
Abstract
Background
Gastric cancer (GC) is known as the fourth leading cause of cancer-related death and the fifth major cancer in the world, and this is a serious threat to general health all over the world. The lack of early detection markers results in a belated diagnosis, i.e. the final stages, which could be associated with the ineffectiveness of the treatment strategies, and naturally, it leads to poor prognosis. Even though a variety of treatments have been developed, there is a trend of studying traditional medicinal plants, due to the worrying side effect of drugs available in the market.
Methods
In this study, pharmacophore generation and 3D-QSAR model were created using 50 compounds with anti-gastric cancer activity (with IC50 had been reported in the previous studies).
Results
Based on three of the best pharmacophoric hypotheses, virtual screening was performed to discover the top anti-gastric cancer compounds from a database of 183,885 compounds. The selected compounds were used for molecular docking with three protein receptors 7BKG, 4F5B, and 4ZT1 to investigate the intermolecular interactions between these ligands and receptors. Finally, 21 lead compounds with the highest amount of docking score ranging from − 13.366 to -6.404 kcal/mol were selected, and then the ADME/Tox properties of these compounds were calculated. All these compounds have a fitness score above 1.8, a molecular weight of less than 500 g/mol, hydrogen bond donors up to 3, hydrogen bond acceptors up to 8.50, and logP of 1.013 to 4.174. Finally, molecular dynamic simulations for top-scoring ligand-receptor complexes were investigated.
Conclusion
These selected lead compounds have the most anti-gastric cancer effects among the 183,885 compounds in the database. Therefore, lead compounds might be considered for gastric cancer therapy in future studies.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gastric cancer ranks as the fourth leading cause of death and the fifth major cancer in the world [1, 2]. GC is a serious threat to general health all over the world and is one of the most common gastrointestinal malignancies in East Asia [3,4,5]. Due to the lack of early detection markers, GC is usually possible to be diagnosed in the final stages, which is associated with a lack of effective treatment strategies that lead to poor prognostication. Surgery is considered the most effective way to treat GC in the early stages of the disease, although chemotherapy and radiotherapy have been used to treat GC-related death, with a 5-year survival rate below 25% [6,7,8].
Tumor marker analysis is an important tool for cancer prevention. Due to important roles of proteins involved at both the cellular and molecular level, proteomics knowledge has been used in research into various types of cancer, for example, gastric cancer to search for new cancer markers and drug targets [9]. Many studies to date have attempted to identify markers that are effective in GC such as the fibulin-5 [10], nicotinamide N-methyltransferase (NNMT) [10, 11], ANXA1 [10], UQCRC1 [10], Her-2 [10, 12, 13], EGFR [14, 15], carcinoembryonic antigen (CEA) [16], alpha-fetoprotein (AFP) [16], carbohydrate antigen (CA) [16], VEGF [17], c-SRC [18], HGF/MET [19, 20], cancer antigen 19‑9 [21, 22], erb‑b2 receptor tyrosine kinase 2 [21], and E-cadherin [6, 23].
Nicotinamide N-methyltransferase (NNMT) protein is the enzyme that catalyzes the N-methylation reaction of nicotinamide, and high NNMT expression has been reported in the GC tissues. This abnormal expression reveals that NNMT is a potential prognostic biomarker and molecular therapeutic target in early and advanced GC [24, 25]. C-Src is a sub-group of nonreceptor protein tyrosine kinases (SFK) with key roles in intracellular signaling. Upregulation of this factor has been found in gastric cancer with the promotion of cancer cell proliferation and metastasis. Src is considered as a promising therapeutic target for the treatment of gastric cancer [26, 27]. Cadherin-1 or E-cadherin (CDH1) belongs to the cadherin family, membrane proteins, which play role in maintaining cell membrane integrity. Loss or downregulation of E-cadherin has been displayed in GC and this alteration may be considered an effective therapeutic approach for GC treatment [28, 29].
Identifying potential targets for metastasis attenuation and developing strong therapeutic drugs is essential for the effective treatment of GC [30]. The main goal and reason of cancer experiments are controlling the cell cycle by inducing cell death by activating a cell cycle blocker or activating apoptosis [31]. Induction of apoptosis in target cells is a key mechanism that should be considered in anti-cancer drug activity tests. An important strategy in the prevention of chemotherapy and also the use of natural compounds is the actuation of apoptotic pathways by inhibition of anti-apoptotic BLC-2 family or activating TRAIL death receptors [32].
Despite the development of therapies used in the treatment of cancer, due to the worrying side effect of drugs available in the market, a wider platform is provided for researchers to study traditional medicinal plants, given the fact that only 1% of 500,000 (about five thousand) plants known to date have been studied, increase the need to discover bioactive drug compositions [31]. In the development of effective drugs with low toxicity to inhibit tumor recurrence and metastasis, natural and biologically active products are widely used in clinical and basic research. Currently, plant-derived anti-cancer drugs that are used clinically include vinblastine, vincristine, paclitaxel, and camptothecin. Due to the diverse source of medical plants, much research has been done on screening natural compounds as molecular targets for cancer prevention which has led to the discovery of anti-cancer agents [32, 33].
