
Abstract
Purpose of Review
This review aims to examine the current definitions of primary and secondary hemolytic uremic syndromes. Specifically, it seeks to determine which external conditions can result in secondary Thrombotic microangiopathy (TMA), which can trigger cases of primary atypical uremic syndromes (aHUS), and the role of complement in the pathogenesis of TMA spectrum disorders.
Recent Findings
Building on the growing insight about the pathogenic role of dysregulation of the alternative complement pathway in primary aHUS, the successful use of complement-blocking treatment in cases of thrombotic microangiopathy with coexisting conditions (secondary TMA), along with the identification of complement mutations in some of these cases, indicates a so far possibly under-appreciated pathogenic role for complement in diagnoses within the TMA spectrum.
Summary
Uncontrolled complement activity and pro-thrombotic environments represent a unifying pathogenic mechanism in aHUS and the TMA spectrum disorders and point towards shared diagnostic and therapeutic pathways.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
TMA Spectrum
Thrombotic microangiopathy (TMA) defines a group of diseases characterized by non-immune microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and organ injury [1]. TMAs that result in acute kidney injury are referred to as hemolytic uremic syndromes (HUS) [2]. HUS must mainly be differentiated from thrombotic thrombocytopenic purpura (TTP), which is a TMA that manifests with more prominent neurological symptoms and is due to the severely reduced function of ADAMTS13 [3]. The differential clinical features help to characterize HUS, but the identification of possibly underlying genetic causes, primary conditions, and triggering events is important for proper treatment and management of the condition. Historically, HUS has been characterized into diarrhea-associated HUS and the less prevalent atypical HUS (aHUS). Diarrhea-associated HUS is caused by Shiga toxin (Stx)–producing Escherichia coli (STEC-HUS; eHUS) and accounts for 90% of cases of HUS [4]. Cases of TMA that are negative for (Stx) infection and have normal ADAMTS13 function (> 10%) can be attributed to atypical HUS (aHUS, unless they occur secondary to an underlying condition (secondary TMA)).
aHUS involves uncontrolled, chronic activation of the complement cascade, resulting in endothelial damage with neutrophil and platelet recruitment that leads to the clinical triad of HUS (above). The complement system is part of innate immunity and involves systemic and membrane-bound factors mainly designed to lyse invasive pathogens or to remove cellular debris. It is equipped with regulatory mechanisms to prevent self-damage. However, when genetic defects, autoantibodies, or factors such as drugs, toxins, or infections are present, the complement system can be dysregulated to eventually attack host cells, thus creating a pro-inflammatory environment with subsequent tissue damage [5].
Approval of the complement blocker eculizumab has revolutionized the management of patients with aHUS. Eculizumab is a humanized monoclonal antibody to complement protein C5 that inhibits initiation of the terminal pathway and formation of the membrane attack complex (MAC; C5b-9), which when not properly controlled results in endothelial cell damage as seen in aHUS [6]. Atypical cases of HUS differ from STX-HUS as there are increased rates of morbidity and mortality, increased recurrence rates following kidney transplantation, and patterns of familial inheritance [7]. Clinically, the incidence of aHUS is 1–2 cases per million, and there is a poor prognosis, with half of patients progressing to end-stage renal disease (ESRD) and 25% of cases resulting in fatality [8].
In addition to TTP, STEC-HUS, and aHUS, an additional group of TMA exists, referred to as cases of secondary TMA. In these cases, the symptoms of HUS develop due to an underlying condition such as infection, chronic disease, pregnancy, or pharmaceutical use [3, 9]. The complement-blocking C5 antibody eculizumab has been shown to be effective in both patients with primary aHUS and also in a number of patients with secondary TMA. In primary aHUS, 40–50% of cases occur without documented complement mutations or autoantibodies but have been proven to respond equally well to eculizumab [10]. In secondary TMA, there is emerging evidence of a pathogenic role for complement with an increasing number of patients found to carry complement mutations known from primary aHUS.
Given the phenomenon of incomplete genetic penetrance, a “multiple hit” hypothesis has been proposed for the clinical presentation of aHUS [11]. A combination of inter-individual differences in genetic mutation type and preceding environmental stressors determines disease penetrance and influences the time to disease manifestation. Epigenetic factors and environmental triggers have been implicated to cause aHUS symptoms in individuals with an underlying complement mutation. Clinical observation points towards precipitating events such as upper respiratory tract infections or immunizations, which in a majority of cases precede the manifestation of aHUS [3].
This article will briefly review the known genetic causes (Table 1)of primary aHUS and will, in addition, focus on the reported secondary causes for TMA as well as triggering events for the manifestation of TMA (Fig. 1).

