
Abstract
Construction of suitable structural models in order to account for chemical short-range orders is the reason behind the difficult multi-scale computational simulation methods for solid solutions. Herein, using Ti–Mo alloys as representative, we used our cluster-plus-glue-atom model to address the chemical short-range orders for body-center cubic lattice. In accordance with the atomic interaction mode, an Mo solute atom would prefer 14 Ti solvent atoms as its nearest neighbors, forming a rhombic-dodecahedral cluster, and some next outer-shell Mo and Ti atoms would serve as the glue atoms, which is formulated as [Mo–Ti14](Mo,Ti)x. The number of glue atoms x corresponds to different spatial distribution of the clusters. One of the formula having good stability is [Mo–Ti14]Mo, i.e., with one Mo as the glue atom. To verify its stability, mechanical properties and electronic density of state are obtained using the first-principles calculations and the Young’s modulus agrees with the experimental values. Also the formulated structural unit [Mo–Ti14]Mo is indeed verified by the cluster expansion method. This work then confirms the existence of simple structural unit covering the nearest neighbors and a few next outer-shell atoms for the Ti–Mo alloy of high structural stability.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Short-range distribution of solute in a substitutional solid solution is due to agglomeration of atoms in the atomic range. However, computer simulations like SQS [1] and EMTO [2] consider solid solution as being disordered randomly as addressing atomic order in short range is extremely difficult due to simultaneous involvement of order and disorder. On the contrary, specific chemical composition of the good quality alloys which are regularly used in our daily life implies that certain structural units must be responsible for the chemistry of the alloys. Clarification of this fundamental issue has recently been possible due to identification of the structural units using the cluster-plus-glue-atom model [3]. For instance, the most widely used brass, Cu–30Zn, is explained using a unit formulated as [Zn–Cu12]Zn4 [4], but such formulism is only based on speculated atomic interaction mode in accordance with their mixing enthalpies. It is therefore significant to justify such short-range model using state-of-the-art ab initio calculations, which constitute the major objective of the present work.
To carry out the calculations, β-Ti bio-alloys having low Young’s modulus [5, 6] were chosen as the model alloy system. The design of the alloys needs to be proper as it contains both BCC stabilizers (i.e., Mo) and destabilizers (i.e., Sn) with low modulus [7,8,9]. There are several methods available for multicomponent alloy design, such as d-electron concept [10], Mo-equivalence (Moeq) [11] and electron concentration criterion (e/a) [12]. Nevertheless, these prevailing methods do not offer precise composition design and the alloy development is still based on empirical exploration.
Herein, we first establish a model for β-Ti–Mo solid solution alloys based on short-range order using our cluster-plus-glue-atom model. The reason behind the choice of this binary system is that it is the base for all kinds of β-Ti bio-alloys. Our main focus was on the composition of monotectoid point Ti88Mo12, due to highest BCC stability of the alloy. Thereafter, mechanical properties and electronic density of states of the formulated structure are calculated using ab initio method, showing good agreement with the experimental data, while the cluster-plus-glue-atom model [Mo–Ti14]Mo is successfully verified using the cluster expansion (CE) method.
2 Cluster-plus-glue-atom model for β-Ti–Mo BCC alloys
Cluster-plus-glue-atom model is particularly useful in dealing with chemical structures of short-range order. The basis of this model is to derive a local structural unit that covers the nearest neighbors (which are forming a coordination polyhedral cluster) along with few outer-shell glue atoms which represent the chemical short-range order and thereby providing key information on the overall structure of the solid solution [3, 13]. The chemical form of the model is [cluster](glue atoms)x, with the alloy structure being divided into two parts, cluster part and glue-atom part, where the cluster characterizes the local structure, and the glue atoms are located between the clusters. The model has been successfully applied to FCC Cu–Zn brass alloys [3]. For instance, the most widely used α-brass Cu–30Zn is precisely formulated as [Zn–Cu12]Zn4 [4], where the square bracketed part represents the nearest-neighbor cluster that is centered by Zn and shelled by Cu, in accordance with their negative enthalpy of mixing(ΔHCu–Zn = − 6 kJ mol−1).