There are many reports of a high effect of apoptosis on gastric cancer cell lines, such as saffron [34], curcumin [35,36,37], quercetin [38, 39], carvacrol [39], berberine [40, 41], gallic acid [42], resveratrol [43, 44], salidroside [45], oleanolic acid [46, 47], anthocyanins [48], stilbenes [32], 6-gingerole [49], ellagic acid [50], and β-sitosterol [50]. These compounds have biological and pharmacological properties including anti-inflammatory, antioxidant, antibacterial, anti-cancer, and anti-growth and they affect many cancer cell lines including gastric cancer by inducing apoptosis and suppressing the proliferation and invasion of cancer cells.
The current project aims to discover new natural anti-gastric cancer compounds in the treatment of gastric cancer. For this aim, a ligand-based pharmacophore hypothesis was generated and a 3D-QSAR model was performed to find common features that can be used to predict the biological properties of ligands, and by using these common features to connect ligands structures and their activities with predicted pIC50. Then the binding of these active compounds to amino acid residues in protein receptors that play an important role in the mechanism of gastric cancer was investigated by molecular docking. In this context, a natural compounds database was created, and by using pharmacophore generation and the 3D-QSAR model, the virtual screening, molecular docking, and molecular dynamic are performed. Ultimately, twenty-one lead compounds were selected from this database.
Methods
Protein preparation
In this study, three protein biomarkers that had an effective and key role in gastric cancer and drug mechanism of action in previously published articles were selected as ligand-protein receptors and their crystal structures were downloaded from the RCSB Protein Data Bank (PDB) (https://www.rcsb.org/pdb). In addition, a list of genes involved in gastric cancer was prepared using previously published articles; then, by determining these genes related to the protein-coding category using the GeneCards database (https://www.genecards.org/), and the identification of these proteins by the UniProt site (https://www.uniprot.org/) was confirmed the same protein biomarkers involved in gastric cancer. The selected structures include PDB IDs 7BKG, 4F5B, and 4ZT1. The structure of these proteins was prepared by using a protein preparation wizard (Maestro version 12.5, 2020). Thus, the addition of hydrogen atoms, the creation of disulfide bonds, the deleting of water molecules beyond 3.00 Å from HET groups, generating HET states using Epik (pH 7 ± 2), filling Missing side chains and loops using prime, and then optimization and minimization using the OPLS3e force field were performed.
Ligand preparation
To convert 2D to 3D structures, the LigPrep module of Maestro version 12.5 (Schrödinger, LLC, New York,) was used, and the settings including OPLS3 force field, ionization states using Epik (pH 7 ± 2) and generating 4 isomers at most per ligand was performed. A database with over 183,885 structures was created. For creating natural compounds database, compounds were downloaded from the AnalytiCon Discovery database (https://www.ac-discovery.com), IBScreenNP database (https://www.ibscreen.com/naturalcompounds), SpecNatural database (https://www.specs.net), and Zinc15 database (http://www.zinc.docking.org/browse/catalogs/naturalproducts).
Developing a pharmacophore model
The PHASE module of Maestro version 12.5 (Schrödinger LLC New York) was used for pharmacophore modeling. A common pharmacophore hypothesis is created by the placement of several pharmacophoric features together that indicates the major binding interactions between the active ligand and the receptor [51]. Pharmacophore features include a hydrogen bond acceptor (A), a hydrogen bond donor (D), a hydrophobic group (H), a negatively charged group (N), a positively charged group (P), and an aromatic ring (R). In the present study, a set of 50 diverse structures with anti-gastric cancer effects were collected from previously published reports (7 of these 50 ligands contain chemotherapeutic drugs used to treat gastric cancer and are FDA approved) with the reported amount of IC50 (The half-maximal inhibitory concentration). Using the following formula, IC50 values were converted to pIC50.
In this study IC50 is the nanomolar (or micromolar) concentration of half maximal inhibitory. All the chemical structures of these ligands and their IC50 values are shown in Table S1 in supplementary data. The Pharm Set column was divided into active and inactive groups, taking into threshold range of pIC50 ≥ 6.5 for active and < 5.5 for inactive. However, only all active ligand conformations are involved in the formation of pharmacophore hypotheses.
Pharmacophore model validation
The validity and significance of pharmacophore models were validated with statistical parameters. In this study evaluating the quality of the pharmacophore model and enrichment calculations were performed using the PHASE application. The validation set consists of two groups, the decoy set (A dataset of 1,000 drug compounds with 400 molecular weight Da (http://www.schrodinger.com/glide_decoy_set)) and the active set (Contains 70 known compounds with anti-cancer effect) that were used for validation. The studied parameters include the enrichment factor, robust initial enhancement, Boltzmann-enhanced discrimination of receiver operating characteristic, and the goodness of hit.
