
Abstract
The anti-cancer agent doxorubicin (DOX) has high cardiotoxicity that is linked to DOX-mediated increase in oxidative stress, mitochondrial iron overload, DNA damage, autophagy, necrosis, and apoptosis, all of which are also associated with secondary tumorigenicity. This limits the clinical application of DOX therapies. Previous studies have attributed DOX-mediated cardiotoxicity to mitochondrial iron accumulation and the production of reactive oxygen species (ROS), which seem to be independent of its anti-tumor DNA damaging effects. Chemo-sensitization of soluble guanylate cyclase (sGC) in the cyclic guanosine monophosphate (cGMP) pathway induces tumor cell death despite the cardiotoxicity associated with DOX treatment. However, sGC–cGMP signaling must be activated during heart failure to facilitate myocardial cell survival. The sGC pathway is dependent on nitric oxide and signal transduction via the nitric oxide–sGC–cGMP pathway and is attenuated in various cardiovascular diseases. Additionally, cGMP signaling is regulated by the action of certain phosphodiesterases (PDEs) that protect the heart by inhibiting PDE, an enzyme that hydrolyses cGMP to GMP activity. In this review, we discuss the studies describing the interactions between cGMP regulation and DOX-mediated cardiotoxicity and their application in improving DOX therapeutic outcomes. The results provide novel avenues for the reduction of DOX-induced secondary tumorigenicity and improve cellular autonomy during DOX-mediated cardiotoxicity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
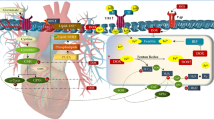
Doxorubicin induces high cardiotoxicity, leading to obstacles in clinical practice. |
The regulation of cyclic guanosine monophosphate (cGMP) is an effective molecular therapeutic target in improving cardiac dysfunction. |
Soluble guanylate cyclase activity is important in the regulation of cGMP in onco-cardiology. |
1 Introduction
Doxorubicin (DOX) is an effective anti-cancer agent; however, its clinical application is limited as a result of its demonstrated cytotoxicity in cardiac cells, which can lead to heart failure [1]. DOX-induced cardiotoxicity can be divided into three categories: acute change, early chronic progressive cardiotoxicity, and late chronic progressive cardiotoxicity. Early onset chronic cardiotoxicity, such as cardiomyopathy that progresses to congestive heart failure (CHF), usually occurs within a year of discontinuing DOX therapy [2]. The enzymatic reduction of the carbonyl groups in the side chains of DOX leads to the formation of the secondary alcohol metabolite doxorubicinol (DOXOL). DOXOL is several times more toxic than DOX [3]. DNA damage and mitochondrial dysfunction have been shown to be crucial to the underlying mechanism of DOX cardiotoxicity [1, 4]. In addition, iron dyshomeostasis and generation of intracellular reactive oxygen species (ROS) were also observed in patients treated with DOX [5]. However, the subcellular mechanism of DOX-induced heart failure and the effectiveness of the therapy are not well understood.
A recent review reported that manipulation of nitric oxide (NO)-cyclic guanosine monophosphate (cGMP) signals could open up new and innovative approaches to cancer treatment, stem cell proliferation, and differentiation [6]. cGMP regulates contractility, accelerates relaxation, and improves the stiffness of cardiac muscle. Most studies on the cardiac role of cGMP/cGMP-dependent protein kinase 1 (cGK-1) signaling have focused on the effects of upregulated cGMP synthesis.
Clinical therapies have been developed using NO donors (or organic nitrate) or natriuretic peptide, and both are routinely used in the treatment of heart disease. In addition, interest in phosphodiesterases (PDEs), which control cGMP degradation, has increased over the past decade [7]. cGMP affects cAMP (cyclic adenosine monophosphate) signaling and regulates heart function through crosstalk regulation via cGMP-regulated PDE (PDE2 or PDE3). In a rat model of right ventricular hypertrophy, acute PDE5 inhibition increased myocardial cAMP (as well as cGMP) and cardiac contraction, which were linked to reduced PDE3 activity. Similar positive anisotropic conduction properties following PDE5 inhibition were observed by in a single study of the human right ventricle [8].
It is critical to identify altered molecular signals involved in DOX-induced heart failure and to develop adjuvant therapy to prevent DOX-induced cardiotoxicity. The recovery of the NO–soluble guanylate cyclase (sGC)–cGMP pathway could provide protective effects for cardiomyocytes. Additionally, increasing sGC-cGMP pathway activity is an important treatment target in heart failure, and research has indicated that DOX administration decreases cardiac sGC activity [9].