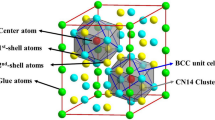
As per literature reports [14,15,16,17], the Ti–Mo BCC alloys are modeled according to following procedure. In BCC Ti–Mo lattice, any Mo atom (the red atom in Fig. 1) is selected as the center of a cluster, and then, solvent Ti atoms are placed in its eight nearest-neighboring sites (the green atoms \(\sqrt 3 a/2\), a being the lattice constant) as well as six second nearest-neighboring sites (the blue atoms, radial distance being a). The reason behind the choice of such a local [Mo–Ti14] rhombic dodecahedron cluster with CN 14 is the negative enthalpy of mixing between Ti and Mo [9, 10] as well as the consideration that the most important interactions fall on the first two sets of nearest neighbors of 14 atoms for BCC alloys [18, 19].

Geometrical configuration of the cluster-plus-glue-atom model for BCC structure, centered by red atom Mo, and shelled by 14 Ti is a rhombic-dodecahedral cluster [Mo–Ti14]. The eight nearest neighbors are shown as green balls, and the six second nearest neighbors are shown as blue balls. The glue atoms locate at the next outer-shell of twelve atoms (gray balls). The Ti–Mo monotectoid alloy Ti88Mo12 is modeled as a [Mo–Ti14] cluster glued with one Mo atom (out of the 12 available gray sites), formulated as [Mo–Ti14]Mo = Ti14Mo2 = Ti87.5Mo12.5
These clusters are then distributed in the BCC lattice without spatial overlapping, to avoid the extension of local cluster-type order to longer range that destroys the stability of the solid solution. In such cases, the distance between two neighboring clusters which is described by vectors must be larger than or equal to vector <331>a/2 [20]. The positions of the glue atoms are supposed to be only at the next outer neighbors of twelve sites (gray atoms in Fig. 1). In cluster-plus-glue-atom model, it is only the glue atoms that can be shared by two clusters. The maximum number of glue atoms is found to be 1–8 as determined experimentally [20], but the maximum cluster packing density is observed for cluster formula with one glue atom. In Ti–Mo binary phase diagram, the composition Ti88Mo12 (at%) of monotectoid reaction corresponds to the stable BCC structure at the lowest temperature. The Ti88Mo12 is thus modeled as a [Mo–Ti14] cluster glued with one Mo atom, formulated as [Mo–Ti14]Mo = Ti14Mo2 = Ti87.5Mo12.5 as per cluster-plus-glue-atom model and whose chemical composition falls close to the experimental one. The glue Mo atom is located in the interstitial sites between the clusters, as shown in Fig. 2.

Parallelepiped super-cell unit satisfying the cluster formula [Mo–Ti14]Mo (monotectoid alloy), defined by linking the [Mo–Ti14] cluster center (red atoms, Mo) as the cell vertices. There is only one Mo glue atom (gray atoms) in the unit. The cluster shell atoms are represented by green and blue atoms
3 Computational Methods
Density functional theory-based calculations are performed using the Blochl’s projector-augmented wave (PAW) [21] method within the generalized gradient approximation (GGA) of Perdew, Burke and Ernzerhof (PBE) parameterization [22], as implemented in the Vienna Ab Initio Simulation Package (VASP) [23]. The electronic wave functions are expanded using a plane-wave basis set with a cutoff energy of 350 eV. The Brillouin zone is sampled employing 5 × 5×6 k-point meshes generated with Monkhorst–Pack scheme [24]. The atomic positions and unit cell parameters are fully relaxed with the conjugate-gradient method.