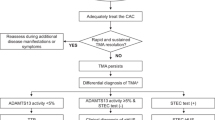
Schematic of TMA diagnoses. Upon presentation of the hallmark features of thrombotic microangiopathy (TMA), which includes thrombocytopenia, hemolytic anemia, and organ injury, further diagnostic measures must be taken to determine the appropriate course of treatment. All TMA are given supportive management, which consists of fluid management, and if the prognosis is severe renal replacement therapy. The first disorder to rule out is thrombotic thrombocytopenic purpura (TTP) due to its high rate of mortality. A diagnosis of TTP is confirmed with enzymatic activity of ADAMTS13 of < 10% and plasma exchange is conducted as the appropriate therapy. Shiga toxin–producing E. coli hemolytic uremic syndrome (STEC-HUS) must be ruled out by testing for Shiga toxin via stool culture, PCR, or serology. After ruling out TTP and STEC-HUS, atypical hemolytic uremic syndrome (aHUS) is the expected diagnoses. The first-line treatment is eculizumab for suspected complement-mediated aHUS. Secondary TMA may or may not respond to eculizumab. Identification of the underlying cause determines the appropriate course of treatment
The Complement System
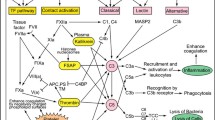
The complement cascade is a component of the innate immune system with both circulating and membrane-bound components designed to protect the individual from invasive pathogens and to remove cellular debris [5]. Complement can be activated through one of the three pathways: classical, lectin, or alternative. The classical pathway is initiated by immunoglobulin binding to antigens resulting in formation of the C1 complex [12]. The lectin pathway is prompted by the presence of mannose-binding lectin (MBL) residues [12]. The alternative pathway is constantly active as C3 is constantly hydrolyzed spontaneously at a low rate (“tick over”) [12]. Each initiation pathway converges with formation of a C3 convertase resulting in protease-mediated cleavage of complement C3 into microbe-opsonizing C3b and inflammatory anaphylatoxin C3a. C3b, in addition to the C3 convertase, forms the C5 convertase, which results in cleavage of complement C5 into anaphylatoxin C5a and C5b, finally resulting in formation of C5b-9 or membrane attack complex (MAC; C5b-9) and cell lysis [12]. Complement is regulated by a series of fluid-phase and surface-bound proteins to prevent host damage by inactivation of complement C3b and accelerating the decay of proteases [5]. In aHUS, the complement, typically mediated by alternative pathway activation, becomes uncontrolled and can result in amplified deposition of complement C3b resulting in endothelial cell injury.
Primary aHUS
Loss of Function Complement Mutations
Mutations in complement-activating or complement-regulating genes can be identified in about 50–60% of aHUS patients [16]. The most common predisposition to aHUS, accounting for 25% of hereditary cases, comes from mutations in the genes encoding products related to complement factor H (FH) [13]. FH, a 150-kDa liver-borne protein circulating in the blood in high concentrations (about 200–500 μg/ml) is considered the most important fluid-phase regulator of complement [14, 15]. The N-terminal domain of FH acts as a fluid-phase cofactor to complement factor I (FI) to aid in inactivation of C3b, as well as accelerate the decay of the C3 convertase [14]. On cell surfaces, the C-terminal domain of FH binds to the glycocalyx of cells, providing protection from self-damage [14]. The majority of FH mutations are missense heterozygous mutations in the C-terminal that result in normal serum FH levels but deletion mutations can occur in other regions resulting in decreased serum levels due to truncation or decreased secretion [17]. In aHUS, these C-terminal mutations eliminate the protective function of FH on the cell surface which results in an unprotected endothelium primed for complement attack. In addition, C-terminal FH mutations result in increased complement activation on platelets and consequently platelet aggregation and clot formation [15]. The prognosis for patients with FH mutations is poor, as 60–70% of patients die or reach end-stage renal disease (ESRD) within a year of onset [13].
Complement factor I (FI) is a serine protease, which together with FH or other cofactors, cleaves and thus inactivates C3b to prevent formation of convertases and decrease complement activation [13]. FI mutations account for 5–10% of aHUS cases [18]. All described mutations in FI are heterozygous and clustered in the serine protease domain of the protein [13]. The prognosis for patients with FI mutations is moderate to severe, as 50% of patients die or progress to ESRD within 2 years and often present with multiple genetic mutations [13].
Ten percent of aHUS mutations occur in the gene that encodes the membrane cofactor protein (MCP/CD46), which is a surface-anchored complement regulator that acts as a cofactor to FI in the process of cleaving and inactivating bound C3b on the host cell [18, 19]. Most MCP/CD46 mutations occur in the extracellular domain of the protein and eliminate protein function [3]. MCP/CD46 mutations are more likely to occur in pediatric patients, confer a lower risk of ESRD than fluid-phase complement mutations, and have positive transplant outcomes [7]. On the contrary, adult onset of MCP/CD46-caused aHUS is associated with significantly worse outcome as compared with other genetic condition [7].