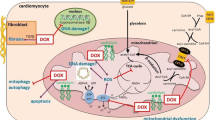
We hypothesized that regulating the cGMP pathway is an effective method for treating heart failure, and thus investigated the role of the cGMP-related pathways, NO–cGMP–sGC, and cGMP-PDEs in alleviating DOX-mediated cardiotoxicity (Fig. 1). Our aim was to understand the relationship between DNA damage, mitochondrial iron overload, energy dysregulation, oxidative stress, autophagy and apoptosis and their effects on both DOX anti-tumor activity and DOX-induced cardiotoxicity. We reviewed recent research articles, and provide the groundwork for an anti-tumor approach to prevent DOX-mediated cardiotoxicity.

Schematic diagrams of cyclic GMP regulations. The activity of sGC and PDE altered by DOX complementarily regulates the concentration of cGMP. It raises the possibility that the NO-sGC-cGMP signaling pathway may be therapeutically beneficial for DOX-induced cardiac toxicity in cardio-oncology. cGMP cyclic guanosine monophosphate, DOX doxorubicin, eNOS endothelial nitric oxide synthease, GMP guanosine monophosphate, NO nitric oxide, PDE phosphodiesterase, PDE5 phosphodiesterase type 5, ROS reactive oxygen species, sGC soluble guanylate cyclase
2 DOX-Induced Heart Failure During Cancer Therapy
DOX can inhibit tumor formation and ultimately retards cancer cell proliferation and division by limiting the proliferation of cancer cells through its prophylactic interference with DNA or RNA structures [2]. A total of 32% of breast cancer patients, 57–70% of elderly lymphoma patients, and 50–60% of childhood cancer survivors are treated with anthracycline therapies [10]. Therefore, any long-term clinical effects of these therapies may have far reaching consequences on the health and survival of cancer patients. Significant evidence has linked increased cardiovascular morbidity and mortality in these patients with conventional anthracycline chemotherapies.
The incidence of cardiac toxicity depends on the method of treatment, with cardiac toxicity ranging from obvious clinical symptoms requiring urgent hospital stays to asymptomatic structural changes in cardiac imaging, new onset arrhythmias, or significant biomarker elevations before symptoms and structural or electrical changes are detected. These observations span frequency and severity, with cardiotoxicity frequently associated with other symptoms of systemic heart failure [10]. In addition, both myocardial and vascular toxicities have been associated with several aspects of myocardial ischemia and pulmonary hypertension caused by prolonged QTc, arrhythmia, and arteriosclerosis. Heart failure with preserved ejection fraction (HFpEF; also called diastolic heart failure) occurs when the left ventricle (LV) is not properly filled with blood during the diastolic phase. Many HFpEF patients have increased LV mass or relative wall thickness and may have concentric remodeling or hypertrophy. The total LV chamber size is usually normal or almost normal, but the myocardial cells themselves may exhibit increased diameters [11]. Since the pathophysiology underlying HFpEF is heterogeneous, with each displaying slightly different phenotypes, it remains poorly understood, with even the etiological definition of HFpEF being variable [12]. Therefore, accurate diagnosis is difficult and there is currently no effective treatment for HFpEF. However, recently novel biomarkers including cardiac stress protein biomarkers (ST2), matrix metalloproteinase-2, and growth differentiation factor-15 have been identified for risk stratification in HFpEF and could be used to develop important therapeutic targets for the treatment of HFpEF [10, 13].
Several mechanisms for cardiac DOX toxicity memory have been proposed [14]. One of the most widely accepted explanations is that the oxidative stress induced by these therapies produces ROS and oxygen free radicals that damage the oxidative respiratory chain. There have been many studies on DOX-induced heart damage and its mechanism, and we summarize the cited studies in Table 1. Mitochondria, which are abundant in myocardial cells, have been identified as major targets of DOX-induced cardiotoxicity [15]. DOX derivatives accumulate in the inner mitochondrial membrane, interfering with the electron transport chain promoting cytochrome C release. In addition, DOX has been shown to decrease the calcium storage capacity of mitochondria by specifically activating the calcium channels, exacerbating calcium overload. Changes in calcium homeostasis cause mitochondrial dysfunction and apoptosis [16].
Dexrazoxane, the only known protective agent, approved by the Food and Drug Administration (FDA), against the cardiotoxic effects of the anthracyclines, has been shown to be a catalytic inhibitor of topoisomerase II (TopII). There are two TopII isomers. TopIIα, highly expressed in cancer cells and required for cell division, is a target for the anti-tumor effect of anthracyclines. However, TopIIβ is only expressed when it is not required for adult myocardial cell division. Since dexrazoxane binds TopIIβ and inhibits DOX-induced DNA double-strand breakage, it is possible to target TopIIβ in cardiomyocytes and induce a cardioprotective effect [15]. Although this effect can be observed in reduced DOX-mediated cardiotoxicity in a TopIIβ knockout mouse model, the long-term effects of DOX treatment on cardiac progenitor cells remains unclear. Dexrazoxane appears to be the most effective drug tested to date. In addition to directly competing with TopII, it can reduce the oxidative stress experienced by myocardial cells produced by the interactions between DOX and iron ions, as well as stimulating the expression of the mitochondrial antioxidant enzymes [16].