Building a pharmacophore-based 3D QSAR model
The PHASE module of Maestro version 12.5 (Schrödinger) was used for building 3D-QSAR models. Models that link molecular descriptors and encode molecular structure information to the target property of molecules are quantitative structure-activity relationships (QSARs) [52]. There are several methods for quantifying the relationship between structure and activity, one of the most important of which can be partial least-squares regression (PLS) [52]. The most important purpose of creating the QSAR model is to predict the biological activity of new structures. A QSAR model can be performed in two forms: atom-based or pharmacophore-based. In the atom-based model, all atoms are deemed in the entire structure of the molecules, but in the pharmacophore-based model, only the pharmacophoric features that can be matched to the hypothesis are considered. The first model is suitable for congeneric series ligands and the second model is suitable for diverse series ligands that have more flexibility [53, 54]. To create the QSAR model, a set of 50 diverse structures, which have the reported amount of pIC50, were randomly divided into two groups, training set, and test set, considering 80% of the training structures. The training set is used to create the QSAR model and the test set is used to validate the created model. The QSAR model must be validated both internally and externally [51]. Externally validation is performed using the predicted activities of the test set compounds. Internally validation of pharmacophoric hypotheses is performed with statistical parameters including correlation coefficient (R2), cross-validation regression coefficient (q2), the standard deviation of regression (SD), statistical significance (P), and variance ratio (F) [52, 55]. The cross-validation regression coefficient was calculated by two factors, the prediction error sum of squares (PRESS) and the sum of squares of deviation of the experimental values from their mean (SSY), according to the following equation:
where Yexp, Ypred and Ymean indicate the experimental activity of the training set compound, the predicted activity of the training set compound and the mean values of the activity of training set compound, respectively [55]. Also, the efficiency of the model was validated by the determination of the coefficient in prediction (r2 test), according to the following equation:
The Ypredtest, Ytest and Ymean show the predicted activity of the test set compound, the experimental activity of the test set compound and the mean values of the activity of test set compound, respectively [55].
Ligand-based virtual screening
The PHASE module of Maestro version 12.5 was used for ligand and database screening. This module with the best pharmacophores matching, creates a 3D database of hit compounds with the best fitness scoring and keeps an inter-site distance matching tolerance of 2.0 Å to study molecular docking against three protein receptors. For hypotheses with 5 sites, the compounds must be matched at least 4 sites. The fitness score represents the alignment of these compounds on the selected hypotheses. The range of fitness score is 0 to 3 and the score of 3 indicates the most ligand alignment on the hypothesis.
Molecular docking
Glide application of Maestro version 12.5 (Schrödinger) was used to study the docking between prepared protein receptors and ligands. For this purpose, in the receptor grid generation panel, the active binding site of protein was created with a dimension of 20 Å to interact with the ligands. To generate an active site for two protein structures with PDB IDs 7BKG and 4F5B used their native ligand and for protein structure with PDB ID: 4ZT1, the site map application was used with the highest score. Grid box dimensions are shown in Table 1. The ligands docking was performed first by the high throughput virtual screening (HTVS) method and then the first 100 of the compounds by extra precision (XP) method with flexible docking and keeping 10% of the best compound after docking.
ADME and molecular properties (absorption, distribution, metabolism, and excretion)
QikProp application of Maestro version 12.5 (Schrödinger) was used to study the physicochemical properties and Drug-likeness calculations of all hit compounds by applying Lipinski’s rule of five, central nervous system activity (CNS), human oral absorption (PCaco), predicted brain/blood partition coefficient (logBB) and polar surface area (PSA).
Molecular dynamics (MD) simulation
MD simulations were applied for top-scoring ligand-receptor complexes to investigate the ligand-receptor interactions and to confirm their stability. Ligand-receptor complexes selected from docking calculations were moved to molecular dynamics simulations by using GROMACS software. Ligand preparation was done using Swiss Param web server by CHARMM force field. All systems were solvated in a triclinic box with TIP3P water molecules as shown in Fig. S1 in supplementary data. Energy minimization by using SD algorithm for 1 ns, equilibration of system by using NVT and NPT ensembles by maintaining temperature at 300 K and pressure 1 bar, Figs. S2 and S3, were done and a total production run, was completed during 100 ns. Finally, by using VMD and Tecplot, gain trajectories were analyzed.
Results and discussion
The generation of pharmacophore and 3D-QSAR model
Many studies have investigated the anti-gastric cancer effect of natural compounds either in vitro on gastric cancer cell lines or in silico. Among these, we can refer to natural compounds such as saffron [34], curcumin [36], quercetin [38], gallic acid [42], carvacrol [39], and anthocyanins [48]. Also, in the other study, the inhibitory effect of kaempferol on Jack bean urease with a highlighted role in creating gastric cancer has been examined using docking and molecular dynamic (MD) simulation [56]. To the best of our knowledge, to date, few studies, or maybe no study has investigated the inhibitory effect on gastric cancer using virtual screening of a large number of compounds at the same time, pharmacophore modeling, and the 3D-QSAR.
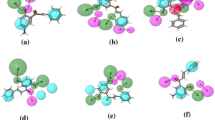
The purpose of this study is to find new natural compounds with the best and most effective anti-gastric cancer properties. So, pharmacophore generation, 3D-QSAR, virtual screening, molecular docking, and molecular dynamic are used to discover these compounds. At first, 183,885 hit compounds were investigated for ADME and physicochemical properties and then 141,173 compounds were selected in ligand and database screening step to be matched with the best pharmacophoric hypotheses. Then 1000 compounds after matching the hypotheses were obtained in the ligand and database screening step which was used for the virtual screening workflow step. Common pharmacophore hypotheses have been created of a set of 23 active ligands in the Pharm Set column that have the maximum and most important structural features required to interact with protein receptors. For this purpose, to match the hypothesis with the active ligands we considered at least 50% matching and the minimum site to be 5 and a maximum site to be 6 to optimum the best feature for creating pharmacophore hypotheses. Eventually, 10 five-feature pharmacophore hypotheses were developed, and the three best pharmacophore hypotheses with the most survival score, site score, vector score, and volume score were chosen (Fig. 1; Table 2).

