
Abstract
Alzheimer’s disease (AD) presents a significant global health challenge, with its prevalence expected to rise sharply in the coming years. Despite extensive research, effective treatments addressing the multifaceted pathophysiology of AD remain elusive. This study investigates the therapeutic potential of twenty-seven prolinamides (P1 – P27), with the focus on their interactions with key proteins implicated in AD pathogenesis. Four of the compounds, namely; 10-((4-nitrophenyl)prolyl)-10 H-phenothiazine (P14), 2-((4-nitrophenyl)prolyl)isoindoline (P19), 1-(4-formylphenyl)-N-(p-tolyl)pyrrolidine-2-carboxamide (P22), and N,1-bis(4-nitrophenyl)pyrrolidine-2-carboxamide (P27) showed promising potential as Alzheimer’s drug. In-silico approaches including molecular docking, molecular dynamic (MD) simulation, post md study, physicochemical and drug-likeness parameters were employed to ascertain the potential of these compounds as inhibitors of certain proteins implicated in the pathophysiology of Alzheimer’s disease. Molecular docking and dynamics simulations demonstrated that P14, P19, P22 and P27 exhibited promising binding affinities towards crucial AD-associated proteins, including Beta-Secretase 1 (BACE1), Butyrylcholinesterase (BuChE), and Tau-tubulin kinase 2 (TTBK2). Structural stability analyses revealed that prolinamides, particularly P22 and P27 for BACE1 and P14 and P19 for BuChE, exhibited greater stability than their reference ligands, indicated by lower RMSD, RoG, and RMSF values. For BuChE, Rivastigmine had a docking score of -7.0 kcal/mol, a binding free energy (ΔGbind) of -22.19 ± 2.44 kcal/mol, RMSD of 1.361 ± 0.162 Å, RMSF of 9.357 ± 3.212 Å, and RoG of 22.919 ± 0.064 Å, whereas P19 exhibited a superior docking score of -10.3 kcal/mol, a significantly better ΔGbind of -33.74 ± 2.84 kcal/mol, RMSD of 1.347 ± 0.132 Å, RMSF of 8.164 ± 2.748 Å, and RoG of 22.868 ± 0.070 Å. Physicochemical and pharmacokinetic assessments affirmed the drug-likeness and bioavailability of P19 notably capable of penetrating the blood-brain barrier. Compounds P19 and P22, emerged as multi-targeted ligands, offering the potential for simultaneous modulation of multiple AD-related pathways. These findings highlight the possibilities of these compounds to be explored as novel therapeutic agents for AD. They also highlight the need for further experimental validation to confirm their efficacy and safety profiles, advancing them toward clinical application in AD management.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a cognitively crippling condition, accounting for at least 60% of cases of dementia in people of age 65 and older (Kumar et al. 2024). This dysfunction gradually reduces a person’s memory and capacity for learning. AD is a neurological disease of global concern. It is estimated that the number of dementia cases worldwide will rise from 57.4 million in 2019 to 152 million by 2050 (Nichols et al. 2022) if not curbed. Numerous pathways have been identified regarding the pathophysiology and treatment of AD. Firstly, acetylcholinesterase (AChE) levels in the brain decline as AD progresses, whereas butyrylcholinesterase (BuChE) activity gradually rises. As a result, BuChE primarily regulates acetylcholine (Ach) levels (de Almeida et al. 2023). Neuroinflammation, oxidative stress, decreased neurogenesis, synaptic pathology, and dysfunction are also present, along with the progressive accumulation of amyloid-β (Aβ) peptides that result in Aβ plaques and highly phosphorylated tau protein that forms tau neurofibrillary tangles (Fronza et al. 2023).
An important enzyme in the process of converting the amyloid precursor protein (APP) into Aβ is beta-secretase 1 (BACE1) (Cervellati et al. 2020). It has been suggested that elevated serum BACE1 activity is an early indicator of AD (Nicsanu et al. 2022). Tau tubulin kinase 2 (TTBK2) is implicated in this process, phosphorylating tau at specific sites (Liao et al. 2015). In AD, the hyperphosphorylation of tau protein is a hallmark pathology. (Liao et al. 2015). Although these pathological processes have been known for decades, no viable treatment has yet been developed to halt the disease’s course through direct manipulation of these events using the pathways identified (Yiannopoulou and Papageorgiou 2020).
Several therapeutics are employed in the treatment of AD, including acetylcholinesterase inhibitors such as tacrine and rivastigmine, as well as N-methyl-D-aspartate receptor antagonists like memantine. Cholinesterase inhibitors often cause gastrointestinal side effects. Common adverse effects of memantine include constipation, headaches, body aches, and dizziness (Kumar et al. 2024). Therefore, there is a need for the discovery of safe yet effective bioactive compounds for the treatment and management of AD, targeting essential proteins involved in the pathogenesis and progression of the disease.
Prolinamides are carboxamide forms of proline, exhibiting the electronic and spatial properties of the amide bonds derived from peptides (Osinubi et al. 2020). They have been extensively studied due to their relevance in various fields of science and medicine. Prolinamides have been used as catalyst in asymmetric aldol reactions (Yadav et al. 2019), and as antitumor agents (Kumar et al. 2018). Osinubi et al. recently reported a series of substituted N-(4´-nitrophenyl)-L-prolinamides and their in vitro anticancer activities (Osinubi et al. 2020). Their findings showed that some compounds outperformed 5-fluorouracil against some cancer strains used for the study. Literature has it that 75% of known drugs could also be used to cure various diseases, even though it is still under investigation. This process is known as ‘drug ‘repurposing’, and it has been explored in the discovery of drugs for different diseases, especially if nothing or few things are known about the disease, such as in the case of COVID-19 (Singh et al. 2020). Herein, in-silico predictions indicated that several of the twenty-three L-prolinamide derivatives reported by Osinubi et al. (Osinubi et al. 2020), along with four additional prolinamide derivatives predicted during the synthesis process, showed potential to inhibit AChE, BuChE, BACE1, and Tau-tubulin kinase-2. Thus, the aim of this study was to explore the therapeutic potential of these prolinamide derivatives as agents for Alzheimer’s disease treatment by employing molecular docking, molecular dynamics (MD) and post-MD simulation analysis, thus validating the potential of these prolinamides as drugs for AD in the future after further research.