Gain of Function Complement Mutations
Gain-of-function mutations have also been reported in the pathology of aHUS. Mutations in C3 account for 2–10% of aHUS cases and can incur pathology by preventing inactivation of C3b [13]. Forty-eight genetic mutations in C3 have been identified in aHUS patients and often cluster at the FH and MCP/CD46 binding sites, with the leading functional consequence being impaired inactivation of C3b and complement overactivation [20]. The prognosis for patients with a gain of function mutation in C3 is severe [20].
Rare mutations have been reported in complement factor B (FB), with a frequency of up to 4% [13, 30]. These gain of function mutations, located at either directly at the C3b binding site or at the area responsible electrostatic repulsion between FB and FH, which is important for complement regulation, have been reported to result in C3 convertase hyperactivity or resistance to convertase-regulating mechanisms such as FH binding [30]. The prognosis for patients with FB mutations is poor, as the majority have progressed to ESRD [13].
FH and FI Autoantibodies
Autoantibodies against complement pathway regulators FH and FI have also been reported in aHUS patients. FH autoantibodies account for 4–14% of all aHUS cases and are reported to account for 25% of pediatric cases [8, 27]. In some cases, a homozygous deletion in the FHR1 and FHR3 genes has been identified leading to speculation of a genetic predisposition, but other reports support an environmental link or an idiopathic development of autoantibodies [28]. In fact, a sub-group of HUS called deficiency of FHR plasma proteins andautoantibody-positive form HUS (DEAP-HUS) is characterized by having both a genetic deletion in the FHR3 and FHR1 genes as well as acquired autoantibodies [29]. DEAP-HUS occurs primarily in children and has a relatively positive prognosis when treated efficiently [29].
Functionally, FH autoantibodies reduce the binding efficacy of FH to both endothelial surfaces and C3b at short consensus repeats (SCR) 19 and 20 of the C-terminal and inhibit the regulatory effects of FH [28]. Anti-FH antibodies have been shown to be the most common cause of aHUS in pediatric and adult patients, with the average age of onset being approximately 8 years [7, 31].
FI autoantibodies are much less prevalent than FH, with an estimated incidence up to 2% of aHUS cases [3]. Specific genetic mutations have not been associated with FI autoantibodies and patients that present have other susceptibility factors for aHUS [21]. Functionally, these autoantibodies diminish the C3b-inactivating regulatory activity of FI [21]. Age of onset is in adulthood for reported patients with FI autoantibodies and each case required a triggering event to initiate aHUS onset [21].
Patients with autoantibodies generally have a high rate of relapse but have a moderate prognosis with 20–35% of cases progressing to ESRD [28]. As autoantibodies present a unique challenge of complement-mediated disease with an autoimmune component, treatment can consist of plasma exchange, eculizumab therapy to control complement, and immunosuppression with prednisolone and other agents to control the antibody load [22, 31].
Coagulation Cascade Related Mutations
Mutations in the gene encoding for thrombomodulin (THBD/CD141), an important component of clotting regulation, have also been reported to cause aHUS. THBD/CD141 initiates protein C activation by thrombin, which prevents downstream thrombin generation and suppresses clot formation, as well as inhibits fibrinolysis through activation of pro-carboxypeptidase B [23]. THBD has an influence on the complement cascade, as it affects coagulation, inflammation, and C3a and C5a activation and can act as a cofactor to FH to aid in C3b inactivation, and it has been shown to negatively regulate each complement activation pathway [24]. The exact prevalence of THBD/CD141 mutations in aHUS is unknown, but it is considered a rare mutation that results in loss of secreted THBD/CD141 protein [3].
Recently, genetic mutations in PLG, a gene encoding plasminogen of the coagulation pathway, has been implicated in aHUS [25]. Plasminogen deficiency results in decreased degradation of clots found in aHUS patients [25].
Non-Complement-Related Genes
Recessive mutations in diacylglycerol kinase epsilon (DGKE) have been shown to result in a severe aHUS phenotype, characterized by chronic proteinuria, typically in children under the age of 1 [26, 32]. The resulting pathology from the loss of DGKE function is postulated to be related to the development of a pro-thrombotic state, as this mutation has not been linked to any effects on the complement cascade [26]. Two-thirds of DGKE HUS patients are reported to relapse, and eculizumab and immunosuppressive therapies have been unsuccessful in treatment, as the pathogenesis is not complement-mediated [32].
Mutations in the inverted formin 2 gene (INF2) have been identified as the cause of aHUS in two families [33]. INF2 mutations are the most common cause of autosomal dominant nephrotic syndrome and have been associated with recessive DGKE mutations [33]. It is hypothesized that in this non-complement-associated mutation, damage occurs directly on endothelial through impaired secretion of VEGF from podocytes [33]. Anti-complement therapy was unsuccessful in all cases of INF2 aHUS [33].