Three best five-feature pharmacophore hypotheses with the distance of pharmacophoric hypothesis features. A AARRR-1, B AARRR-2, C AARRR-3. Note:(A), hydrogen bond acceptors (Pink sphere with arrows); (R), aromatic ring (yellow open circle) and all distances are in Å units
A good pharmacophoric hypothesis can discriminate between active and inactive ligands. Here the best-developed hypothesis is AARRR, which shows the two groups of the hydrogen acceptor (AA) and three groups of the aromatic ring (RRR) come together. The distance of pharmacophoric hypotheses features is shown in Table 3.
Before the virtual screening, generated pharmacophore hypotheses should be validated using the enrichment factor. The results are shown in Table 4. According to these results, the pharmacophore hypothesis AARRR-2 has shown the highest EF1%, BEDROC, and ROC, which indicates that the prediction ability of hypothesis AARRR-2 is more compared to other hypotheses. Figure 2 shows the active and inactive ligands alignment on pharmacophoric hypothesis AARRR-2.

The alignment of a all active and b all inactive ligands and c most active and d most inactive ligand on the best pharmacophoric hypothesis AARRR-2
The QSAR models were created for three of the top-ranked hypotheses using the atom-based partial least square regression (PLS) method. To generate good 3D-QSAR models, QSAR must be validated. Internal validation of three pharmacophore hypotheses was performed using statistical parameters based on PLS calculations. The statistical parameters of the developed 3D-QSAR models for three of the best pharmacophore hypotheses are shown in Table 5. Although the high value of R2 (squared correlation coefficient), the low value of SD (Standard deviation), the high value of F (variance ratio), and the lowest value of RMSE (root-mean-square error) are the hallmarks of all three hypotheses, however, hypothesis 2 (AARRR-2) has a better prediction ability than the other two. Hypothesis 2 (AARRR-2) showed the value of R2 = 0.94, F = 242, SD = 0.34 and RMSE = 0.98. These statistical parameters indicate the robustness of the developed 3D-QSAR model and pharmacophoric hypothesis. The Scatter plot of the actual and predicted biological activity of the training and the test sets is shown in Fig. 3. It reveals the linear regression model of predicted pIC50 values versus the real activity of training and test sets for the third PLS factor. The effectiveness of the model was determined from the calculated correlation coefficient and Q2 for the randomly selected test set. Therefore, it confirmed the selected model has a good predictive ability.

Scatter plot of actual and predicted biological activity of the training and the test set
The validated hypothesis AARRR-2 obtained from the 3D-QSAR was used to generate the contour map. Contour maps can help understand the importance of functional groups at specific points in a biological activity pathway. These insights can be obtained by comparing the contour maps of ligands with the most and least activity. The results of the hydrogen-bond donor, negative ionic, and positive ionic contour map on the most and least active ligands are shown in Fig. 4. Blue and red cubes show favorable and unfavorable regions of hydrogen bond donor effect, respectively, while pink and green cubes indicate favorable and unfavorable regions of negative ionic effect, and purple and yellow cubes indicate favorable and unfavorable regions of positive ionic effect.

Hydrogen-bond donor effect of a least active and b most active (blue, favorable; red, unfavorable), negative ionic effect of c least active and d most active (pink, favorable; green, unfavorable), positive ionic effect of e least active and f most active (purple, favorable; yellow, unfavorable)
Molecular docking studies
Molecular docking studies were performed using the virtual screening workflow in the Glide application of Maestro version 12.5 (Schrödinger) to investigate the intermolecular interactions between the ligand and the receptor. At first, the HTVS (high throughput virtual screening) method was used for docking, which resulted in 254 compounds, that all of these ligands had a molecular weight of less than 500 g/mol, matched ligand sites above 4, and a fitness score above 1.8. This great fitness score indicates that the ligands are well-matched to the pharmacophoric hypotheses in the ligand and database screening step. These ligands were docked with nicotinamide N-methyltransferase (PDB ID: 7BKG), cadherin-1 (PDB ID: 4ZT1), and proto-oncogene tyrosine-protein kinase Src (PDB ID: 4F5B) receptors. Then, for further analysis, the first 100 compounds obtained from HTVS (with docking score higher than − 7 kcal/mol) were investigated with XP (extra precision) method. Finally, 21 compounds of Glide XP were obtained, and the docking score of these lead compounds was from − 13.366 to -6.404 kcal/mol. The docking score, fitness score, ΔG Bind, amino acid residues involved in the interaction, and predicted pIC50 using the QSAR model of lead compounds are listed in Table 6 and their 2D chemical structure is shown in Fig. 5.

The 2D chemical structures of all the lead compounds are presented
The NA-1 compound with the highest docking score (-13.366 kcal/mol) showed the highest interaction with the nicotinamide N-methyltransferase (PDB ID: 7BKG) receptor compared to other compounds in Table 6. Analysis of this ligand docking results showed that the interactions between the ligand and the active site of the protein were hydrogen bonding and pi-pi stacking. 2D and 3D interactions between the NA-1 compound and nicotinamide N-methyltransferase (PDB ID: 7BKG) receptor are shown in Fig. 6. Important interactions include hydrogen bonding with amino acid residues ASN 90, LEU 164, and TYR 204 and pi-pi stacking with amino acid residues TYR 11 and TYR 204. Also, the study of the first 9 compounds in Table 6, which have docking score values of -13.366 to -10.207 kcal/mol and have the highest interaction with receptor nicotinamide N-methyltransferase (PDB ID: 7BKG) shows that the amino acid residues involved in interaction include TYR 11, ASP 85, ASN 90, VAL 143, LEU 164 and TYR 204.