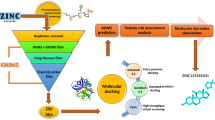
After identifying these aforementioned protein targets for compounds 10-((4-nitrophenyl)prolyl)-10 H-phenothiazine (P14), 2-((4-nitrophenyl)prolyl)isoindoline (P19), 1-(4-formylphenyl)-N-(p-tolyl)pyrrolidine-2-carboxamide (P22), and N,1-bis(4-nitrophenyl)pyrrolidine-2-carboxamide (P27), we theoretically predicted the interactions between these compounds and protein targets using molecular docking and molecular dynamic (MD) simulations. The post-MD analyses were also carried out to calculate parameters such as binding energy, root mean square fluctuation (RMSF), root mean square deviation (RMSD), and the radius of gyration (RoG). These parameters give an in-depth insight into the interaction between the potential drug candidates and the protein targets (See Fig. 1).

2D representation of our prolinamides of interest
Methodology
Molecular docking procedure
The docking analysis was done to determine the compounds’ affinity for specific proteins associated with Alzheimer’s disease in humans. In this procedure, BACE1 (5DQC) (Ghosh et al. 2016), acetylcholinesterase (5HF6) (Franklin et al. 2016), butyrylcholinesterase (6EP4) (Rosenberry et al. 2017), and Tau-tubulin kinase 2 (7Q8Y) (Nozal et al. 2022), 3D crystallographic structures were downloaded from the Protein Data Bank. Prior to the docking process, any co-crystallized ligands, heteroatoms, or water molecules from the complexes derived from X-ray crystallography were eliminated. Also, the 3D structures of the prolinamides P1-P27 and the reference compounds were prepared using the Open Babel software (‘O’Boyle et al., 2011).
Auto-Dock vina (Rauf et al. 2015) was utilized for planning, executing, and evaluating the docking procedures. The receptor grid boxes were meticulously set with dimensions detailed in Table 1 to cover the binding pocket of the proteins, and dockings were done with an exhaustiveness of 8. Similarly, the ligand underwent preparation using the default settings, and their predicted binding energies were recorded. Subsequently, Discovery Studio 2021 Client was used to visualize the interaction of protein-ligand complexes.
Molecular docking validation
To validate docking outcomes, the procedure followed Warren et al.‘s protocol (Warren et al. 2006). The aim was to accurately replicate binding orientation and molecular interactions with co-crystallized ligands.
For this, original ligands were dissociated, prepared and redocked into the protein’s active site using AutoDock vina. Resulting complexes were superimposed onto X-ray structures with co-crystallized ligands. Interactions in both complexes were analyzed with Biovia Discovery Studio.
Molecular dynamics (MD) simulation of the best ligand candidate and its receptors
Four prolinamides out of twenty-seven were selected for molecular dynamic simulations using the GPU version of Amber 18 software and the FF18SB force field (Nair and Miners 2014). Ligand atomic partial charges were generated with ANTECHAMBER using the General Amber Force Field (GAFF) and Restrained Electrostatic Potential (RESP) algorithms (Wang et al. 2004). Systems were neutralized by adding hydrogen atoms to the protein residues and counter ions (Cl− and Na+) via LeaP, then immersed in TIP3P water in a truncated octahedral box extending 8 Å from the protein atoms. Initially, 1000 steps of steepest descent and 1000 steps of conjugate gradient minimization were applied to both ligand-bound and unbound proteins. Proteins were immobilized using position restraints (force constant of 500 kcal mol− 1 Å−2), followed by another 1000 steps of both minimization methods. Systems were then heated to 300 K for 50 ps under NVT conditions with weak positional restraints (force constant of 10 kcal mol − 1 Å−2), equilibrated at 300 K for 500 ps under NPT conditions at 1 atm using isotropic position scaling with a 2 ps relaxation time, and temperature control via Langevin dynamics (collision frequency of 1 ps− 1) (Loncharich et al. 1992). The molecular dynamic simulation ran for 100 ns, with each step interval set to 2 fs. The MD simulation ran for 100 ns with a 2 fs step interval, and results were analyzed using the CPPTRAJ program in Amber18.
Validation of MD Simulation
The root mean square deviation (RMSD), radius of gyration (RoG), and root mean square fluctuation (RMSF) of the protein-ligand complexes and ligands within the binding pocket were analyzed using the CPPTRAJ script in the AMBER 18 package. Data were processed and visualized using OriginPro software (Seifert 2014).
Binding free energy calculations
Free binding energy was calculated using the Molecular Mechanics/Poisson–Boltzmann Surface Area method (MM/PBSA) to estimate and compare the binding affinity of the protein-ligand complex systems. Binding free energy was averaged over 100,000 snapshots from the 100 ns trajectory. The computed free binding energy (G) for each molecular species (complex, ligand, and receptor) can be represented as follows:
The term Egas refers to the gas-phase energy, which includes internal energy (Eint), electrostatic energy (Eelec), and Van der Waals energy (Evdw), and is directly estimated from the FF14SB force field. The solvation-free energy (Gsol) is calculated from the polar (GGB) and non-polar (GSA) contributions. GSA is derived from the solvent-accessible surface area (SASA) using a water probe radius of 1.4 Å, while GGB is obtained by solving the GB equation which follows the Gibbs-Bogoliubov variational principle to approximate the free energy of complex molecular systems in molecular dynamics (Kuzemsky 2015). S and T denote the total entropy and temperature of the solute, respectively.