The cluster expansion method [25] is derived from the well-known Ising Hamiltonian. For a binary substitutional alloy, we firstly introduce a spin-like occupation variable σi to each site: σi = − 1(+1) if site i is occupied by an A (B) atom. Here, vector σ represents a particular configuration of parent lattice containing all occupation variables for each site. Thus, the properties correlated with particular configuration of a lattice can be conveniently calculated using the spin-like occupation variable, for example, energy as below [26]:
where α is a cluster comprised of a group of sites, mα denotes the number of sites per lattice of symmetrically equivalent to clusters α, and Jα is so-called effective cluster interaction (ECI). The summation is over all nonequivalent clusters α as per asymmetry operation of the space group of the parent lattice, whereas the average is taken over all clusters of the lattice with symmetry equivalence. After knowing all ECIs, the property correlated with atomic configuration can be predicted by employing Eq. 1, whereas the unknown ECIs can be determined by the Alloy Theoretic Automated Toolkit (ATAT) code [27], which constructs cluster expansion by fitting the energies of some configurations based on first-principles calculations. It is also implemented in VASP with PAW pseudo-potentials, and the exchange-correlation interactions are also parameterized by PBE functional for GGA. High energy cutoff (30% higher than the maximum cutoff of both potentials) along with 5 × 5 × 5 k-point Monkhorst–Pack meshes ensured numerical convergence.
4 Results and Discussion
4.1 Verification of the cluster-based formula
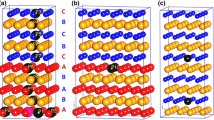
The formation energy of Ti–Mo alloys for each generated structure is searched by CE method with ATAT as a function of Mo concentration and is shown in Fig. 3. The “known str” denotes structures whose energy has been calculated from first principles. The “predicted” denotes structures that are predicted by the cluster expansion, but still not confirmed by first-principles-based calculations. There are three stable structures for the alloy with 12.5% Mo as described in Fig. 4. The first, second and third nearest neighbors of the central Mo atom or Ti atom corresponding to three different structures are listed in Table 1. There are 8 nearest-neighboring Ti atoms and 6 s nearest-neighboring Ti atoms centering any Mo atom in the structural unit [MoTi14]Mo, forming a two-shell cluster according to the cluster-plus-glue-atom model. Glue Mo atoms are located at the next outer-shell (third nearest neighbors).

Energies predicted from the cluster expansion as a function of composition for each structure generated in Ti–Mo alloy

Structures of Ti87.5Mo12.5 obtained by ATAT (a′, b′, c′ show the 1st, 2nd and 3rd nearest neighbor of Mo atom for a, b, c)
We consequently observe the expected structural model in the searched results. The first, second and third nearest neighbors in structure a completely agree with the cluster-plus-glue-atom model, with one [Mo–Ti14] cluster being matched with one Mo glue atom (the apparent two Mo atoms at the third nearest-neighbor shell are actually shared by two adjacent [Mo–Ti14] clusters). In structure b, there is no Mo atom located at the first, second and third nearest neighbors with respect to a central Mo, but they are located with respect to central a Ti atom. Two Mo atoms are also located in the nearest neighbors, and thus, the structural unit in terms of cluster-plus-glue-atom model is [Ti–Mo2Ti12]Ti. For structure c, two Mo atoms are located at the first-neighbor if centered by any Mo atom, but when centered by Ti atom, the formula can be expressed as [Ti–Mo2Ti12]Mo and [Ti–Ti14]Mo. The respective formation energies of the structures a, b and c are − 0.1048 eV, − 0.1013 eV and − 0.0821 eV. Thereby, structure a is found to have the lowest formation energy among the three structures and is the most stable one. It perfectly agrees with the cluster-plus-glue-atom model of BCC structure. Therefore, the formula is confirmed to be a reliable method to describe and design stable solid solution alloys.