Secondary TMA
With recent learnings and deeper insight into HUS, historical terminology defining the disease is reaching its limit. Cases presented as secondary TMA have been defined as having no known genetic complement mutation with symptoms due to a precipitating condition. However, many cases of secondary TMA have been shown to respond to anti-complement therapy eculizumab, and in certain cases, an underlying complement mutation has been revealed with genetic testing. It is established that aHUS can be triggered by certain events such as vaccination, pneumococcal infection, and certain drugs, and a “multiple hit” hypothesis has been proposed to explain this phenomenon. This hypothesis suggests that the onset of disease is a consequence of mild or severe disposition in synergy with epigenetic or triggering external factors or conditions, as the presence of one or multiple known genetic mutations does not ensure aHUS manifestation [11, 13]. With current definitions, it is unclear whether certain conditions confer a secondary TMA or trigger a manifestation of aHUS. With further mechanistic insight and increased clinical evidence, it may become clear that predispositions can create complement-amplifying conditions that precipitate an episode of secondary TMA or can add a second hit to a complement-primed system due to genetic mutation. The remainder of this review will present reported cases of HUS that are due to an external condition and examine the role of complement in these cases being a secondary TMA or a trigger to a primary aHUS.
Infections
It has previously been reported that infections, especially caused by Streptococcus pneumoniae, human immunodeficiency virus (HIV), and influenza A can lead to the development of HUS [34,35,36]. Historically, these infectious cases have been referred to as secondary TMA, but recent reports indicate certain infections act as triggers to reveal an underlying complement mutation. Successful treatment with eculizumab has been seen in infectious HUS cases with and without known mutations, indicating a pathogenic role for the complement system. Three cases of HIV-induced TMA or HUS have been successfully treated with eculizumab [37,38,39]. In all reported cases, no known complement mutation was present, yet after unsuccessful treatment with plasma exchange, eculizumab was administered and recovery occurred [37,38,39]. As no complement mutation was present but anti-complement therapy was successful, it is likely that HIV induces cases of complement-amplified secondary TMA.
Recent literature points to influenza B as a trigger for primary aHUS. In 2017, van Hoeve et al. reported three separate cases of HUS triggered by influenza B infection, all revealing an underlying complement mutation; however, all cases occurred before the rise of eculizumab. The article postulates two potential hypotheses for influenza B pathogenesis, with either an altered immune response occurring due TNF-α activation or presence of the Thomsen-Friedenreich antigen overactivating a complement system primed by genetic mutation in the complement cascade [40]. Additional cases have since been reported, both with a pathogenic MCP/CD46 mutation as the underlying cause [41, 42].
In addition to influenza B, Shigella flexneri, Mycoplasma pneumoniae, and Enterococcal infections have all been reported as infectious triggers that revealed an underlying complement mutation resulting in aHUS pathology [43,44,45,46]. Cytomegaloviruses (CMVs) have recently been implicated in aHUS, with two case reports proving success of eculizumab treatment. However, the reports indicate no known complement mutations in these cases, indicating this is a secondary TMA with speculation that the virus is directly damaging endothelial cells [47, 48].
Other newly reported infectious causes of secondary TMA that recovered with eculizumab treatment without a known complement mutation include Enterococcus raffinosus, Clostridium difficile, and varicella [49,50,51]. These new infectious cases reported here underscore the importance of researching into additional pathological and/or genetic mechanisms of aHUS. In certain infectious cases with no known complement mutation, complement activity was detected with elevated CH50, FH, or FI or low C3, indicating there may be unknown genetic mutations or alternate mechanisms mediating the development of TMA [37, 47, 49,50,51]. The evidence for multiple new infectious causes of aHUS indicates that further testing may be needed upon suspicion of aHUS and provide support for a multiple-hit hypothesis of aHUS.
Chronic Diseases
TMA development has been associated with certain chronic diseases, including systemic lupus erythematosus, metabolic disorders, and various types of cancer. Systemic lupus erythematosus (SLE) is an autoimmune disorder targeting multiple organ systems that results in an increased risk for thrombotic events, including TMA in up to 10% of cases [52, 53]. A systematic review and three recent case reports have shown success of anti-complement treatment with eculizumab in cases of SLE with TMA, cases of SLE and lupus nephritis, and cases of SLE and anti-phospholipid syndrome [53,54,55,56,57,58,59]. Mutations in the complement cascade are rare in these conditions, with only two of the cases reported having mutations in FHR3-FHR1 and C3 [53]. The pathological mechanism of TMA in these conditions is currently unknown; however, reported complement cascade mutations indicate that SLE symptoms create a complement amplifying environment that results in the development of TMA.
TMA has been implicated as a rare complication of cancer and its associated treatments [60]. Complement has been shown to be upregulated in ovarian and lung cancers, indicating a system primed for complement overactivation [60, 61]. Additionally, cancer is associated with increased generation of thrombin, which has been shown to efficiently cleave complement C5 without the requirement of a convertase [62]. This pro-complement environment can lead to the development of HUS, especially when coupled with cancer treatments such as chemotherapy.