Screening of the prolinamides of interest for Physicochemical, Drug-Likeness, and Pharmacokinetics properties of the best candidates
Some compounds may interact with various molecules, leading to false positive responses (Dahlin et al. 2015). To address this issue, compounds known as PAINS (pan-assay interference compounds) were identified using false positive remover software. Interestingly, none of our ligands has this property. SwissADME (Daina et al. 2017) and ADMETLab 2.0 (Xiong et al. 2021) were used to estimate the physicochemical and pharmacokinetics of our ligands of interest after the result of docking and dynamic simulation studies. To evaluate these drug-likeness characteristics, the candidate molecule’s SMILES format was entered into the input pane of the online server.
Results and discussion
Molecular Docking
Binding Affinity
The docking results for the prolinamides and the respective standard inhibitors against BACE1 (5DQC), acetylcholinesterase (5HF6), butyrylcholinesterase (6EP4), and Tau-tubulin kinase 2 (7Q8Y) are presented in Supplementary Tables S1. Compared to the reference compound, Verubecestat (Pubchem CID: 51352361), two prolinamides—1-(4-formylphenyl)-N-(p-tolyl)pyrrolidine-2-carboxamide (P22) and N,1-bis(4-nitrophenyl)pyrrolidine-2-carboxamide (P27) exhibited the highest binding energy for BACE1. Additionally, two other prolinamides, 10-((4-nitrophenyl)prolyl)-10 H-phenothiazine (P14) and 2-((4-nitrophenyl)prolyl)isoindoline (P19), demonstrated higher binding energy than rivastigmine (PubChem CID: 77991), the standard inhibitor of BuChE. Furthermore, two prolinamides 2-((4-nitrophenyl)prolyl)isoindoline (P19) and 1-(4-formylphenyl)-N-(p-tolyl)pyrrolidine-2-carboxamide (P22) showed higher binding affinity than 9IV (Pubchem SID: 461502151), the standard inhibitor of Tau-tubulin kinase. However, for acetylcholinesterase, no prolinamide exhibited a higher binding affinity than the reference compound, donepezil (PubChem CID: 3152). The prolinamides with higher binding affinities for the binding pockets of their respective proteins, along with their reference ligands, underwent a 100 ns molecular dynamic simulation (MDS) trajectory. Since none of the prolinamides showed a higher binding affinity than donepezil for acetylcholinesterase, further interactions of prolinamides with acetylcholinesterase were not pursued.
Molecular Docking Validation and Analysis of Protein-Ligand interactions following docking
Molecular docking is a key computational technique used to predict the preferred orientation of a ligand as it binds to a target protein, facilitating the understanding of molecular interactions and binding affinities. By comparing the docking interactions of hit molecules with those of available ligand-bound crystal structures, we aim to ensure the reliability and accuracy of our docking simulations. Figure 2; Table 2, confirm the validity of the molecular docking in this study, showing that the search volumes selected in this study are similar to those of the co-crystalized ligand.

3D representation of the superimposition of the co-crystallized and re-docked ligand in the binding site of (A) BACE1 (B) AChE (C) BuChE (D) TTBK2
For BACE 1 (PDB: 5DQC), the co-crystallized ligand SCHEMBL15299610 [PubChem CID: 89836206] interacts with residues such as GLY11, GLN12, GLY13, TYR14, LEU30, ASP32, GLY34, SER35, SER36, VAL69, PRO70, TYR71, THR72, GLN73, PHE108, ILE110, ILE126, TRP115, TYR198, ILE226, ASP228, SER229, GLY230, THR232, ASN233, ARG235, THR321, LYS321, SER325, VAL332, and ALA335. The re-docked ligand SCHEMBL15299610 shows consistent interactions with key residues GLY11, LEU30, ASP32, GLY34, SER35, TYR71, THR72, GLN73, PHE108, ILE110, TRP115, ILE118, TYR198, LYS224, ILE226, ASP228, GLY230, THR231, THR232, ASN233, ARG235, ARG307, LYS321, SER325, THR329, and VAL332. This indicates that the re-docked ligand retains many of the critical interactions seen in the co-crystallized form, suggesting reliable docking.
In the case of AChE (PDB: 5HF6), the co-crystallized ligand N-ACETYL-beta-D-GLUCOSAMINE [PubChem CID: 24139] interacts primarily with ASN265, THR267, and GLU268. The re-docked ligand N-ACETYL-beta-D-GLUCOSAMINE interacts with residues GLY163, SER164, ARG165, GLU166, ARG245, ASN265, ASP266, and THR267, while retaining the interaction with ASN265. This supports the docking accuracy, even though some new interactions are observed.
For BuChE (PDB: 6EP4), the co-crystallized ligand decamethonium [PubChem CID: 2968] interacts with residues such as ASP70, TRP82, GLY116, TYR128, GLU197, SER198, PRO285, ALA328, PHE329, TYR332, TRP430, HIS438, GLY439, TYR440, and ILE442. The re-docked ligand decamethonium retains most interactions, including ASP70, SER79, TRP82, GLY115, GLY116, TYR128, GLU197, SER198, ALA328, PHE329, TYR332, TRP430, HIS438, GLY439, and TYR440, closely mirroring the co-crystallized interactions. This validates the docking procedure.