3 DOX-Induced Genome Instability
There are three interrelated properties in clinical DOX chemotherapy: cardiotoxicity, anti-tumor activity, and secondary malignancies [1, 17]. Genome instability is an underlying issue since the generation of ROS and cytotoxic protein-linked DNA breaks (PDBs), primarily double-strand breaks (DSBs), are caused by DOX binding to DNA-TopII and mitochondria, respectively [1, 18, 19]. DOX interacts with TopII, which affects the tension and topologic features of genomic DNA, resulting in DNA damage and inducing DSBs. In the latter, DOX binds to mitochondria via oxidative phosphorylation (OXPHOS) complex-I [19] and induces mitochondrial iron accumulation, generating ROS via the Fenton reaction [20]. These ROS produce DNA damage, which induces the DNA-damage-response pathways that suppress transcription factors known to be critical in the regulation of mitochondrial biogenesis, which may induce mitochondrial dysfunction [1, 4]. In addition, it has been reported that sGC activity decreases during DOX-mediated cardiomyopathy, and that this reduced sGC activity exacerbated ROS formation and cardiac dysfunction [9].
The mitochondria are also an important target of DOX toxicity. Frataxin (FXN) is a nuclear encoded mitochondrial protein that maintains mitochondrial iron homeostasis and modulates complexes I and III of the electron transport chain system [21], including the binding surfaces for iron and ferrochelatase [22]. FXN is also a direct target of p53, which can become dysregulated in tumor cells and downregulated in cardiomyocytes following DOX treatment [18, 23]. MtFt, an iron-sequestering protein, plays a role in iron storage and has a functional ferroxidase center with ferroxidase activity. MtFt prevents the production of ROS through the Fenton reaction, linking it to DOX-iron mediated oxidative stress [20]. DOX interacts with metal ions, especially iron, leading to the formation of DOX-iron (III) complexes. Iron also plays an important independent role in the production of harmful free radicals, which can have a detrimental effect on the myocardium [15, 24]. MtFt is highly expressed in heart tissue and decreases ROS production to inhibit mitochondrial damage [20]. Overexpression of MtFt leads to cellular iron redistribution and translocation into the mitochondria, even in tumor cells [20, 25]. Importantly, tumor growth is inhibited by the cytosolic iron deprivation caused by MtFt expression [25]. MtFt acts as a tumor suppressor protein, although its expression depends on tissue type. Furthermore, the inhibition of mitochondrial metabolism leads to p53 genetic inactivation via an ROS-dependent mechanism in cancer cells [26], and ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis [27], which can stimulate cellular proliferation, cell migration, and invasion, contributing to carcinogenesis [27].
Chromosomal DNA is essential for maintaining homeostasis and promoting cellular survival. Autophagy and biogenesis are tightly linked and have been shown to exhibit conserved responses to DOX-induced stress or damage. These processes exert some control on cellular turnover and act to restore cellular homeostasis and genomic stability [4, 28]. However, electron microscopy has revealed that the autophagic process in response to DOX is initiated by an increase in lysosomes followed by rare autophagy-lysosome formation [28], which supports the generally accepted belief that DOX impairs the autophagic process. Autophagy failure promotes genomic instability resulting from gene amplification and aneuploidy, both of which are associated with tumorigenesis [27, 29]. Although autophagy, programmed as a bulk degradation process, is part of the programmed cell death response, it can also suppress tumor growth by limiting genomic instability and oncogenic signaling [29]. Depending on the context and cell type, autophagy is an essential autonomous mechanism for restoring and maintaining cell health. In addition, autophagy is also important for recycling ferritin, and autophagy defects can alter iron homeostasis, which increases the cell’s susceptibility to oxidative stress. In contrast, autophagy could be helpful in reversing DOX-induced iron dyshomeostasis via its functional role in ferritin recycling [30].
4 Damage to cGMP-Dependent Pathway During Heart Failure
NO plays a fundamental role in activating sGC, which produces circulating GMP, a potent activator of protein kinase G1α (PKG1α), which plays an essential role in normal cardiovascular function by inhibiting vasoconstriction, inflammation, hypertrophy, and fibrosis. cGMP is a ubiquitous second messenger found throughout the cells in the cardiovascular system. Acute elevations of cGMP generally exert anisotropic conduction properties as well as negative metabolism, while chronic elevations prevent and reverse heart hypertrophy [31]. A few studies found that the sGC/cGMP pathway is mediated in onco-cardiology therapy (Table 2). Thus, reduced sGC activity is an important contributor to coronary microvascular damage, myocardial cell stiffness, and interstitial fibrosis [32, 33]. sGC is a heterodimeric enzyme composed of α and β subunits containing NO-sensitive heme cofactors. Small molecule stimulants bind and act on sGC in the presence of these heme cofactors to enhance NO signaling [34]. Changes in sGC activity are recognized as a major feature of heart failure syndrome as reflected by the overproduction of ROS, which reduces NO bioavailability [35]. Therefore, in order to increase cGMP, many studies have focused on the use of sGC activators or stimulators [36]. A recent study by Ritchie and colleagues demonstrated that sGC activator BAY 58-2667 induces myocardial protective effects in vitro, limiting myocardial hypertrophy [37]. In addition, the most recently published paper revealed that BAY 60-2770, an activator of oxidized sGC, acts as a cardioprotector by mitigating autophagy and mitochondrial membrane potential loss associated with oxidative stress in DOX-induced cardiac injury models. These new results highlight the therapeutic potential of sGC signaling to prevent DOX-induced cardiomyopathy [38].