While an association between the use of chemotherapy drugs and TMA has been previously reported, a recent case has shown a significant improvement in patient health by using anti-complement therapy [63]. A patient receiving gemcitabine treatment developed the clinical triad of TMA symptoms and the condition only improved upon treatment with eculizumab [63]. This patient did not have an underlying genetic complement mutation, indicating that chemotherapy drugs can create a pro-complement environment required to develop a secondary TMA [63]. In addition to chemotherapy, cancer patients may also be treated with VEGF inhibitors to prevent cancer-associated angiogenesis [64]. As VEGF is important in the function of the glomerular fenestrated endothelium, inhibition can result in TMA as seen in both patient cases as well as in an animal model [64, 65]. Complement activation and the use of complement therapy have not been implicated in these cases as discontinuation of anti-VEGF therapy is typically sufficient for symptom improvement. However, VEGF has been shown to increase expression of complement regulators, such as FH, so overactive complement may also contribute to the renal damage seen in VEGF-inhibitor-induced TMA [66].
TMA is considered a rare side effect of sickle cell disease (SCD). SCD is a red blood cell disorder that causes erythrocytes to become rigid and adhesive, leading to hemolysis and vascular membrane damage in a pro-inflammatory and pro-coagulant environment [67]. It has been shown that SCD increases resting alternative pathway activation, leading to a triggering pro-complement environment [68, 69], as seen with increased levels of FB, C3a, C4a, and sC5b-9 at resting state, and even higher levels during painful vaso-occlusive crisis [68,69,70,71,72]. Moreover, the hemolysis seen in SCD (as well as in TMA) results in the release of heme into the bloodstream. Heme has been shown to activate the alternative complement pathway in serum and on endothelial cell surfaces and enhance C3 deposition on sickled RBCs, leading to even more hemolysis [73]. In addition, SCD-derived TMA has been reported to have been successfully treated with eculizumab, since a SCD patient with an underlying CFB mutation responded to this treatment [74].
Metabolic disorders have been linked with HUS development, especially the cobalamin C (CblC) deficiency. CblC deficiency is a subtype of methylmalonic academia (MMA), which is a disease that causes metabolite accumulation and can lead to multiple organ failure [75]. CblC deficiency is caused by a mutation in the methylmalonic acidura and homocystinuria type C (MMACHC) gene [75]. HUS manifestation has been reported in five recent reports of individuals with CblC deficiency [75,76,77,78,79]. All cases presented with a mutation in the MMACHC gene but no mutations in known predisposing complement genes were identified [76,77,78,79]. Two cases responded successfully to eculizumab therapy for treatment of TMA symptoms [77, 78].
A rare case of aHUS due to diabetic ketoacidosis has also been reported, with an underlying CFB mutation being discovered and eculizumab therapy improving the condition [80]. This metabolic condition represents a triggering event that added the second hit necessary for aHUS manifestation.
Transplantation
Certain pharmaceuticals, especially those associated with post-transplantation immunosuppression, have been reported to trigger TMA. The use of calcineurin inhibitors (CNI) like tacrolimus and cyclosporine A, which suppress immune activity by preventing T cell–mediated cytokine upregulation and immune cell proliferation, has resulted in a reported TMA incidence of 1–4% following transplantation [81, 82]. Complement dysregulation has been implicated with CNI usage. A proposed mechanism for pathogenesis speculates that complement becomes activated, leading to downregulation of fibrinolytic factors on endothelial cells of the graft tissue [83]. Cyclosporine has also been shown to have an effect on complement regulatory proteins, resulting in cascade overactivation [84]. Once TMA is identified, the clinical course typically discontinues the use of CNI; however, three recent kidney transplant cases demonstrate the necessity of eculizumab to stabilize renal function [83, 85, 86]. In each of those cases, genetic testing for known complement mutations was not reported, indicating that CNI usage is causing an environment of hyperactive complement activation leading to secondary TMA [83, 85, 86]. Tacrolimus-induced HUS has been reported in patients undergoing heart transplantation, indicating that the diagnosis is not renal transplant specific, but rather results in systemic complement activation [87, 88]. Additionally, it has been reported that individuals with FH and FI mutations are predisposed to the development of de novo TMA post-kidney transplant, with the proposed mechanism linking reduced complement activation as a primer for endothelial damage from agents such as CNI and mTOR inhibitors [89, 90].