Regarding TTBK2 (PDB: 5HF6), the co-crystallized ligand 9IV [PubChem SID: 461502151] interacts with residues including ILE27, GLY28, GLY29, GLY30, ILE35, ALA48, LYS50, CYS78, MET94, GLN95, LEU96, GLN97, ASN100, ALA102, ASP103, SER145, and LEU162. The re-docked ligand 9IV retains interactions with many of the same residues, such as ILE27, GLY28, GLY30, GLY31, GLY33, PHE32, GLU34, ILE35, ALA48, LYS50, VAL51, LEU61, CYS78, MET94, GLN95, LEU96, GLN97, ASN100, SER145, LEU162, and ASP163. The consistency in interactions between the co-crystallized and re-docked ligands indicates reliable docking results.
Molecular dynamic simulation studies
Since molecular docking primarily assesses the geometric fit of molecules at the active site of a protein, a molecular dynamics simulation study was conducted on the prolinamides. This study aimed to evaluate their binding affinities and energies and to perform a more detailed structural analysis (Obakachi et al. 2022; Oladipo et al. 2024).
We utilized the MM/PBSA method, a computational technique described by Ylilauri and Pentikäinen in 2013, to estimate the binding free energies (ΔGbind) between a ligand and a protein/enzyme (Ylilauri and Pentikäinen 2013). After conducting 100 ns molecular dynamic simulations on the molecules, we estimated the binding energy of prolinamides and referenced drugs at the active sites of proteins. The results which are presented in Table 3 show the thermodynamic binding free energy profiles of various prolinamides and reference compounds toward three different enzymes: Beta-Secretase 1 (BACE-1), Butyrylcholinesterase (BuChE), and Tau-tubulin kinase 2 (TTBK2). These profiles include several energy components: Van der Waals energy (Evdw), electrostatic energy (Eelec), gas-phase free energy (ΔGgas), solvation-free energy (ΔGsol), and binding free energy (ΔGbind).
For Beta-Secretase 1 (BACE-1), the reference compound Verubecestat exhibits a ΔGbind of -34.2816 ± 4.8124 kcal/mol, indicating a moderately strong binding affinity. Among the prolinamides, P22 shows a binding free energy of -25.8303 ± 5.2845 kcal/mol, lower than Verubecestat’s, the reference compound. In contrast, P27 has a slightly stronger binding affinity with a ΔGbind of -36.1877 ± 3.7713 kcal/mol. This suggests that P27 might be a more effective inhibitor of BACE-1 than the reference compound Verubecestat.
In the case of Butyrylcholinesterase (BuChE), Rivastigmine serves as the reference compound with a ΔGbind of -22.1872 ± 2.4416 kcal/mol, demonstrating moderate binding affinity. The prolinamides P14 and P19 show significantly stronger binding affinities, with ΔGbind values of -34.1186 ± 3.0861 kcal/mol and − 33.7397 ± 2.8442 kcal/mol, respectively. These results indicate that P14 and P19 could potentially be more potent BuChE inhibitors than Rivastigmine.
For Tau-tubulin kinase 2 (TTBK2), the reference compound 9IV exhibits a strong binding affinity with a ΔGbind of -38.8444 ± 3.4714 kcal/mol. The prolinamide P19 shows a comparable binding affinity with a ΔGbind of -39.2187 ± 5.4971 kcal/mol, suggesting similar efficacy. Notably, P22 demonstrates the strongest binding affinity among all tested compounds for TTBK2, with a ΔGbind of -48.7026 ± 3.6359 kcal/mol, indicating it might be the most effective inhibitor for this enzyme.
Overall, the table reveals that Van der Waals and electrostatic energy significantly contribute to the binding free energy. The gas-phase free energy combines these interactions to reflect the total gas-phase interaction strength. Solvation-free energy represents the energy change due to solvation effects and can offset or enhance the binding free energy. By analyzing the binding free energy values, we can assess the relative effectiveness of the prolinamides in inhibiting their target enzymes compared to the reference drugs. Generally, the prolinamides exhibit competitive or superior binding affinities in each case, suggesting their potential as effective inhibitors.
Protein-ligand interaction
To provide a clearer and better understanding of the interactions between the prolinamides of interest and the amino acid residues at the protein binding pocket, ligand-protein interaction plots were generated for the prolinamides against the proteins following the MD simulation, as shown in Fig. 3. Verubucestat exhibited hydrogen bonds with the THR72, GLN73, and ARG235 residues of BACE1, while also showing amide-π and π-π stacked interactions with the TYR71 and GLY230 residues. Additionally, ASP32 displayed a halogen bond with verubucestat, along with Van der Waals interactions involving the PHE108, ILE118, ASP228, THR232, ASN233, LYS321, and SER325 residues of BACE1. P22 formed hydrogen bonds with the PRO70, THR72, ASP228 (carbon-hydrogen), and GLY230 (carbon-hydrogen) residues of BACE1. It also exhibited alkyl and π-alkyl interactions with the ILE126, ILE226, and VAL332 residues. Furthermore, TYR198 and ASP228 showed π-π T-shaped and π-anion interactions, respectively, with P22, while Van der Waals interactions were observed with ASP32, SER35, GLN73, ILE118, ARG128, LYS224, GLY230, THR231, and THR329 residues. P27 exhibited hydrogen bonds with the PRO73, THR231 (π-donor), and ARG235 (carbon-hydrogen) residues of BACE1, and a π-alkyl interaction with TYR71. Additionally, Van der Waals interactions were present between P27 and the following BACE1 residues: GLY11, GLN12, GLY13, TYR14, LEU30, GLY74, SER229, GLY230, ALA335, THR232, THR72, ASP32, ILE118, PHE108, ILE110, and TRP115.