Elevated intracellular cGMP levels in cardiac myocytes affect several different pathways, including the activation of PKG, inhibition or stimulation of PDE activity, and changes in cAMP levels. Both sGC and cGMP are important targets for PDE proteins. Four PDEs (PDE1a, c; PDE2; PDE3a; and PDE5a) were found to contribute to the regulation of hydrolysis and/or signal transduction in myocytes. PDE1 is a calcium-calmodulin (CaM)-dependent double esterase, PDE2 is a cGMP-stimulated cAMP esterase, and PDE3a is a cAMP esterase that can be competitively inhibited by cGMP. PDE5a is the only selective cGMP esterase in this group [39]. High concentrations of cGMP possibly induce contractile changes through cross-activation of protein kinase A (PKA), as demonstrated in smooth muscle cells [40]. Activation of PDE was observed at low (less than 1 μM) concentrations of cGMP [41]. In hypertrophic right ventricular myocardia, PDE5 is upregulated, PKG activity is inhibited, and cGMP is preferentially shifted to inhibition of PDE3. This increases cAMP and intracellular calcium levels, PKA activation, and contractility. Increased PDE5 expression tends to adversely affect LV remodeling in mice after myocardial infarction. LV systolic and diastolic disorders were shown to be more pronounced in PDE5-TG mice, which overexpress PDE5 within cardiomyocytes specifically, than in wild-type mice. This was related to improved hypertrophy and decreased contractile function in myocardial cells isolated from remote myocardia [42]. The combination of cGMP and PDE reduces the activity of the enzymes that facilitate cAMP accumulation in myocardial cells. Initial investigations in frog, guinea pig, and human cardiomyocytes have shown that inhibition of PDE initiated by cGMP stimulates transmembrane calcium transport [41].
5 Cardioprotective Effect of PDE Pathways in Tumor Therapy
At least four different PDE isoforms account for the hydrolysis of cGMP in cardiac tissues. Among these, PDE5 is the most highly expressed in vascular smooth muscle [7, 31]. PDE5 inhibitors directly increase cGMP levels [43], which can increase the chemotherapy efficacy of DOX in cancer, such as prostate cancer, and protects against cardiac dysfunction [44]. PDE5 inhibitors effect their function via PKG1α signaling.
These studies led to the discovery of a novel application of the PDE5 inhibitor sildenafil in cardiovascular medicine. Sildenafil has been approved for use in pulmonary hypertension, but its efficacy in HFpEF patients has not been demonstrated. cGMP inhibition of myocardial remodeling following inhibition of PDE5 by sildenafil has been shown to be an attractive option for the treatment of lung and systemic hypertension and heart failure [35]. In a single-center study of 44 patients with HFpEF, pulmonary function with central hemodynamics, LV function, and PDE5 inhibitory function significantly improved, without inducing significant changes in the plasma cGMP levels between sildenafil and placebo groups. Thus, upregulation of PDE5 in HFpEF has not been clearly shown to be the underlying mechanism for reduced cGMP signaling, and reduced cGMP production has been shown to be more critical in HFpEF than PDE5 [11].
NO is the key initiator of the cGMP pathway, and many studies have investigated it as a therapeutic target for HFpEF. Several studies have reported that the introduction of an organic nitrate NO donor improves the health of the cardiac environment via cGMP-independent pathways, and may be applicable as a clinical intervention. Interestingly, simultaneous administration of a PDE5 inhibitor and NO donor results in large increases in cGMP [31]. Conversely, low endogenous NO-cGMP production significantly limits or impairs the effectiveness of PDE5 inhibitors [32]. cGMP is produced by sGCs activated by NO and particulate GC downstream of sodium diuretic peptides [45]. Therefore, activation of sGCs is required to induce cGMP. The higher efficacy of sGC stimulants compared to PDE5 inhibitors suggests that direct targeting of sGCs may be more beneficial than targeting downstream components of cGMP production [32, 46].
6 sGCs and hsp90 in Tumor Therapy
More recently developed sGCs modulators that stimulate and activate sGCs in an NO-independent manner have been shown to have broader therapeutic potential. sGC modulators include sGC stimulators and sGC activators. These sGC stimulators bind to the heme-containing sGCs, and their activity is dependent on the presence of heme, while the sGC activators preferentially bind to oxidized sGCs and act independently of the heme group [32].