The 2D (right) and 3D (left) receptor-ligand interaction of NA-1 compound with nicotinamide N-methyltransferase (PDB ID: 7BKG) active site. Important amino acid residues involved in the binding are shown in 2D and 3D interactions
Next, we used the native ligand (UOZ) for the nicotinamide N-methyltransferase (PDB ID: 7BKG) receptor as a control for molecular docking. This native ligand with the docking score values of -7.889 kcal/mol showed the interactions include hydrogen bonding with amino acid residue SER 213 and pi-pi stacking with amino acid residues TYR 204 and TYR 24. The comparison of native ligand and receptor interactions with our selected ligands shows the same active site and interactions.
The NA-10 compound with a docking score of -9.219 kcal/mol showed the highest interaction with the cadherin-1 (PDB ID: 4ZT1) receptor (Table 6). Analysis of the docking results of the NA-10 compound with cadherin-1 (PDB ID: 4ZT1) receptor showed that the important protein-ligand interactions include hydrogen bonding with amino acid residues LEU B:21, SER A:8, and SER B:8 and pi-pi stacking with amino acid residue TRP B:59. 2D and 3D interactions between the NA-10 compound and cadherin-1 (PDB ID: 4ZT1) receptor are shown in Fig. 7. Also, the study of the 8 compounds that have the most interaction with the cadherin-1 (PDB ID: 4ZT1) receptor (-9.219 to -7.695 kcal/mol docking score) shows that the interactions are hydrogen bonding and pi-pi stacking, and the amino acid residues involved in the interaction include SER A:8, SER B:8, LEU A:21, LEU B:21, PRO A:6, PRO B:6, and TRP B:59.

The 2D (right) and 3D (left) receptor-ligand interaction of NA-10 compound with cadherin-1 (PDB ID: 4ZT1) active site. Important amino acid residues involved in the binding are shown in 2D and 3D interactions
Finally, the NA-18 compound (docking score: -7.620 kcal/mol) showed the most interactions with proto-oncogene tyrosine-protein kinase Src (PDB ID: 4F5B) receptor (Table 6). The most important of these interactions include hydrogen bonding with amino acid residues ARG 158, THR 182, ASN 201, HIE 204, SER 180, and GLU 181 and and pi-cation with amino acid residue LYS 198. 2D and 3D interactions between the NA-18 compound and proto-oncogene tyrosine-protein kinase Src (PDB ID: 4F5B) receptor are shown in Fig. 8. The study of the last 4 compounds in Table 6 (-7.620 to -6.404 kcal/mol docking score) which had the most interaction with proto-oncogene tyrosine-protein kinase Src (PDB ID: 4F5B) receptor showed that the NA-19 to the NA-21 compounds had hydrogen bonding interactions. The amino acid residues involved in these protein-ligand interactions include amino acid residues ARG 158, ARG 178, SER 180, GLU 181, THR 182, ASN 201, HIE 204, and LEU 206.

The 2D (right) and 3D (left) receptor-ligand interaction of NA-18 compound with proto-oncogene tyrosine-protein kinase Src (PDB ID: 4F5B) active site. Important amino acid residues involved in the binding are shown in 2D and 3D interactions
Also, the molecular docking study of the native ligand (PTR) with proto-oncogene tyrosine-protein kinase Src (PDB ID: 4F5B) receptor (-8.269 kcal/mol docking score) showed that the most interaction includes hydrogen bonding with amino acid residues HIE 204, ARG 178, ARG 158, SER 180, and GLU 181. So, the active site and the interactions are the same for our selected ligands with the native ligand.
ADME/Tox studies
QikProp application of Schrödinger software was used to predict the pharmacokinetic properties and Drug-likeness calculations of 21 lead compounds. Lipinski’s rule of five (Molecular Weight ≤ 500 g/mol, hydrogen bond donors ≤ 5, hydrogen bond acceptors ≤ 10, octanol-water partition coefficient ≤ 5), central nervous system activity (CNS), human oral absorption, PCaco, brain/blood partition coefficient (logBB) and polar surface area (PSA) were calculated for these 21 lead compounds. The results are reported in Table 7. As it is shown in Table 7, the molecular weight of all lead compounds is below 500 g/mol, hydrogen bond donors are up to 3, hydrogen bond acceptors are up to 8.50 and the octanol-water partition coefficient of all compounds 1.013 to 4.174 is estimated. Also, all compounds have a polar surface area of less than 140 Å, except for the NA-21 compound (with PSA = 143.487 Å). Percent human oral absorption is one of the important factors in predicting pharmacokinetic properties. This study predicted 100% oral absorption for 8 compounds (NA-4, NA-5, NA-8, NA-9, NA-10, NA-13, NA-14, and NA-15), and oral absorption is above 70% for most of the remaining compounds.
Molecular dynamics studies
Molecular dynamics simulations were performed for the most active candidate on each target receptor (NA-1, NA-10, and NA-18). During MD simulation, all systems are checked for structural and movements stabilities inside the active site, Fig. 9. After simulations, average of important parameters, for checking system stabilities during 100 nanoseconds are summarized in Table 8. H-bond interactions, explained what residues from receptors interact with ligands and which one has the most important role, Table 9. Results of energy minimization, total production run steps for three complexes (7BKG/ NA-1, 4ZT1/ NA-10, 4F5B/ NA-18) and finally RMSD plots for most important hydrogen bonding for ensuring stability and working reliability, after simulation of 100 ns, were shown in Figs. 10 and 11, respectively.