Rivastigmine displayed carbon-hydrogen bonds with the THR120 and HIS438 residues of BuChE, along with π-π stacked interactions with the TRP82 and HIS438 residues. Van der Waals interactions were observed with the GLY116, TYR128, GLY121, PHE329, ASP70, TRP430, SER79, TYR332, ALA328, GLY439, TYR440, GLU197, and ILE442 residues of BuChE. P14 formed a hydrogen bond with the GLY117 residue and exhibited π-π interactions with the TRP82, PHE329, and HIS438 residues of BuChE. It also showed alkyl and π-alkyl interactions with the ALA328 and TYR332 residues, and Van der Waals interactions with the TRP430, SER79, ASP70, ASN83, THR120, TYR128, ILE442, GLY439, GLY115, LEU286, GLU197, ALA199, and SER198 residues of BuChE. P19 exhibited hydrogen bonds with the GLY117, SER198, and PRO285 (carbon-hydrogen) residues of BuChE, while showing π-π interactions with the TRP82 and PHE329 residues. Additionally, the TYR332 residue exhibited a π-alkyl interaction with P19. Van der Waals interactions were observed with the TYR128, ASP70, GLY115, ALA199, TRP231, PHE398, LEU286, GLY116, THR120, HIS438, GLY439, ILE442, and GLU197 residues of BuChE.
9IV exhibited hydrogen bonds with the GLY30, GLN95, and GLN97 residues of TTBK2, along with π-alkyl interactions with the ILE35, ALA48, LYS50, and CYS78 residues. The PHE32, LEU162, and ASP163 residues of TTBK2 displayed amide-π stacked, π-sigma, and π-anion interactions, respectively. Van der Waals interactions were observed with the ILE27, LEU96, GLY31, MET94, LEU61, GLY33, GLU34, VAL51, GLY28, SER145, and ASN100 residues of TTBK2. P19 formed hydrogen bonds with the LYS50 (carbon-hydrogen), GLN97, and SER145 (carbon-hydrogen) residues of TTBK2. Additionally, the ILE35, LEU61, and LEU162 residues exhibited π-alkyl bonds with P19. The PHE32 and ASP163 residues displayed amide-π stacked and π-anion interactions, respectively. Van der Waals interactions were observed with the GLU52, VAL51, GLU34, ASN146, ALA48, LEU96, ALA148, GLY30, GLY33, and GLY31 residues of TTBK2. P22 exhibited hydrogen bonds with the LYS50, CYS78, and LEU162 residues of TTBK2. It also displayed π-alkyl/alkyl interactions with the ILE35, ALA48, and LEU162 residues. Additionally, ILE35 and ASP163 exhibited π-sigma and π-anion/cation interactions, respectively. Van der Waals interactions were observed with the MET94, GLN97, LEU96, ILE27, ASN100, SER145, GLY28, GLY30, GLY31, GLU34, VAL51, LEU61, PHE32, GLY33, and VAL60 residues of TTBK2.
The interaction plot of BACE1, BuChE, and TTBK2 complexes, as seen in Fig. 3, illustrates the distinct protein-ligand interactions of the prolinamides of interest and reference ligands.

2D interaction plots of protein-ligand interactions of selected prolinamides and the reference compounds with the target proteins implicated in Alzheimer’s disease. Protein-ligand interaction of BACE1 with: (A). Verubecestat (B). P22 (C). P27. Protein-ligand interaction of BuChE with (D). Rivastigmine (E). P14 (F). P19 Protein-ligand interaction of TTBK2 with (G). 9IV (H). P19 (I). P22
Molecular Dynamics Simulation
Ligand-binding effects on protein’s structural stability
To investigate the structural stability of the prolinamides of interest on their protein, we analyzed the root mean square deviation (RMSD), the radius of gyration (RoG), and root mean square fluctuation (RMSF) of alpha carbon (Cα) atoms for the bound system of the prolinamides versus with their reference ligands. This analysis was performed for three proteins: Beta-Secretase 1 (BACE-1), Butyrylcholinesterase (BuChE), and Tau-tubulin kinase 2 (TTBK2 in complex with their various selected Ligands). These parameters were monitored throughout 100 ns molecular dynamic simulations, and their average values were calculated, as shown in Table 4. The corresponding plots are displayed in Fig. 4.
For BACE1 complexes, the binding of the prolinamides P22 and P27 to Beta-Secretase 1 (BACE1), as shown in Table 4; Fig. 4, resulted in lower average RMSD values compared to the binding of the reference ligand, Verubecestat. This indicates that the P22 and P27 complexes were more structurally stable than Verubecestat. Specifically, the RMSD values for Verubecestat, P22, and P27 were 1.775 ± 0.382 Å, 1.408 ± 0.167 Å, and 1.346 ± 0.144 Å, respectively, implying greater stability of P22 and P27 than verubecestat. Similarly, the RoG results values of 21.155 ± 0.071 Å for Verubecestat, 20.845 ± 0.071 Å for P22, and 20.960 ± 0.066 Å for P27 showed that P22 and P27 had lower average RoG values compared to Verubecestat, further supporting their increased stability. The RMSF analysis also revealed that P22 (9.271 ± 2.956 Å) and P27 (113 ± 3.201 Å) exhibited lower fluctuations than Verubecestat (13.203 ± 4.710 Å), suggesting that these prolinamides have a greater influence on the residue stability of BACE1.