Studies have shown that sGC is one of the more than 200 chaperone heat shock protein 90 (hsp90) client proteins. sGC requires hsp90 for heme insertion during NO-active enzyme maturation [47], and hsp90 inhibitors are considered a potential treatment avenue for various cancers [48]. Additionally, tumor suppressor p53 is one of the hsp90 client proteins [49, 50]. However, one type of sGC activator, BAY 60-2770 (4-(((4-carboxybutyl) (2-(5-fluoro-2-((4′-(trifluoromethyl) biphenyl-4-yl) methoxy) phenyl) ethyl) amino) methyl) benzoic acid) [51], can overcome DOX-induced oxidized stress and activate sGC-cGMP-based signal cascades, bypassing the requirements for active hsp90 [47]. Therefore, it is unlikely that BAY 60-2770 supports hsp90-mediated tumor growth. sGC expression is significantly lower in embryonic stem cells and glioma specimens as well as in hepatoma and breast cancer; restoring sGC expression blocks the aggressive expansion of both glioma and breast cancer [6]. sGC consists of a large α and small heme-binding β subunit, with the β-subunit described as essential for sGC function [47, 52]. Previous reports have indicated that sGCα1 is a potential regulator of sGC activity, and that the α-subunit maintains sGCs in an auto-inhibited basal state [52]. If this sGC activator bypasses active hsp90 and heme insertion and overcomes allosteric inhibition by the α-subunit [47], this process could be an innovative approach to cancer therapy using DOX, although this approach would be cell-type dependent.
7 sGC-cGMP and TopI-Mediated Transcriptional Regulation
The sGC-cGMP signal pathway is poorly understood, but it has been linked to the activation of topoisomerase I (TopI), which might mediate PDB repair [53]. Additionally, TopI promotes RNA maturation and regulates gene expression [54]. TopI expression (as opposed to TopIIα, which is a target of DOX in tumor cells) is proliferation independent and is similar in cycling and non-cycling cells. TopIIα, which is expressed only in the S phase of the cell cycle, has been shown to increase in response to tumor tissue proliferation [55]. In comparison, disruption of TopIIα or TopIIβ results in embryonic or perinatal lethality and is not associated with TopI [53]. As discussed above, DOX can serve toxicity to TopIIα and TopIIβ. While cGMP targets TopI, sGC-cGMP signaling does not activate TopII. This indicates that it is unlikely that the DOX-TopII interaction is impaired by DOX target effects. Next, we focused on the effect of sGC activators on DOX-induced secondary malignancies related to topoisomerase in transcription-associated carcinogenesis. Previous studies have shown that TopIIα and TopIIβ drive oncogenic translocations in TopII poison-induced secondary malignancies; however, in contrast to TopII, TopI has been implicated in gene deletion, and its role in carcinogenesis is not yet defined [55]. Taken together, these results support a causative role for TopII poisons in carcinogenic translocation, but do not support the same conclusion for TopI. The 180 kDa form of TopII was shown to be involved in the main steps preceding repair-specific DNA incision in UV-irradiated human fibroblasts. Because TopII inhibitors, particularly DOX, block UV-induced DNA repair, the strongest intercalators causes the most pronounced inhibition of repair. However, TopI inhibitors did not markedly diminish DNA repair synthesis [56]. Therefore, sGC activators could activate TopI-mediated transcription, regulating gene expression [54] and PDB repair attenuating DOX-associated secondary malignancies.
8 Study Limitations
As with other analytical reviews, our review currently has limitations. All of the studies used in the analysis were very high quality, but there were some differences in the subject, method, or mechanism among each experiment. It was not possible to define the extent to which these differences could be related to the variation in the trial design or the target model. Bias could not be completely ruled out, and there is the potential for a hasty generalization error. Nevertheless, the direction of the study results was consistent, and the details can be found in the included tables. In addition, only studies on heart failure among cardiotoxic diseases that may occur when anthracycline drugs are used during chemotherapy were included. It should be confirmed whether this study can be applied to other cardiotoxic diseases that may develop, such as cardiac ischemia, hypertension, and arrhythmia. A systematic study of these will be needed.
9 Conclusion
Biochemical changes in myocardial cells induced by DOX pose a major obstacle to researchers and the development of DOX-reliant clinical therapies in the treatment of various cancers. Autophagy and mitochondrial damage are important mechanisms underlying DOX-induced cardiotoxicity [4, 56]. In particular, mitochondria, which play an important role in cardiomyocytes, are not only affected by DOX-induced DNA damage and transcriptional changes [19], but also induce iron accumulation resulting from reduced availability of MtFt and FXN [18, 26]. These factors can increase ROS in cardiomyocytes or activate p53 gene expression, resulting in apoptosis [50, 57]. These cellular pathways are important for homeostasis, and subsequent genomic instability and aberrations in these pathways can alter cancer development.