Ligands movement from 0-ns to 100-ns. a 7BKG/ NA-1, b 4ZT1/ NA-10, c 4F5B/ NA-18.

Potential plots for a1, a2) 7BKG/ NA-1, b1, b2) 4ZT1/ NA-10, c1, c2) 4F5B/ NA-18, after minimization and total production run steps, respectively

RMSD plots for most important hydrogen bonding of a 7BKG/ NA-1, b 4ZT1/ NA-10, c 4F5B/ NA-18, respectively, after simulation of 100 ns
Conclusions
This study aimed to identify new scaffolds with anti-gastric cancer properties against three protein receptors 7BKG, 4F5B, and 4ZT1. For this purpose, pharmacophoric hypotheses and 3D-QSAR models were generated, using these models, virtual screening was done to discover these new scaffolds. Fifty compounds with anti-gastric cancer properties were used to develop 3D pharmacophore models. Based on three of the best pharmacophore models, virtual screening and finally 3D-QSAR models were performed and the biological activity of hit compounds was approximately predicted. All the selected compounds from this stage had a molecular weight of less than 500 g/mol and a fitness score above 1.8 and matched more than 4 sites with pharmacophore models. Then the molecular docking of these compounds with three receptors 7BKG, 4F5B, and 4ZT1 was done with HTVS and XP methods. Finally, 21 compounds with high docking scores were selected, and ADME properties were calculated for them. Molecular dynamics simulations were performed for top-scoring ligand with their receptors (NA-1, NA-10, and NA-18). The results of our study showed that three pharmacophore models can determine the characteristics of gastric cancer inhibitors and show the relationship between the structure and activity of these compounds using 3D-QSAR models.
Data availability
The data will be available upon request.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. 2018;68(6):394–424.
Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Piñeros M, Znaor A, et al. Cancer statistics for the year 2020: an overview. Int J Cancer. 2021;149(4):778–89.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. 2021;71(3):209–49.
Zaafouri H, Jouini R, Khedhiri N, Khanchel F, Cherif M, Mesbahi M, et al. Comparison between signet-ring cell carcinoma and non-signet-ring cell carcinoma of the stomach: clinicopathological parameters, epidemiological data, outcome, and prognosis—a cohort study of 123 patients from a non-endemic country. World J Surg Oncol. 2022;20(1):1–12.
Liu D, Zhang R, Chen S, Sun B, Zhang K. Analysis of gastric microbiome reveals three distinctive microbial communities associated with the occurrence of gastric cancer. BMC Microbiol. 2022;22(1):1–13.
Jiang X-j, Lin J, Cai Q-h, Zhao J-f, Zhang H-j. CDH17 alters MMP-2 expression via canonical NF-κB signalling in human gastric cancer. Gene. 2019;682:92–100.
Yang Y, Chen Y, Hu Y, Feng Y, Mao Q, Xue W. Outcomes of laparoscopic versus open total gastrectomy with D2 lymphadenectomy for gastric cancer: a systematic review and meta-analysis. Eur J Med Res. 2022;27(1):1–9.
Shi J, Li N, Tang Y, Jiang L, Yang L, Wang S, et al. Total neoadjuvant therapy for locally advanced gastric cancer and esophagogastric junction adenocarcinoma: study protocol for a prospective, multicenter, single-arm, phase II clinical trial. BMC Gastroenterol. 2022;22(1):1–9.
Lee HJ, Venkatarame Gowda Saralamma V, Kim SM, Ha SE, Vetrivel P, Kim EH, et al. Comparative proteomic profiling of tumor-associated proteins in human gastric cancer cells treated with pectolinarigenin. Nutrients. 2018;10(11): 1596.
Wang X, Zhi Q, Liu S, Xue S-L, Shen C, Li Y, et al. Identification of specific biomarkers for gastric adenocarcinoma by ITRAQ proteomic approach. Sci Rep. 2016;6(1):1–15.
Zhu AK, Shan YQ, Zhang J, Liu XC, Ying RC, Kong WC. Exosomal NNMT from peritoneum lavage fluid promotes peritoneal metastasis in gastric cancer. Kaohsiung J Med Sci. 2021;37(4):305–13.
Chen Y, Ye B, Wang C, Nie Y, Qin J, Shen Z. PLOD3 contributes to HER-2 therapy resistance in gastric cancer through FoxO3/Survivin pathway. Cell Death Discovery. 2022;8(1):1–12.
Zhao S, Bi Y, Wang Z, Zhang F, Zhang Y, Xu Y. Accuracy evaluation of combining gastroscopy, multi-slice spiral CT, Her-2, and tumor markers in gastric cancer staging diagnosis. World J Surg Oncol. 2022;20(1):1–10.
Lordick F, Kang Y-K, Chung H-C, Salman P, Oh SC, Bodoky G, et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): a randomised, open-label phase 3 trial. Lancet Oncol. 2013;14(6):490–9.
Wang Y, Yin X, Chen L, Yin Z, Zuo Z. Discovery and evaluation of cytisine N-isoflavones as novel EGFR/HER2 dual inhibitors. Bioorg Chem. 2022;127: 105868.
Xu S, Chen W, Wang Y, Zhang Y, Xia R, Shen J, et al. N6-methyladenosine-related lncRNAs identified as potential biomarkers for predicting the overall survival of asian gastric cancer patients. BMC Cancer. 2022;22(1):1–18.