For Butyrylcholinesterase (BuChE), the prolinamides P14 (1.337 ± 0.126 Å) and P19 (1.347 ± 0.132 Å) demonstrated lower RMSD values than the reference ligand, Rivastigmine (1.361 ± 0.162 Å), indicating greater stability in binding (Table 4; Fig. 4). In addition, the RoG values of 22.919 ± 0.064 Å for Rivastigmine, 22.767 ± 0.058 Å for P14, and 22.868 ± 0.070 Å for P19 implied that P14 and P19 had lower RoG values compared to Rivastigmine, suggesting more compact and stable complexes. More so, the lower RMSF values of 8.172 ± 2.872 Å for P14 and 8.164 ± 2.748 Å for P19 than Rivastigmine (8.172 ± 2.872 Å) showed that P14 and P19 both had lower fluctuations than Rivastigmine, thus, indicating enhanced residue stability of the former.
In the case of Tau-tubulin kinase 2 (TTBK2), the RMSD values for 9IV, P19, and P22 were 1.646 ± 0.222 Å, 2.075 ± 0.271 Å, and 1.794 ± 0.217 Å, respectively. Both P19 and P22 showed higher RMSD values than the reference ligand, 9IV, indicating less stability (Table 4; Fig. 4). The RoG values of 19.948 ± 0.132 Å for 9IV, 19.999 ± 0.124 Å for P19, and 20.016 ± 0.117 Å for P22 were also obtained. These RoG results showed that the reference ligand had lower average RoG values than P19 and P22, suggesting that the reference ligand forms more compact complexes than P19 and P22. Interestingly, RMSF values of 9.387 ± 3.240 Å for 9IV, 8.569 ± 2.859 Å for P19, and 8.238 ± 3.093 Å for P22 showed that P19 and P22 had lower fluctuations than 9IV, suggesting P19 and P22’s potential for higher binding affinity than 9IV despite having higher RMSD value.

Comparative RMSD, RoG, and RMSF plots of alpha C atoms in the Alzheimer’s disease-related protein system, estimated over 100 ns of molecular dynamic simulations. The plots for (A) RMSD, (B) RoG, and (C) RMSF of alpha carbon (Cα) atoms of the BACE1 system, estimated over 100 ns molecular dynamic simulations, are shown. The plots for (D). RMSD (E). RoG and (F). RMSF of alpha carbon (Cα) atoms of the BuChE system, estimated over 100 ns molecular dynamic simulations, are shown. The plots for (G). RMSD (H). RoG and (I). RMSF of alpha carbon (Cα) atoms of the TTBK2 system, estimated over 100 ns molecular dynamic simulations, are shown
Therefore, the structural stability analysis of the inhibitor complexes reveals that for BACE1, the prolinamides P22 and P27 exhibited greater stability than Verubecestat, making them promising candidates for BACE1 inhibition. For BuChE, P14 and P19 demonstrated higher stability than Rivastigmine, indicating their potential as effective BuChE inhibitors. In the case of TTBK2, while P19 and P22 had higher RMSD values than the reference ligand 9IV, their lower RMSF values suggest they might still be potent inhibitors due to higher binding affinity. Overall, as shown in Table 4; Fig. 4, the prolinamides, particularly P22 and P27 for BACE1 and P14 and P19 for BuChE show promise as more stable and potentially more effective inhibitors than their respective reference ligands.
Evaluation of Physicochemical and Pharmacokinetic properties of selected prolinamides for Alzheimer’s Disease Treatment
The physicochemical and pharmacokinetic properties of the prolinamides of interest are presented in Table 5. The prolinamides studied exhibit molecular weights ranging from 336.34 to 417.48 g/mol, which are within the acceptable range for good drug candidates. The estimated topological polar surface area (TPSA) values for the compounds P14, P19, P22, and P27 fall within the standard range of ≤ 140 Ų, indicating their potential for effective cellular permeation, especially after further optimization. Additionally, the LogP and LogS values of all the prolinamides are within the acceptable thresholds (≤-6 for LogS and < 5 for LogP), suggesting that these compounds can permeate the intestinal epithelium surface, thus establishing their bioavailability. High gastrointestinal absorption predicted for all prolinamides further supports their potential as effective drug candidates. Compound P19, notably among others, was predicted to penetrate the blood-brain barrier (BBB), making it a particularly promising candidate for AD treatment. Also, its favourable physicochemical and pharmacokinetic properties, including its ability to penetrate the BBB and its high affinity for BuChE and TTBK2, justify its candidacy as a novel compound for AD treatment. The selected prolinamides also demonstrate compliance with Lipinski’s Rule of Five, enhancing their drug-likeness. While P14, P22, and P27 show high gastrointestinal absorption, they do not permeate the BBB, which may limit their effectiveness for central nervous system targets. However, they could still be valuable for peripheral targets. These findings highlight the promising potential of prolinamides, particularly P19, as multi-targeted therapeutic agents for AD.
Conclusion
This study underscores the therapeutic potential of prolinamides as multi-targeted agents for AD treatment. The binding affinities of these compounds to key proteins implicated in AD, such as Beta-Secretase 1 (BACE1), Butyrylcholinesterase (BuChE), and Tau-tubulin kinase 2 (TTBK2), were thoroughly analyzed using molecular dynamic simulation studies. Among the twenty-seven prolinamides examined, our findings reveal that P19 exhibited a superior binding affinity for both BuChE and TTBK2 compared to their reference ligands, while P22 demonstrated a higher affinity for both BACE1 and TTBK2 compared to their respective reference ligands. The structural stability analyses of the inhibitor complexes showed that the prolinamides, particularly P22 and P27 for BACE1 and P14 and P19 for BuChE, exhibited greater stability than their reference ligands. This was evidenced by their lower RMSD, RoG, and RMSF values, indicating their potential as effective inhibitors. Recent advances in molecular medicine have highlighted the amyloid-β (Aβ) pathway as central to AD pathophysiology, emphasizing the need for multi-targeted drugs. The compounds identified in this study, particularly P19 and P22, emerged as promising multi-targeted ligands due to their favourable binding affinities and stability profiles. P19’s ability to penetrate the blood-brain barrier further enhances its therapeutic potential.
