pharmaceutical policies: effects of restrictions on reimbursement. Cochrane Database Syst Rev. 2010(8):CD008654.
Lee IH, Bloor K, Hewitt C, Maynard A. International experience in controlling pharmaceutical expenditure: influencing patients and providers and regulating industry—a systematic review. J Health Serv Res Policy. 2015;20(1):52–9.
Vogler S, Zimmermann N. Glossary of pharmaceutical terms. In: WHO Collaborating Centre for Pharmaceutical Pricing and Reimbursement Policies, ed. WHO Collaborating Centre for Pharmaceutical Pricing and Reimbursement Policies; 2016. http://whocc.goeg.at.
Verordnung über die Krankenversicherung (KVV). In: Bundesrat DS, ed. vol 832.102. Systematische Sammlung des Bundesrechts (SR). 1995.
Hughes DA. Value-based pricing: incentive for innovation or zero net benefit? PharmacoEconomics. 2011;29(9):731–5.
Ferrario A, Kanavos P. Managed entry agreements for pharmaceuticals: the European experience. 2013. http://eprints.lse.ac.uk/50513/.
Klemp M, Fronsdal KB, Facey K, Forum HTP. What principles should govern the use of managed entry agreements? Int J Technol Assess Health Care. 2011;27(1):77–83.
Garrison LP Jr, Towse A, Briggs A, et al. Performance-based risk-sharing arrangements-good practices for design, implementation, and evaluation: report of the ISPOR good practices for performance-based risk-sharing arrangements task force. Value Health. 2013;16(5):703–19.
Marshall AD, Cunningham EB, Nielsen S, et al. Restrictions for reimbursement of interferon-free direct-acting antiviral drugs for HCV infection in Europe. Lancet Gastroenterol Hepatol. 2018;3(2):125–33.
Flume M, Bardou M, Capri S, et al. Feasibility and attractiveness of indication value-based pricing in key EU countries. J Mark Access Health Policy. 2016. https://doi.org/10.3402/jmahp.v4.30970.
Spezialitätenliste: Avastin. BAG; 2018. http://www.spezialitaetenliste.ch/. Accessed 1 Aug 2018.
Persson U, Norlin JM. Multi-indication and combination pricing and reimbursement of pharmaceuticals: opportunities for improved health care through faster uptake of new innovations. Appl Health Econ Health Policy. 2018;16(2):157–65.
PubMed. PubMed search. 2018; https://www.ncbi.nlm.nih.gov/pubmed/. Accessed 31 Aug 2018.
Cochrane Effective Practice and Organisation of Care (EPOC). What study designs should be included in an EPOC review? EPOC Resources for review authors. Oslo: Norwegian Knowledge Centre for the Health Services; 2016.
Persson U, Jonsson B. The end of the international reference pricing system? Appl Health Econ Health Policy. 2016;14(1):1–8.
World Bank Country and Lending Groups. 2016. https://datahelpdesk.worldbank.org/knowledgebase/articles/906519-world-bank-country-and-lending-groups. Accessed 31 Dec 2016.
Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ (Clinical research ed.). 2009;339:b2535.
Drummond MF, Jefferson TO. Guidelines for authors and peer reviewers of economic submissions to the BMJ. The BMJ Economic Evaluation Working Party. BMJ (Clinical research ed.). 1996;313(7052):275–83.
Drummond M, Sculpher M, Torrance G, O’Brien BJ, Stoddart G, editors. Methods for the economic evaluation of health care programmes. 3rd ed. Oxford: Oxford University Press; 2005.
Cochrane Effective Practice and Organisation of Care (EPOC). Suggested risk of bias criteria for EPOC reviews. EPOC Resources for review authors. Oslo: Norwegian Knowledge Centre for the Health Services; 2015.
Ofman JJ, Sullivan SD, Neumann PJ, et al. Examining the value and quality of health economic analyses: implications of utilizing the QHES. JMCP. 2003;9(1):53–61.
Philips Z, Bojke L, Sculpher M, Claxton K, Golder S. Good practice guidelines for decision-analytic modelling in health technology assessment: a review and consolidation of quality assessment. PharmacoEconomics. 2006;24(4):355–71.
Tacconelli E. Systematic reviews: CRD’s guidance for undertaking reviews in health care. Heslington, York: University of York NHS Centre for Reviews & Dissemination; 2009. ISBN 978-1-900640-47-3.
Evers S, Goossens M, de Vet H, van Tulder M, Ament A. Criteria list for assessment of methodological quality of economic evaluations: Consensus on Health Economic Criteria. Int J Technol Assess Health Care. 2005;21(2):240–5.
Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401–6.
Cochrane Effective Practice and Organisation of Care (EPOC). Reporting the effects of an intervention in EPOC reviews. EPOC Resources for review authors. 2017:24. https://epoc.cochrane.org/resources/epoc-resources-review-authors. https://epoc.cochrane.org/sites/epoc.cochrane.org/files/public/uploads/Resources-for-authors2017/how_to_report_the_effects_of_an_intervention.pdf.