Ohtsu A, Shah MA, Van Cutsem E, Rha SY, Sawaki A, Park SR, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J Clin Oncol. 2011;29(30):3968–76.
Chu X-d, Lin Z-b, Huang T, Ding H, Zhang Y-r, Zhao Z, et al. Thrombospondin-2 holds prognostic value and is associated with metastasis and the mismatch repair process in gastric cancer. BMC Cancer. 2022;22(1):1–13.
Iveson T, Donehower RC, Davidenko I, Tjulandin S, Deptala A, Harrison M, et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: an open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study. Lancet Oncol. 2014;15(9):1007–18.
Anestis A, Zoi I, Karamouzis MV. Current advances of targeting HGF/c-Met pathway in gastric cancer. Ann Transl Med. 2018;6(12):247.
Yuan J, Tan L, Yin Z, Tao K, Wang G, Shi W, et al. Glis2 redundancy causes chemoresistance and poor prognosis of gastric cancer based on co–expression network analysis. Oncol Rep. 2019;41(1):191–201.
Kim HK, Reyzer ML, Choi IJ, Kim CG, Kim HS, Oshima A, et al. Gastric cancer-specific protein profile identified using endoscopic biopsy samples via MALDI mass spectrometry. J Proteome Res. 2010;9(8):4123–30.
Wang L, Chen J, Zuo Q, Wu C, Yu T, Zheng P, et al. Calreticulin enhances gastric cancer metastasis by dimethylating H3K9 in the E-cadherin promoter region mediating by G9a. Oncogenesis. 2022;11(1):1–11.
Zhang L, Song M, Zhang F, Yuan H, Chang W, Yu G, et al. Accumulation of nicotinamide N-methyltransferase (NNMT) in cancer-associated fibroblasts: a potential prognostic and predictive biomarker for gastric carcinoma. J Histochem Cytochem. 2021;69(3):165–76.
Pozzi V, Campagna R, Sartini D, Emanuelli M. Nicotinamide N-methyltransferase as promising tool for management of gastrointestinal neoplasms. Biomolecules. 2022;12(9): 1173.
Okamoto W, Okamoto I, Yoshida T, Okamoto K, Takezawa K, Hatashita E, et al. Identification of c-Src as a potential therapeutic target for gastric Cancer and of MET activation as a cause of resistance to c-Src inhibitionc-src as a therapeutic target for gastric Cancer. Mol Cancer Ther. 2010;9(5):1188–97.
Nam H-J, Im S-A, Oh D-Y, Elvin P, Kim H-P, Yoon Y-K, et al. Antitumor Activity of Saracatinib (AZD0530), a c-Src/Abl kinase inhibitor, alone or in combination with chemotherapeutic agents in gastric cancerantitumor effects of saracatinib in gastric Cancer. Mol Cancer Ther. 2013;12(1):16–26.
Malpeli G, Barbi S, Innamorati G, Alloggio M, Filippini F, Decimo I, et al. Landscape of druggable molecular pathways downstream of genomic CDH1/Cadherin-1 alterations in gastric Cancer. J Personalized Med. 2022;12(12): 2006.
Yan Q, Wu Y, Li D, Li Y. A-Kinase anchoring protein 9 promotes gastric cancer progression as a downstream effector of cadherin 1. J Oncology 2022:2830634.
Wanga X-z, Gu J-l, Gao M, Bian Y, Liang J-y, Wen H-m, et al. Peperomin E induces promoter hypomethylation of metastatic-suppressor genes and attenuates metastasis in poorly differentiated gastric Cancer. Cell Physiol. 2018;50(6):2341–64.
Ngabire D, Seong Y, Patil MP, Niyonizigiye I, Seo YB, Kim G-D. Induction of apoptosis and G1 phase cell cycle arrest by Aster incisus in AGS gastric adenocarcinoma cells. Int J Oncol. 2018;53(5):2300–8.
Trinh TA, Lee D, Park S, Kim SH, Park JG, Kim JH, et al. Stilbenes contribute to the anticancer effects of Rheum undulatum L. through activation of apoptosis. Oncol Lett. 2019;17(3):2953–9.
Shukla S, Mehta A. Anticancer potential of medicinal plants and their phytochemicals: a review. Brazilian J Bot. 2015;38(2):199–210.
Ashktorab H, Soleimani A, Singh G, Amin A, Tabtabaei S, Latella G, et al. Saffron: the golden spice with therapeutic properties on digestive diseases. Nutr Cancer. 2019;11(5):943.
Alibeiki F, Jafari N, Karimi M, Peeri Dogaheh H. Potent anti-cancer effects of less polar curcumin analogues on gastric adenocarcinoma and esophageal squamous cell carcinoma cells. Sci Rep. 2017;7(1):1–9.
Ham I-H, Wang L, Lee D, Woo J, Kim TH, Jeong HY, et al. Curcumin inhibits the cancer–associated fibroblast–derived chemoresistance of gastric cancer through the suppression of the JAK/STAT3 signaling pathway. Int J Oncol. 2022;61(1):1–12.
Mohammadi A, Khanbabaei H, Zandi F, Ahmadi A, Haftcheshmeh SM, Johnston TP et al. Curcumin: a therapeutic strategy for targeting the Helicobacter pylori-related diseases. Microb Pathog. 2022:105552.