4.2 Young’s modulus
The elastic properties of [Mo–Ti14]Mo alloy are calculated by DFT method from the cluster-plus-glue-atom model. The XRD pattern of [MoTi14]Mo alloy [14] indicates stable BCC β-phase at room temperature. Mo has been demonstrated as an effective β-Ti stabilizer with 5 at% Mo being enough for stabilizing BCC β-Ti solid solution [14]. This value is close to the Mo concentration of 6.25% in the as-formulated structure of [Mo–Ti14]Ti, i.e., instead of Mo in the monotectoid formula, the cluster is glued with one glue Ti. For [Mo–Ti14]Mo alloy, the equilibrium density upon relaxation is 5.12 g/cm3, and it agrees well with our own measured value of 5.17 g/cm3 [16] on Ti87.5Mo12.5. The mechanical stability of the structures a, b and c is illustrated by calculating the elastic constants of the structures a, b and c. The elastic stiffness constants Cij (in the Voigt notation) are listed in Table 2. We can see that all the three structures satisfy the criteria of mechanical stability [28] and hence are mechanically stable. The experimental Young’s modulus of Ti87.5Mo12.5 alloy prepared by suction casting is 99 GPa [16], thus agreeing nicely with our DFT calculated value of 103.4 GPa. This coincidence gives a direct validation for the cluster-plus-glue-atom model of Ti–Mo monotectoid alloy.
4.3 Electron density of states
As shown in Fig. 5, the d states of Ti atoms dominate the total electron density of the states of Ti–Mo monotectoid alloy rendering the contribution from Mo atoms negligible. Low Mo content in the alloy (12.5% only), coupled with strong shielding effect over the central Mo atom by the surrounded solvent atoms of Ti, is the reason behind this dominance of Ti atoms.

Total and partial electronic densities of the states of [Mo–Ti14]Mo
The charge density difference [29] and quantity plots of structures a, b and c are calculated in Figs. 6 and 7. The results indicate that charge has transferred between Mo and Ti atoms, and charge densities of 0.0199 e−/Å3 and − 1.2876 e−/Å3 for structure a, 0.0234 e−/Å3 and − 1.2890 e−/Å3 for structure b, and 0.0233 e−/Å3 and − 1.2901 e−/Å3 for structure c. As shown in Fig. 6, the red zone denotes the increased charge, while the blue zone denotes the decreased charge. The charge density accumulation between the Mo and Ti atoms indicates the strong orbital hybridization and bonding of these atoms.

Difference charge density for structures a, b, c. Isovalues: 0.005 and − 0.005 a.u.

Quantity plots of the difference charge density for structures a, b, c. Isovalues: 0.005 and − 0.005 a.u.
5 Conclusion
BCC Ti–Mo alloys have been described by an idealized local unit, [MoTi14](Mo,Ti)x with short-range order as per cluster-plus-glue-atom model where with respect to center Mo atom, Ti atoms cover the first eight and second six nearest-neighboring shell atoms. In the next outer shell which is the third nearest neighbors, there are some Mo atoms with Ti atoms serving as the glue atoms. Varying number of glue atoms x corresponds to different spatial distribution of the clusters. The cluster packing density reaches the maximum in the formula [MoTi14]Mo where x = 1, signifying that the structure having one Mo atom as the glue atom possesses superior structural stability and peculiar properties. The formulated structure has Young’s modulus value calculated as 100.4 GPa and which agrees very well with that of the experimental value 99 GPa. Successful verification of the atomic structure [MoTi14]Mo via CE method supports the application of cluster-plus-glue-atom model in the investigation of nearest-neighbor configurations in solid solutions.