Armeni P, Jommi C, Otto M. The simultaneous effects of pharmaceutical policies from payers’ and patients’ perspectives: Italy as a case study. Eur J Health Econ. 2016;17(8):963–77.
Augurzky B, Gohlmann S, Gress S, Wasem J. Effects of the German reference drug program on ex-factory prices of prescription drugs: a panel data approach. Health Econ. 2009;18(4):421–36.
Barros PP, Nunes LC. The impact of pharmaceutical policy measures: an endogenous structural-break approach. Soc Sci Med. 2010;71(3):440–50.
Grootendorst PV, Marshall JK, Holbrook AM, Dolovich LR, O’Brien BJ, Levy AR. The impact of reference pricing of nonsteroidal anti-inflammatory agents on the use and costs of analgesic drugs. Health Serv Res. 2005;40(5 Pt 1):1297–317.
Grootendorst P, Stewart D. A re-examination of the impact of reference pricing on anti-hypertensive drug plan expenditures in British Columbia. Health Econ. 2006;15(7):735–42.
Morgan S, Cunningham C. The effect of evidence-based drug coverage policies on pharmaceutical r&d: a case study from british columbia. Healthc Policy. 2008;3(3):e128–53.
Stargardt T. The impact of reference pricing on switching behaviour and healthcare utilisation: the case of statins in Germany. Eur J Health Econ. 2010;11(3):267–77.
Yfantopoulos J. Pharmaceutical pricing and reimbursement reforms in Greece. Eur J Health Econ. 2008;9(1):87–97.
Kalo Z, Abonyi-Toth Z, Bartfai Z, Voko Z. Pitfalls associated with the therapeutic reference pricing practice of asthma medication. BMC Pulm Med. 2012;12:35.
Kanavos PG, Vandoros S. Determinants of branded prescription medicine prices in OECD countries. Health Econ Policy Law. 2011;6(3):337–67.
Kyle MK. Pharmaceutical price controls and entry strategies. Rev Econ Stat. 2007;89(1):88–99.
Leopold C, Mantel-Teeuwisse AK, Seyfang L, et al. Impact of external price referencing on medicine prices—a price comparison among 14 European countries. South Med Rev. 2012;5(2):34–41.
Stargardt T. Modelling pharmaceutical price changes in Germany: a function of competition and regulation. Appl Econ. 2011;43(29):4515–26.
von der Schulenburg F, Vandoros S, Kanavos P. The effects of drug market regulation on pharmaceutical prices in Europe: overview and evidence from the market of ACE inhibitors. Health Econ Rev. 2011;1(1):18.
Atella V, Bhattacharya J, Carbonari L. Pharmaceutical price controls and minimum efficacy regulation: evidence from the United States and Italy. Health Serv Res. 2012;47(1 Pt 1):293–308.
Kaiser U, Mendez SJ, Ronde T, Ullrich H. Regulation of pharmaceutical prices: evidence from a reference price reform in Denmark. J Health Econ. 2014;36:174–87.
Koenig P, Macgarvie M. Regulatory policy and the location of bio-pharmaceutical foreign direct investment in Europe. J Health Econ. 2011;30(5):950–65.
Comanor WS, Schweitzer SO, Riddle JM, Schoenberg F. Value based pricing of pharmaceuticals in the US and UK: does centralized cost effectiveness analysis matter? Rev Ind Organ. 2018;52(4):589–602.
Galizzi MM, Ghislandi S, Miraldo M. Effects of reference pricing in pharmaceutical markets: a review. PharmacoEconomics. 2011;29(1):17–33.
Lee JL, Fischer MA, Shrank WH, Polinski JM, Choudhry NK. A systematic review of reference pricing: implications for US prescription drug spending. Am J Manag Care. 2012;18(11):e429–37.
Morgan S, Hanley G, Greyson D. Comparison of tiered formularies and reference pricing policies: a systematic review. Open Med. 2009;3(3):e131–9.
Puig-Junoy J. What is required to evaluate the impact of pharmaceutical reference pricing? Appl Health Econ Health Policy. 2005;4(2):87–98.
Relakis J, Maniadakis N, Kourlaba G, Shen J, Holtorf AP. Systematic review on the impacts of strict pharmaceutical price controls. Value Health. 2013;16(7):A486.
Remuzat C, Urbinati D, Mzoughi O, El Hammi E, Belgaied W, Toumi M. Overview of external reference pricing systems in Europe. J Mark Access Health Policy. 2015. https://doi.org/10.3402/jmahp.v3.27675.
Zhou W, Cui X, Qian Y. Review of reference pricing effects on pharmaceuticals. Value Health. 2014;17(7):A787.