Administration of DOX reduces sGC activity in the heart, accelerating ROS formation and exacerbating heart failure [9]. Additionally, DOX promotes PDE (an enzyme involved in other cGMP signaling pathways) activation, inducing the degradation of cGMP in cardiomyocytes [39, 42]. If the cGMP concentration in myocardial cells falls below a certain level, normal activity of the heart may not be achieved. sGC or PDE-cGMP signaling is important in innovative cancer treatments [6].
In conclusion, cGMP signaling pathways are a very important part of DOX oncology. Such interactions can attenuate DOX-induced cardiotoxicity and potentially reduce secondary malignant tumors that may occur during DOX treatment in clinic, increasing the efficacy of DOX chemotherapy. This article focused on integrating multiple pathways to evaluate approaches to avoid DOX-mediated cardiac toxicity. As a result, DOX provides information on signaling pathways that regulate cGMP as a potential and innovative approach to cancer therapy. This provides additional information that can be incorporated into clinical practice and the prevention of DOX-induced cardiomyopathy.
10 Clinical Perspective
DOX is used as a very effective anticancer agent, but it directly causes high cardiac toxicity and presents serious obstacles to clinical treatment. When cGMP is regulated in the heart and the concentration exceeds a certain level, heart function can be maintained normally. Therefore, the control of cGMP can be an effective therapeutic target in the event of heart abnormalities. The activities of sGC and PDE, which are closely related to the cGMP regulation pathway, will play a very important role in cardio-oncology. If a combination therapy with drugs that activates sGC or inhibits PDE is performed in clinical treatment to avoid heart damage, the use of DOX is smoother by mitigating the cardiac toxicity caused by cGMP. Treatment effect can be expected in chemotherapy for cancer patients.
References
Sawyer DB. Anthracyclines and heart failure. N Engl J Med. 2013;368(12):1154–6.
Torres VM, Simic VD. Doxorubicin-induced oxidative injury of cardiomyocytes—do we have right strategies for prevention. Cardiotoxic Oncol Treat. 2012:1–43.
Salvatorelli E, Menna P, Chello M, Covino E, Minotti G. Modeling human myocardium exposure to doxorubicin defines the risk of heart failure from low-dose doxorubicin. J Pharmacol Exp Ther. 2017;362(2):263–70.
Jean SR, Tulumello DV, Riganti C, Liyanage SU, Schimmer AD, Kelley SO. Mitochondrial targeting of doxorubicin eliminates nuclear effects associated with cardiotoxicity. ACS Chem Biol. 2015;10(9):2007–15.
Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Prasad SVN, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Investig. 2014;124(2):617–30.
Bian K, Murad F. What is next in nitric oxide research? From cardiovascular system to cancer biology. Nitric Oxide. 2014;43:3–7.
Lee DI, Kass DA. Phosphodiesterases and cyclic GMP regulation in heart muscle. Physiology. 2012;27(4):248–58.
Takimoto E. Cyclic GMP-dependent signaling in cardiac myocytes. Circ J. 2012:CJ-12-0664.
Vandenwijngaert S, Swinnen M, Walravens A-S, Beerens M, Gillijns H, Caluwé E, et al. Decreased soluble guanylate cyclase contributes to cardiac dysfunction induced by chronic doxorubicin treatment in mice. Antioxid Redox Signal. 2017;26(4):153–64.
McGowan JV, Chung R, Maulik A, Piotrowska I, Walker JM, Yellon DM. Anthracycline chemotherapy and cardiotoxicity. Cardiovasc Drugs Ther. 2017;31(1):63–75.
Greene SJ, Gheorghiade M, Borlaug BA, Pieske B, Vaduganathan M, Burnett JC Jr, et al. The cGMP signaling pathway as a therapeutic target in heart failure with preserved ejection fraction. J Am Heart Assoc. 2013;2(6):
Bozkurt B, Coats AJ, Tsutsui H, Abdelhamid CM, Adamopoulos S, Albert N, et al. Universal definition and classification of heart failure: a report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure: Endorsed by the Canadian Heart Failure Society, Heart Failure Association of India, Cardiac Society of Australia and New Zealand, and Chinese Heart Failure Association. Eur J Heart Fail. 2021;23(3):352–80.
Dong J, Chen H. Cardiotoxicity of anticancer therapeutics. Front Cardiovasc Med. 2018;5:9.
Wallace KB, Sardão VA, Oliveira PJJCR. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ Res. 2020;126(7):926–41.
Han X, Zhou Y, Liu W. Precision cardio-oncology: understanding the cardiotoxicity of cancer therapy. NPJ Precis Oncol. 2017;1(1):1–11.
Mitry MA, Edwards JG. Doxorubicin induced heart failure: phenotype and molecular mechanisms. IJC Heart Vasc. 2016;10:17–24.