Shang HS, Lu HF, Lee CH, Chiang HS, Chu YL, Chen A, et al. Quercetin induced cell apoptosis and altered gene expression in AGS human gastric cancer cells. Environ Toxicol. 2018;33(11):1168–81.
Sathianarayanan S, Aparna V, Biswas R, Anita B, Sukumaran S, Venkidasamy B. A new approach against Helicobacter pylori using plants and its constituents: a review study. Microb Pathog. 2022:168:105594.
Zhang Q, Wang X, Cao S, Sun Y, He X, Jiang B, et al. Berberine represses human gastric cancer cell growth in vitro and in vivo by inducing cytostatic autophagy via inhibition of MAPK/mTOR/p70S6K and akt signaling pathways. Biomed Pharmacother. 2020;128: 110245.
Li L-L, Peng Z, Hu Q, Xu L-J, Zou X, Huang D-M, et al. Berberine retarded the growth of gastric cancer xenograft tumors by targeting hepatocyte nuclear factor 4α. World J Gastrointest Oncol. 2022;14(4): 842.
Tsai C-L, Chiu Y-M, Ho T-Y, Hsieh C-T, Shieh D-C, Lee Y-J, et al. Gallic acid induces apoptosis in human gastric adenocarcinoma cells. Anticancer Res. 2018;38(4):2057–67.
Kim S, Kim W, Kim D-H, Jang J-H, Kim S-J, Park S-A, et al. Resveratrol suppresses gastric cancer cell proliferation and survival through inhibition of PIM-1 kinase activity. Arch Biochem. 2020;689: 108413.
Wang L-Y, Zhao S, Lv G-J, Ma X-J, Zhang J-B. Mechanisms of resveratrol in the prevention and treatment of gastrointestinal cancer. World J Clin Cases. 2020;8(12):2425.
Qi Z, Tang T, Sheng L, Ma Y, Liu Y, Yan L, et al. Salidroside inhibits the proliferation and migration of gastric cancer cells via suppression of src–associated signaling pathway activation and heat shock protein 70 expression. Mol Med Rep. 2018;18(1):147–56.
Ayeleso TB, Matumba MG, Mukwevho E. Oleanolic acid and its derivatives: biological activities and therapeutic potential in chronic diseases. Molecules. 2017;22(11): 1915.
Li Y, Xu Q, Yang W, Wu T, Lu X. Oleanolic acid reduces aerobic glycolysis-associated proliferation by inhibiting yes-associated protein in gastric cancer cells. Gene. 2019;712: 143956.
Atnip A, Giusti MM, Sigurdson GT, Failla ML, Chitchumroonchokchai C, Bomser JA, et al. The NCI-N87 Cell line as a gastric epithelial model to study cellular uptake, trans-epithelial transport, and gastric anti-inflammatory properties of anthocyanins. Nutr Cancer. 2020;72(4):686–95.
Mathur S, Pareek S, Verma R, Shrivastava D, Bisen PS. Therapeutic potential of ginger bio-active compounds in gastrointestinal cancer therapy: the molecular mechanism. Nutrire. 2022;47(2):1–17.
Singh MP, Chawla V, Kaushik D, Chawla PA, Sisodia SS. Pharmacodynamic stance of phytoconstituents as a gastric Ulcer Protective mechanism: an overview. Curr Mol Med. 2022;22(5):431–41.
Bhansali SG, Kulkarni VM. Pharmacophore generation, atom-based 3D-QSAR, docking, and virtual screening studies of p38-α mitogen activated protein kinase inhibitors: pyridopyridazin-6-ones (part 2). Res Rep Med Chem. 2014;4:1.
Consonni V, Ballabio D, Todeschini R. Comments on the definition of the Q 2 parameter for QSAR validation. J Chem Inf Model. 2009;49(7):1669–78.
Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J Comput Aided Mol Des. 2006;20(10):647–71.
Abbasi M, Sadeghi-Aliabadi H, Amanlou M. Prediction of new Hsp90 inhibitors based on 3, 4-isoxazolediamide scaffold using QSAR study, molecular docking and molecular dynamic simulation. DARU J Pharm Sci. 2017;25:1–16.
Alam S, Khan F. 3D-QSAR, docking, ADME/Tox studies on Flavone analogs reveal anticancer activity through tankyrase inhibition. Sci Rep. 2019;9(1):1–15.
Zolghadr L, Behbehani GR, PakBin B, Hosseini SA, Divsalar A, Gheibi N. Molecular dynamics simulations, molecular docking, and kinetics study of kaempferol interaction on Jack bean urease: comparison of extended solvation model. Food Sci Nutr. 2022;10(11):3585–97.
Acknowledgements
We thank gratefully Dr. Mansour Zahedi and Dr. Bahare Noohi’s efforts and technical support of the Chemistry Computational Center at Shahid Beheshti University. We’re also grateful to Kosar Jalali for her diligent work editing this manuscript.
Author information
Authors and Affiliations
Contributions
Zeinab Jalali did the molecular modeling part and prepared the draft of the manuscript. Samad N Ebrahimi supervised the project. Samad N Ebrahimi and Hassan Rezadoost finalized the manuscript. All authors have approved the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors did not have any conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 2.37 MB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jalali, Z., Nejad Ebrahimi, S. & Rezadoost, H. Identifying natural products for gastric cancer treatment through pharmacophore creation, 3D QSAR, virtual screening, and molecular dynamics studies. DARU J Pharm Sci 31, 243–258 (2023). https://doi.org/10.1007/s40199-023-00480-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40199-023-00480-0