As AD progresses, a decrease in acetylcholinesterase (AChE) levels and an increase in BuChE activity shift the regulation of acetylcholine to BuChE. Compounds P14 and P19 demonstrated inhibitory effects on BuChE, positioning them as promising candidates for AD treatment. Elevated levels of BACE1 activity in late-onset AD patients further highlight the potential of compounds P22 and P27 as therapeutic agents. The prolinamides studied here also possess desirable physicochemical properties and pharmacokinetic profiles, making them suitable for further development. The potential for modifying these compounds to enhance their pharmacokinetic properties opens new avenues for expanding therapeutic options for AD.
In conclusion, prolinamides present a promising class of compounds for the treatment of AD. Through comprehensive computational analyses, they have demonstrated favourable interactions with key AD-related proteins and exhibited desirable stability and drug-likeness properties. These findings support the potential of prolinamides as novel therapeutic candidates for AD. Further experimental validation is warranted to confirm their efficacy and safety profiles, advancing them toward clinical application in AD management.
Data availability
No datasets were generated or analysed during the current study.
References
Cervellati C, Trentini A, Rosta V, Passaro A, Bosi C, Sanz JM, Bonazzi S, Pacifico S, Seripa D, Valacchi G, Guerini R, Zuliani G (2020) Serum beta-secretase 1 (BACE1) activity as candidate biomarker for late-onset Alzheimer’s disease. GeroScience 42(1):159–167. https://doi.org/10.1007/s11357-019-00127-6
Dahlin JL, Nissink JW, Strasser JM, Francis S, Higgins L, Zhou H, Zhang Z, Walters MA (2015) PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J Med Chem 58(5):2091–2113. https://doi.org/10.1021/jm5019093
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717. https://doi.org/10.1038/srep42717
de Almeida RBM, Barbosa DB, do Bomfim MR, Amparo JAO, Andrade BS, Costa SL, Campos JM, Cruz JN, Santos CBR, Leite FHA, Botura MB (2023) Identification of a novel dual inhibitor of acetylcholinesterase and butyrylcholinesterase: in Vitro and in Silico studies. Pharmaceuticals 16(1):Article1. https://doi.org/10.3390/ph16010095
Franklin MC, Rudolph MJ, Ginter C, Cassidy MS, Cheung J (2016) Structures of paraoxon-inhibited human acetylcholinesterase reveal perturbations of the acyl loop and the dimer interface. Proteins Struct Funct Bioinform 84(9):1246–1256. https://doi.org/10.1002/prot.25073
Fronza MG, Alves D, Praticò D, Savegnago L (2023) The neurobiology and therapeutic potential of multi-targeting β-secretase, glycogen synthase kinase 3β and acetylcholinesterase in Alzheimer’s disease. Ageing Res Rev 90:102033. https://doi.org/10.1016/j.arr.2023.102033
Ghosh AK, Reddy BS, Yen Y-C, Cárdenas EL, Rao KV, Downs D, Huang X, Tang J, Mesecar AD (2016) Design of potent and highly selective inhibitors for human β-secretase 2 (memapsin 1), a target for type 2 diabetes. Chem Sci 7(5):3117–3122. https://doi.org/10.1039/C5SC03718B
Kumar RS, Almansour AI, Arumugam N, Mohammad F, Alshahrani WS, Kotresha D, Altaf M, Azam M, Menéndez JC (2018) Highly functionalized pyrrolidine analogues: stereoselective synthesis and caspase-dependent apoptotic activity. RSC Adv 8(72):41226–41236
Kumar A, Sidhu J, Goyal A, Tsao JW (2024) Alzheimer Disease. In StatPearls. StatPearls Publishing. http://www.ncbi.nlm.nih.gov/books/NBK499922/
Kuzemsky AL (2015) Variational principle of Bogoliubov and generalized mean fields in many-particle interacting systems. Int J Mod Phys B 29(18):1530010. https://doi.org/10.1142/S0217979215300108
Liao J-C, Yang TT, Weng RR, Kuo C-T, Chang C-W (2015) TTBK2: A Tau Protein Kinase beyond Tau Phosphorylation. BioMed Research International, 2015, 575170. https://doi.org/10.1155/2015/575170
Loncharich RJ, Brooks BR, Pastor RW (1992) Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N’-methylamide—PubMed. https://doi.org/10.1002/bip.360320508
Nair PC, Miners JO (2014) Molecular dynamics simulations: From structure function relationships to drug discovery. In Silico Pharmacology, 2. https://doi.org/10.1186/s40203-014-0004-8
Nichols E, Steinmetz JD, Vollset SE, Fukutaki K, Chalek J, Abd-Allah F, Abdoli A, Abualhasan A, Abu-Gharbieh E, Akram TT, Hamad HA, Alahdab F, Alanezi FM, Alipour V, Almustanyir S, Amu H, Ansari I, Arabloo J, Ashraf T, Vos T (2022) Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the global burden of Disease Study 2019. Lancet Public Health 7(2):e105–e125. https://doi.org/10.1016/S2468-2667(21)00249-8