Harake D, Franco VI, Henkel JM, Miller TL, Lipshultz SE. Cardiotoxicity in childhood cancer survivors: strategies for prevention and management. Future Cardiol. 2012;8(4):647–70.
Mouli S, Nanayakkara G, AlAlasmari A, Eldoumani H, Fu X, Berlin A, et al. The role of frataxin in doxorubicin-mediated cardiac hypertrophy. Ame J Physiol Heart Circ Physiol. 2015;309(5):H844–59.
Yadav N, Kumar S, Marlowe T, Chaudhary A, Kumar R, Wang J, et al. Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents. Cell Death Dis. 2015;6(11):e1969-e.
Maccarinelli F, Gammella E, Asperti M, Regoni M, Biasiotto G, Turco E, et al. Mice lacking mitochondrial ferritin are more sensitive to doxorubicin-mediated cardiotoxicity. J Mol Med. 2014;92(8):859–69.
Huang ML-H, Becker EM, Whitnall M, Rahmanto YS, Ponka P, Richardson DR. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc Natl Acad Sci. 2009;106(38):16381–6.
Bencze KZ, Yoon T, Millán-Pacheco C, Bradley PB, Pastor N, Cowan J, et al. Human frataxin: iron and ferrochelatase binding surface. Chem Commun. 2007;18:1798–800.
Shimizu R, Lan NN, Tai TT, Adachi Y, Kawazoe A, Mu A, et al. p53 directly regulates the transcription of the human frataxin gene and its lack of regulation in tumor cells decreases the utilization of mitochondrial iron. Gene. 2014;551(1):79–85.
Panjrath GS, Patel V, Valdiviezo CI, Narula N, Narula J, Jain D. Potentiation of doxorubicin cardiotoxicity by iron loading in a rodent model. J Am Coll Cardiol. 2007;49(25):2457–64.
Nie G, Chen G, Sheftel AD, Pantopoulos K, Ponka P. In vivo tumor growth is inhibited by cytosolic iron deprivation caused by the expression of mitochondrial ferritin. Blood. 2006;108(7):2428–34.
Bartesaghi S, Graziano V, Galavotti S, Henriquez NV, Betts J, Saxena J, et al. Inhibition of oxidative metabolism leads to p53 genetic inactivation and transformation in neural stem cells. Proc Natl Acad Sci. 2015;112(4):1059–64.
Galluzzi L, Morselli E, Kepp O, Vitale I, Rigoni A, Vacchelli E, et al. Mitochondrial gateways to cancer. Mol Aspects Med. 2010;31(1):1–20.
Kawaguchi T, Takemura G, Kanamori H, Takeyama T, Watanabe T, Morishita K, et al. Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovasc Res. 2012;96(3):456–65.
White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. AACR; 2015.
Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16(11):1069–79.
Castro LR, Verde I, Cooper DM, Fischmeister R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation. 2006;113(18):2221.
Sandner P, Stasch JP. Anti-fibrotic effects of soluble guanylate cyclase stimulators and activators: a review of the preclinical evidence. Respir Med. 2017;122:S1–9.
Morbidelli L, Pyriochou A, Filippi S, Vasileiadis I, Roussos C, Zhou Z, et al. The soluble guanylyl cyclase inhibitor NS-2028 reduces vascular endothelial growth factor-induced angiogenesis and permeability. 2010;298(3):R824–R32.
Hall KC, Bernier SG, Jacobson S, Liu G, Zhang PY, Sarno R, et al. sGC stimulator praliciguat suppresses stellate cell fibrotic transformation and inhibits fibrosis and inflammation in models of NASH. Proc Natl Acad Sci. 2019;116(22):11057–62.
Michalak M, Armstrong PW. Exploring new cardiovascular pathways: are soluble guanylate cyclase stimulators the right direction?: Am Heart Assoc; 2018.
Boerrigter G, Lapp H, Burnett JC. Modulation of cGMP in heart failure: a new therapeutic paradigm. cGMP: generators, effectors and therapeutic implications. Springer; 2009. p. 485-506.
Irvine JC, Ganthavee V, Love JE, Alexander AE, Horowitz JD, Stasch J-P, et al. The soluble guanylyl cyclase activator BAY 58-2667 selectively limits cardiomyocyte hypertrophy. PLoS One. 2012;7(11).
Xiao-Xiao Z, Cho H, Lee S, Woo JS, Song M-Y, Cheng XW, et al. BAY60-2770 attenuates doxorubicin-induced cardiotoxicity by decreased oxidative stress and enhanced autophagy. Chemico Biol Interact. 2020:109190.
Zhang M, Kass DA. Phosphodiesterases and cardiac cGMP: evolving roles and controversies. Trends Pharmacol Sci. 2011;32(6):360–5.
Layland J, Li JM, Shah AM. Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol. 2002;540(2):457–67.
Kojda G, Kottenberg K. Regulation of basal myocardial function by NO. Cardiovasc Res. 1999;41(3):514–23.