TMA is a known side effect of hematopoietic stem cell transplantation (HSTC), occurring in 25–35% of patients [91]. In 2013, Jodele et al. reported six cases of TMA following HSTC transplantation with 83% of these patients testing positive for the FHR1 and FHR3 deletions, and all patients presenting with FH autoantibodies [91]. It is postulated that the anti-FH antibodies prime the patient system for susceptibility to viral or chemotherapy-triggered secondary TMA [91]. In a follow-up study, it was reported that 67% of patients had a complete response to eculizumab [92]. In 2016, a study was published examining genetic susceptibility to TMA following HCST and 65% of patients were found to have an underlying complement mutation, supporting the idea that HSCT, in addition to post-transplant consequences like CNI use or infection, can trigger complement-mediated TMA [93]. Additionally, a cord-blood transplant in a patient resulted in the clinical triad of aHUS symptoms [94]. A diagnosis of HUS unmasked a deletion in the FHR1 gene and the patient was successfully treated with eculizumab, indicating that this was a case of post-transplant-triggered aHUS [94].
Individuals with complement mutations, especially encoding for CFH and CFI, have been shown to be predisposed to de novo TMA development after kidney transplantation [89]. This type of TMA tends to develop within the first year of transplantation and is not related to HUS-induced TMA [89]. As the incidence for de novo TMA development is low at 1–14% after transplantation, it is hypothesized that a complement mutation increases endothelial susceptibility to damage from agents such as CNI or mTOR inhibitors [90]. Currently, no other complement mutations have been linked to de novo TMA development post-transplant or have any complement-blocking therapies used therapeutically in patients with known complement mutations, without HUS presentation, undergoing kidney transplant.
Pregnancy
TMAs are considered rare side effects of pregnancy, occurring in 1 in 25,000 pregnancies, typically with post-natal onset and a high likelihood of end-stage renal disease [95, 96]. Historically, it has been difficult to distinguish if pregnant women are suffering from TTP, preeclamptic hemolysis, elevated liver enzymes, and low platelet count (HELLP) syndrome, or aHUS [97]. TMA in pregnancy has been considered a secondary TMA, with a significant role for complement supported by successful treatment with eculizumab. A study from 2010 estimated that 20% of female HUS was due to pregnancy and that 80% of cases occurred in the postpartum, as this period results in increased inflammation, infections, and hemorrhage leading to complement activation [98]. A recent study indicated the risk is higher for women that delivered via caesarian section, as this leads to heavier bleeding due to increased damage to the endothelium [99]. The role of complement in pathogenesis of pregnancy-associated HUS is supported by the success of eculizumab treatment. Multiple reports state that patients initially treated with plasma exchange therapy, as ADAMTS13 levels must be tested to rule out TTP, do not show improvement [97, 100,101,102]. However, four recent cases have been reported with mutations in genes for C3 and MCP/CD46 with a rapid response to eculizumab therapy [100,101,102,103]. Interestingly, a rare case of aHUS in the first trimester of pregnancy has been reported with eculizumab treatment occurring throughout the remainder of the pregnancy with no adverse effect on fetal health or delivery [104]. This evidence indicates pregnancy can be both a trigger for aHUS and result in secondary TMA, as the perinatal period creates an environment of increased inflammation and hemorrhage.
Hypertension
Secondary TMA can be induced by severe cases of hypertension [105]. The mechanism of pathology in these cases is speculated to be due to shear stress on renal vasculature, or alterations in renin-angiotensin system activation [105, 106]. In a study of 21 patients with renal TMA and malignant hypertension, it was determined that these patients have a reduced incidence of thrombocytopenia and better renal outcomes than patients with aHUS [107]. A role for the AP has recently been implicated as an underlying cause for cases of treatment-resistant hypertension associated TMA. Sixty-seven percent of patients studied showed defective complement genetics as well as elevated circulating and surface levels of complement activation products [105]. Additionally, both patients treated with eculizumab in that study showed a positive response [105]. The prominent historical intervention for hypertension-induced secondary TMA has been blood pressure management, but a prevalence of complement-mediated cases indicates a need for genetic testing and potential eculizumab treatment in individuals with treatment-resistant cases [108].
Conclusion
HUS can be developed due to a genetic predisposition resulting in complement overactivation, an external condition leading to a pro-complement environment, or likely a combination of both factors. The “multiple hit hypothesis” of aHUS that has been previously presented is supported by cases presented in this review that demonstrate a triggering condition unmasking a previously unknown defect in the complement cascade. Alternatively, cases in this review were presented with no known complement mutation, but rather due to hyperactivation of the complement cascade, leading to a secondary TMA. Some external conditions, such as CblC deficiency and cytomegalovirus infection, have only been implicated in cases of secondary TMA, whereas others, such as Influenza B infection, have been shown to trigger a primary manifestation of complement-mediated aHUS. In turn, infections and pregnancy have been implicated as both triggers to primary aHUS and external causes of secondary TMA (Table 2). Perhaps with further research into alternative genetic predispositions, some cases of secondary TMA may be recategorized as complement-mediated aHUS. The efficacy of anti-complement therapy eculizumab in both primary aHUS and secondary TMA cases without a known mutation infers that there are alternative methods of complement overactivation and identification of complement-amplifying conditions and mechanistic insights will aid diagnostic and therapeutic efforts.