Nicsanu R, Cervellati C, Benussi L, Squitti R, Zanardini R, Rosta V, Trentini A, Ferrari C, Saraceno C, Longobardi A, Bellini S, Binetti G, Zanetti O, Zuliani G, Ghidoni R (2022) Increased serum Beta-secretase 1 activity is an early marker of Alzheimer’s Disease. J Alzheimer’s Disease 87(1):433–441. https://doi.org/10.3233/JAD-215542
Nozal V, Martínez-González L, Gomez-Almeria M, Gonzalo-Consuegra C, Santana P, Chaikuad A, Pérez-Cuevas E, Knapp S, Lietha D, Ramírez D, Petralla S, Monti B, Gil C, Martín-Requero A, Palomo V, de Lago E, Martinez A (2022) TDP-43 modulation by Tau-Tubulin Kinase 1 inhibitors: a New Avenue for future amyotrophic lateral sclerosis therapy. J Med Chem 65(2):1585–1607. https://doi.org/10.1021/acs.jmedchem.1c01942
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: an open chemical toolbox. J Cheminform 3:1–14
Obakachi VA, Kehinde I, Kushwaha ND, Akinpelu OI, Kushwaha B, Merugu SR, Kayamba F, Kumalo HM, Karpoormath R (2022) Structural based investigation of novel pyrazole-thiazole hybrids as dual CDK-1 and CDK-2 inhibitors for cancer chemotherapy. Mol Simul 48(8):687–701
Oladipo SD, Luckay RC, Olofinsan KA, Obakachi VA, Zamisa SJ, Adeleke AA, Badeji AA, Ogundare SA, George BP (2024) Antidiabetes and antioxidant potential of Schiff bases derived from 2-naphthaldehye and substituted aromatic amines: synthesis, crystal structure, Hirshfeld surface analysis, computational, and invitro studies. Heliyon, 10(1)
Osinubi A, Izunobi J, Bao X, Asekun O, Kong J, Gui C, Familoni O (2020) Synthesis and in vitro anticancer activities of substituted N-(4′-nitrophenyl)-l-prolinamides. Royal Soc Open Sci 7(9):200906. https://doi.org/10.1098/rsos.200906
Rauf MA, Zubair S, Azhar A (2015) Ligand docking and binding site analysis with pymol and autodock/vina. Int J Basic Appl Sci 4(2):168
Rosenberry TL, Brazzolotto X, Macdonald IR, Wandhammer M, Trovaslet-Leroy M, Darvesh S, Nachon F (2017) Comparison of the binding of reversible inhibitors to human butyrylcholinesterase and acetylcholinesterase: a crystallographic, kinetic and calorimetric study. Molecules 22(12) Article 12. https://doi.org/10.3390/molecules22122098
Seifert E (2014) OriginPro 9.1: scientific data analysis and graphing software-software review. J Chem Inf Model 54(5):1552. https://doi.org/10.1021/ci500161d
Singh TU, Parida S, Lingaraju MC, Kesavan M, Kumar D, Singh RK (2020) Drug repurposing approach to fight COVID-19. Pharmacol Rep 72:1479–1508
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general amber force field. J Comput Chem 25(9):1157–1174. https://doi.org/10.1002/jcc.20035
Warren GL, Andrews CW, Capelli A-M, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS (2006) A Critical Assessment of Docking Programs and Scoring functions. J Med Chem 49(20):5912–5931. https://doi.org/10.1021/jm050362n
Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, Yin M, Zeng X, Wu C, Lu A (2021) ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res 49(W1):W5–W14
Yadav GD, Deepa, Singh S (2019) Prolinamide-Catalysed Asymmetric Org Transformations ChemistrySelect 4(19):5591–5618
Yiannopoulou KG, Papageorgiou SG (2020) Current and future treatments in Alzheimer Disease: an update. J Cent Nerv Syst Disease 12:1179573520907397. https://doi.org/10.1177/1179573520907397
Ylilauri M, Pentikäinen OT (2013) MMGBSA as a Tool to understand the binding affinities of filamin–peptide interactions. J Chem Inf Model 53(10):2626–2633. https://doi.org/10.1021/ci4002475
Author information
Authors and Affiliations
Contributions
Conceptualization, S.D.O., S.O.O, A.D.O, A.A.B; methodology; S.D.O., S.O.O, A.D.O, A.A.B., V.A; software, S.D.O., S.O.O, A.D.O, A.A.B., O.T.A., O.F., V.A., O.B.A.; validation, S.D.O., S.O.O, A.D.O, A.O.B., O.T.A., O.F., V.A., O.B.A.; formal analysis; S.D.O., S.O.O, A.D.O, A.O.B., O.T.A., O.F., V.A.,; resources’ S.D.O, S.O.O, A.O.B. A.D.O.,; data curation, S.D.O., S.O.O, A.D.O, A.O.B., O.T.A., O.F., V.A., O.B.A.; writing original draft preparation; S.D.O., S.O.O, A.D.O, A.O.B., O.T.A., O.F., V.A., O.B.A., writing review and editing, S.D.O., S.O.O, A.D.O, A.O.B., O.T.A., O.F., V.A., O.B.A.; visualization, S.D.O., S.O.O, A.D.O, A.O.B., O.T.A., O.F., V.A., supervision, S.D.O, O.T.A., O.F., project administration, S.D.O., O.T.A., O.F., V.A funding acquisition, S.D.O., A.D.O.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Olalekan, S.O., Obakachi, V.A., Badeji, A.A. et al. Exploring the therapeutic potential of prolinamides as multi-targeted agents for Alzheimer’s disease treatment: molecular docking and molecular dynamic simulation studies. In Silico Pharmacol. 12, 80 (2024). https://doi.org/10.1007/s40203-024-00250-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40203-024-00250-z