Gronde TV, Uyl-de Groot CA, Pieters T. Addressing the challenge of high-priced prescription drugs in the era of precision medicine: a systematic review of drug life cycles, therapeutic drug markets and regulatory frameworks. PLoS One. 2017;12(8):e0182613.
Cochrane Effective Practice and Organisation of Care (EPOC). Interrupted time series (ITS) analyses. EPOC resources for review authors. 2013. https://epoc.cochrane.org/resources/epoc-resources-review-authors. https://epoc.cochrane.org/sites/epoc.cochrane.org/files/public/uploads/Resources-for-authors2017/interrupted_time_series_analyses.docx.
Ramsay CR, Matowe L, Grilli R, Grimshaw JM, Thomas RE. Interrupted time series designs in health technology assessment: lessons from two systematic reviews of behavior change strategies. Int J Technol Assess Health Care. 2003;19(4):613–23.
Shojania KG, Grimshaw JM. Evidence-based quality improvement: the state of the science. Health affairs (Project Hope). 2005;24(1):138–50.
Eccles M, Grimshaw J, Campbell M, Ramsay C. Research designs for studies evaluating the effectiveness of change and improvement strategies. Qual Saf Health Care. 2003;12(1):47–52.
Ferrario A, Araja D, Bochenek T, et al. the implementation of managed entry agreements in Central and Eastern Europe: findings and implications. PharmacoEconomics. 2017;35(12):1271–85.
Piatkiewicz TJ, Traulsen JM, Holm-Larsen T. Risk-sharing agreements in the EU: a systematic review of major trends. PharmacoEcon Open. 2018;2(2):109–23.
Ferrario A, Kanavos P. Dealing with uncertainty and high prices of new medicines: a comparative analysis of the use of managed entry agreements in Belgium, England, the Netherlands and Sweden. Soc Sci Med. 2015;124:39–47.
Vogler S, Vitry A, Babar ZU. Cancer drugs in 16 European countries, Australia, and New Zealand: a cross-country price comparison study. Lancet Oncol. 2016;17(1):39–47.
Leopold C, Vogler S, Mantel-Teeuwisse AK, de Joncheere K, Leufkens HG, Laing R. Differences in external price referencing in Europe: a descriptive overview. Health Policy. 2012;104(1):50–60.
Banzi R, Moja L, Pistotti V, Facchini A, Liberati A. Conceptual frameworks and empirical approaches used to assess the impact of health research: an overview of reviews. Health Res Policy Syst. 2011;9:26.
Cohen G, Schroeder J, Newson R, et al. Does health intervention research have real world policy and practice impacts: testing a new impact assessment tool. Health Res Policy Syst. 2015;13:3.
EFPIA. EFPIA response to draft opinion on innovative payment models for high-cost innovative medicines. 2017. https://www.efpia.eu/media/288630/final_efpia-response-to-exph-draft-opinion-7_12_2017_wir.pdf.
European Parliament. European Parliament resolution of 2 March 2017 on EU options for improving access to medicines (2016/2057(INI)). European Parliament; 2017. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:52017IP0061.
Godman B, Malmstrom RE, Diogene E, et al. Are new models needed to optimize the utilization of new medicines to sustain healthcare systems? Expert Rev Clin Pharmacol. 2015;8(1):77–94.
Adamski J, Godman B, Ofierska-Sujkowska G, et al. Risk sharing arrangements for pharmaceuticals: potential considerations and recommendations for European payers. BMC Health Serv Res. 2010;10:153.
HABERMAS, Jürgen. Verwissenschaftlichte Politik und öffentliche Meinung. Rentsch, 1964. In: (PUBLISHER) Bretscher, Willy. (1964). Humanität und politische Verantwortung. Erlenbach-Zürich.
Jasanoff S. States of knowledge : the co-production of science and the social order. London: Routledge; 2005.
Author information
Authors and Affiliations
Contributions
All of the authors (DJW and SB) fulfilled the authorship criteria and contributed to the design of the study, review of the search protocol, analysis and interpretation of the results, and writing and review of the manuscript. DJW prepared the search protocol, performed the database searches, screenings, categorisations, and assessments and prepared the draft of the manuscript. All of the authors are aware of the submission and are in agreement with the manuscript.
Corresponding author
Ethics declarations
Funding
No funding was received for the research performed or the writing of the manuscript.
Conflict of interest
DJW is an external PhD student at the University of Lucerne and was employed at the pharmaceutical company Takeda Pharma while performing this research. Neither the former employer of the first author nor any other private or public entity supported or influenced this research in any relevant way. SB has no conflicts of interest to declare. All of the authors submitted a signed Conflict of Interest disclosure form.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wettstein, D.J., Boes, S. Effectiveness of National Pricing Policies for Patent-Protected Pharmaceuticals in the OECD: A Systematic Literature Review. Appl Health Econ Health Policy 17, 143–162 (2019). https://doi.org/10.1007/s40258-018-0437-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40258-018-0437-z