Abbreviations
- ADAMTS13:
-
A disintegrin and metalloproteinase with a thrombospondin type 1 motif member 13
- AP:
-
Alternative pathway (complement system)
- aHUS:
-
Atypical hemolytic uremic syndrome
- CMV:
-
Cytomegaloviruses
- CP:
-
Classical pathway (complement system)
- DEAP-HUS:
-
Deficiency of CFHR plasma proteins and autoantibody-positive form of _______hemolytic uremic syndrome
- DGKE:
-
Diacylglycerol kinase epsilon
- ESRD:
-
End-stage renal disease
- FB:
-
Factor B
- FH:
-
Factor H
- FHR:
-
Factor H–related protein
- FI:
-
Factor I
- HELLP:
-
Hemolysis, elevated liver enzymes, and low platelet count
- HSCT:
-
Hematopoietic stem cell transplantation
- HUS:
-
Hemolytic uremic syndrome
- INF2:
-
Inverted formin-2
- LP:
-
Lectin pathway (complement system)
- MAC:
-
Membrane attack complex
- MAHA:
-
Microangiopathic hemolytic anemia
- MBL:
-
Mannose-binding lectin
- MCP:
-
Membrane cofactor protein (CD46)
- SCR:
-
Short consensus repeat
- STEC-HUS/eHUS:
-
Shiga toxin–producing E. coli mediated HUS
- Stx:
-
Shiga toxin
- THBD:
-
Thrombomodulin (CD141)
- TMA:
-
Thrombotic microangiopathy
- TTP:
-
Thrombotic thrombocytopenic purpura
References
George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654–66.
Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16(4):1035–50.
Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol. 2013;33(6):508–30.
Mayer CL, Leibowitz CS, Kurosawa S, Stearns-Kurosawa DJ. Shiga toxins and the pathophysiology of hemolytic uremic syndrome in humans and animals. Toxins. 2012;4(11):1261–87.
Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344(14):1058–66.
Keating GM. Eculizumab: a review of its use in atypical haemolytic uraemic syndrome. Drugs. 2013;73(18):2053–66.
Schaefer F, Ardissino G, Ariceta G, Fakhouri F, Scully M, Isbel N, et al. Clinical and genetic predictors of atypical hemolytic uremic syndrome phenotype and outcome. Kidney Int. 2018;94(2):408–18.
Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clinical journal of the American Society of Nephrology : CJASN. 2010;5(10):1844–59.
Brocklebank V, Wood KM, Kavanagh D. Thrombotic microangiopathy and the kidney. Clin J Am Soc Nephrol. 2018;13(2):300–17.
Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169–81.
Riedl M, Fakhouri F, Le Quintrec M, Noone DG, Jungraithmayr TC, Fremeaux-Bacchi V, et al. Spectrum of complement-mediated thrombotic microangiopathies: pathogenetic insights identifying novel treatment approaches. Semin Thromb Hemost. 2014;40(4):444–64.
Sarma JV, Ward PA. The complement system. Cell Tissue Res. 2011;343(1):227–35.
Kavanagh D, Goodship T. Genetics and complement in atypical HUS. Pediatr Nephrol. 2010;25(12):2431–42.
Ferreira VP, Pangburn MK, Cortes C. Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol. 2010;47(13):2187–97.
Stahl AL, Vaziri-Sani F, Heinen S, Kristoffersson AC, Gydell KH, Raafat R, et al. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood. 2008;111(11):5307–15.
Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(17):1676–87.
Atkinson JP, Goodship THJ. Complement factor H and the hemolytic uremic syndrome. J Exp Med. 2007;204:1245–8.
Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. 2010;31(6):E1445–60.
Richards A, Kathryn Liszewski M, Kavanagh D, Fang CJ, Moulton E, Fremeaux-Bacchi V, et al. Implications of the initial mutations in membrane cofactor protein (MCP; CD46) leading to atypical hemolytic uremic syndrome. Mol Immunol. 2007;44(1–3):111–22.
Schramm EC, Roumenina LT, Rybkine T, Chauvet S, Vieira-Martins P, Hue C, et al. Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood. 2015;125(15):2359–69.
Kavanagh D, Pappworth IY, Anderson H, Hayes CM, Moore I, Hunze EM, et al. Factor I autoantibodies in patients with atypical hemolytic uremic syndrome: disease-associated or an epiphenomenon? Clin J Am Soc Nephrol. 2012;7(3):417–26.
Noone D, Waters A, Pluthero FG, Geary DF, Kirschfink M, Zipfel PF, et al. Successful treatment of DEAP-HUS with eculizumab. Pediatr Nephrol. 2014;29(5):841–51.
Weiler H, Isermann BH. Thrombomodulin. J Thromb Haemost. 2003;1(7):1515–24.