References
A. van de Walle, P. Tiwary, M. de Jong, D.L. Olmsted, M. Asta, A. Dick, D. Shin, Y. Wang, L.Q. Chen, Z.K. Liu, CALPHAD 42, 13 (2013)
L. Vitos, in Computational Quantum Mechanics for Materials Engineers: The EMTO Method and Applications (Springer, London, 2007)
H.L. Hong, Q. Wang, C. Dong, P.K. Liaw, Sci. Rep. 4, 7065 (2014)
D.D. Dong, Dissertation, Dalian University of Technology (2017)
M. Niinomi, T. Hattori, K. Morikawa, T. Kasuga, A. Suzuki, H. Fukui, S. Niwa, Mater. Trans. 43, 2970 (2002)
M. Geetha, A.K. Singh, R. Asokamani, A.K. Gogia, Prog. Mater Sci. 54, 397 (2009)
Y.L. Hao, S.J. Li, S.Y. Sun, C.Y. Zheng, Q.M. Hu, R. Yang, Appl. Phys. Lett. 87, 091906 (2005)
D.Q. Martins, W.R. Oso’rio, M.E.P. Souza, R. Caram, A. Garcia, Electrochim. Acta 53, 2809 (2008)
L.J. Xu, Y.Y. Chen, J. Alloys Compd. 453, 320 (2008)
M. Abdel-Hady, K. Hinoshita, M. Morinaga, Scr. Mater. 55, 477 (2006)
P.J. Bania, in Beta Titanium Alloys in the 1990 s (TMS, Warrendale, 1993)
E.W. Collings, in Physical Metallurgy of Titanium Alloys (ASM, Metals Park, 1984)
Y. Zhang, Q. Wang, H.G. Dong, C. Dong, H.Y. Zhang, X.F. Sun, Acta Metall. Sin. (Engl. Lett.) 31, 127 (2018)
C. Zhang, H. Tian, C.P. Hao, J.J. Zhao, Q. Wang, E.X. Liu, C. Dong, J. Matter. Sci. 48, 3138 (2013)
C.P. Hao, Q. Wang, R.T. Ma, Y.M. Wang, J.B. Qiang, C. Dong, Acta Phys. Sin. 60, 116101 (2011)
Q. Wang, C. Ji, Y.M. Wang, J.B. Qiang, C. Dong, Metall. Mater. Trans. A 44, 1872 (2013)
B.B. Jiang, Q. Wang, D.H. Wen, F. Xu, G.Q. Chen, C. Dong, L. Sun, P.K. Liaw, Mater. Sci. Eng., A 687, 1 (2017)
J. Singh, P. Singh, S. Rattan, S. Prakash, Phys. Rev. B 49, 932 (1994)
H. Sharma, S. Prakash, Pramana 68, 655 (2007)
C. Pang, B.B. Jiang, Y. Shi, Q. Wang, C. Dong, J. Alloys Compd. 652, 63 (2015)
G. Kresse, D. Joubert, Phys. Rev. B 59, 1758 (1999)
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996)
G. Kresse, J. Furthmuller, Phys. Rev. B 54, 11169 (1996)
H.J. Monkhorst, J.D. Pack, Phys. Rev. B 13, 5188 (1976)
J.M. Sanchez, F. Ducastelle, D. Gratias, Physica 128A, 334 (1984)
A. van de Walle, G. Ceder, J. Phase Equilib. 23, 348 (2002)
A. van de Walle, M. Asta, G. Ceder, CALPHAD 26, 539 (2002)
Z.J. Wu, E.J. Zhao, H.P. Xiang, X.F. Hao, X.J. Liu, J. Meng, Phys. Rev. B 76, 054115 (2007)
J.Y. Dai, J.M. Yuan, P. Giannozzi, Appl. Phys. Lett. 95, 232105 (2009)
Acknowledgements
The work was financially supported by the National Key Research and Development Program of China (Grant No. 2016YFB0101206) and the Natural Science Foundation of China (Grant No. 11674045).
Author information
Authors and Affiliations
Corresponding author
Additional information
Available online at http://springerlink.bibliotecabuap.elogim.com/journal/40195.
Rights and permissions
About this article

Cite this article
Jiang, F., Pang, C., Zheng, Z. et al. First-Principles Calculations for Stable β-Ti–Mo Alloys Using Cluster-Plus-Glue-Atom Model. Acta Metall. Sin. (Engl. Lett.) 33, 968–974 (2020). https://doi.org/10.1007/s40195-020-01006-2
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40195-020-01006-2