Kukreja RC, Salloum FN, Das A. Cyclic guanosine monophosphate signaling and phosphodiesterase-5 inhibitors in cardioprotection. J Am Coll Cardiol. 2012;59(22):1921–7.
Booth L, Roberts JL, Poklepovic A, Gordon S, Dent P. PDE5 inhibitors enhance the lethality of pemetrexed through inhibition of multiple chaperone proteins and via the actions of cyclic GMP and nitric oxide. Oncotarget. 2017;8(1):1449.
Das A, Durrant D, Mitchell C, Mayton E, Hoke NN, Salloum FN, et al. Sildenafil increases chemotherapeutic efficacy of doxorubicin in prostate cancer and ameliorates cardiac dysfunction. Proc Natl Acad Sci. 2010;107(42):18202–7.
Lee KH, Lee S-R, Cho H, Woo JS, Kang JH, Jeong Y-M, et al. Cardioprotective effects of PKG activation by soluble GC activator, BAY 60-2770, in ischemia-reperfusion-injured rat hearts. PloS One. 2017;12(7).
Nakamura T, Zhu G, Ranek MJ, Kokkonen-Simon K, Zhang M, Kim GE, et al. Prevention of PKG-1α oxidation suppresses antihypertrophic/antifibrotic effects from PDE5 inhibition but not sGC stimulation. Circ Heart Fail. 2018;11(3):e004740.
Ghosh A, Stasch J-P, Papapetropoulos A, Stuehr DJ. Nitric oxide and heat shock protein 90 activate soluble guanylate cyclase by driving rapid change in its subunit interactions and heme content. J Biol Chem. 2014;289(22):15259–71.
Barrott JJ, Haystead TA. Hsp90, an unlikely ally in the war on cancer. FEBS J. 2013;280(6):1381–96.
Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16(7):393–405.
Park SJ, Kostic M, Dyson HJ. Dynamic interaction of Hsp90 with its client protein p53. J Mol Biol. 2011;411(1):158–73.
Knorr A, Hirth-Dietrich C, Alonso-Alija C, Härter M, Hahn M, Keim Y, et al. Nitric oxide-independent activation of soluble guanylate cyclase by BAY 60-2770 in experimental liver fibrosis. Arzneimittelforschung. 2008;58(02):71–80.
Purohit R, Fritz BG, The J, Issaian A, Weichsel A, David CL, et al. YC-1 binding to the β subunit of soluble guanylyl cyclase overcomes allosteric inhibition by the α subunit. Biochemistry. 2014;53(1):101–14.
Mattern M, Nambi P, Bartus J, Mirabelli C, Crooke S, Johnson R. Regulation of topoisomerase I and II activities by cyclic nucleotide-and phospholipid-dependent protein kinases. Effects of interactions between the two transduction pathways. Receptor. 1991;1(3):181–90.
Skourti-Stathaki K, Proudfoot NJ. A double-edged sword: r loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014;28(13):1384–96.
Popanda O, Thielmann HW. The function of DNA topoisomerases in UV-induced DNA excision repair: studies with specific inhibitors in permeabilized human fibroblasts. Carcinogenesis. 1992;13(12):2321–8.
Lee KH, Cho H, Lee S, Woo JS, Cho BH, Kang JH, et al. Enhanced-autophagy by exenatide mitigates doxorubicin-induced cardiotoxicity. Int J Cardiol. 2017;232:40–7.
Broz DK, Mello SS, Bieging KT, Jiang D, Dusek RL, Brady CA, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27(9):1016–31.
Acknowledgements
We would like to acknowledge the contributions of each of our team members, as without their individual contributions, this article would not have reached fruition.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (2017R1C1B5075748) with additional funding from the Ministry of Education (2020R1F1A1076495).
Competing interests
Haneul Cho, Xiao–Xiao Zhao, Sora Lee, Jong Shin Woo, Min-Young Song, Xian Wu Cheng, Kyung Hye Lee, and Weon Kim declare no relationship with industry or other relevant entities that might pose a conflict of interest in connection with this article.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
No other person’s work, such as a figure or table, was used in this article. We do not have any content requiring a request for permission.
Authors’ contributions
HC and XXZ participated in drafting the manuscript. SL participated in acquisition of data and table creation. JSW supported research funding. MYS and KHL participated in concept design and analysis. XWC participated in critical revision of the manuscript for important intellectual content. WK participated in analysis as a supervisor. All authors read and approved the final manuscript.
Rights and permissions
About this article

Cite this article
Cho, H., Zhao, XX., Lee, S. et al. The sGC-cGMP Signaling Pathway as a Potential Therapeutic Target in Doxorubicin-Induced Heart Failure: A Narrative Review. Am J Cardiovasc Drugs 22, 117–125 (2022). https://doi.org/10.1007/s40256-021-00487-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40256-021-00487-5