Delvaeye M, DeVriese A, Moons M, Esmon N, Esmon C, Conway EM. Regulation of complement activation by thrombomodulin. Blood: American Society of Hematology; 2009. p. 5127.
Bu F, Maga T, Meyer NC, Wang K, Thomas CP, Nester CM, et al. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2014;25(1):55–64.
Lemaire M, Fremeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet. 2013;45(5):531–6.
Hofer J, Janecke AR, Zimmerhackl LB, Riedl M, Rosales A, Giner T, et al. Complement factor H-related protein 1 deficiency and factor H antibodies in pediatric patients with atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2013;8(3):407–15.
Hofer J, Giner T, Jozsi M. Complement factor H-antibody-associated hemolytic uremic syndrome: pathogenesis, clinical presentation, and treatment. Semin Thromb Hemost. 2014;40(4):431–43.
Zipfel PF, Mache C, Muller D, Licht C, Wigger M, Skerka C. DEAP-HUS: deficiency of CFHR plasma proteins and autoantibody-positive form of hemolytic uremic syndrome. Pediatr Nephrol. 2010;25(10):2009–19.
Marinozzi MC, Vergoz L, Rybkine T, Ngo S, Bettoni S, Pashov A, et al. Complement factor B mutations in atypical hemolytic uremic syndrome—disease-relevant or benign? J Am Soc Nephrol. 2014;25(9):2053–65.
Sinha A, Gulati A, Saini S, Blanc C, Gupta A, Gurjar BS, et al. Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int. 2014;85(5):1151–60.
Azukaitis K, Simkova E, Majid MA, Galiano M, Benz K, Amann K, et al. The phenotypic spectrum of nephropathies associated with mutations in diacylglycerol kinase epsilon. J Am Soc Nephrol. 2017;28(10):3066–75.
Challis RC, Ring T, Xu Y, Wong EKS, Flossmann O, Roberts ISD, et al. Thrombotic microangiopathy in inverted formin 2–mediated renal disease. J Am Soc Nephrol. 2017;28:1084–91.
Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2016;31(1):15–39.
Spinale JM, Ruebner RL, Kaplan BS, Copelovitch L. Update on Streptococcus pneumoniae associated hemolytic uremic syndrome. Curr Opin Pediatr. 2013;25(2):203–8.
Watanabe T. Renal complications of seasonal and pandemic influenza A virus infections. Eur J Pediatr. 2013;172(1):15–22.
Freist M, Garrouste C, Szlavik N, Coppo P, Lautrette A, Heng AE. Efficacy of eculizumab in an adult patient with HIV-associated hemolytic uremic syndrome: a case report. Medicine. 2017;96(51):e9358.
Saab KR, Elhadad S, Copertino D, Laurence J. Thrombotic microangiopathy in the setting of HIV infection: a case report and review of the differential diagnosis and therapy. AIDS Patient Care STDs. 2016;30(8):359–64.
Jin A, Boroujerdi-Rad L, Shah G, Chen JL. Thrombotic microangiopathy and human immunodeficiency virus in the era of eculizumab. Clin Kidney J. 2016;9(4):576–9.
van Hoeve K, Vandermeulen C, Van Ranst M, Levtchenko E, van den Heuvel L, Mekahli D. Occurrence of atypical HUS associated with influenza B. Eur J Pediatr. 2017;176(4):449–54.
Mittal N, Hartemayer R, Jandeska S, Giordano L. Steroid responsive atypical hemolytic uremic syndrome triggered by influenza B infection. J Pediatr Hematol Oncol. 2018.
Kobbe R, Schild R, Christner M, Oh J, Loos S, Kemper MJ. Case report - atypical hemolytic uremic syndrome triggered by influenza B. BMC Nephrol. 2017;18(1):96.
Brocklebank V, Wong EKS, Fielding R, Goodship THJ, Kavanagh D. Atypical haemolytic uraemic syndrome associated with a CD46 mutation triggered by Shigella flexneri. Clin Kidney J. 2014;7(3):286–8.
Miklaszewska M, Zachwieja K, Drozdz D, Pallinger E, Takacs B, Szilagyi A, et al. Hemolytic uremic syndrome with mycoplasma pneumoniae infection and membrane cofactor protein mutation - case report. Przegl Lek. 2016;73(11):862–4.
Omura T, Watanabe E, Otsuka Y, Yoshida Y, Kato H, Nangaku M, et al. Complete remission of thrombotic microangiopathy after treatment with eculizumab in a patient with non-Shiga toxin-associated bacterial enteritis: a case report. Medicine. 2016;95(27):e4104.
Lee MD, Tzen CY, Lin CC, Huang FY, Liu HC, Tsai JD. Hemolytic uremic syndrome caused by enteroviral infection. Pediatr Neonatol. 2013;54(3):207–10.
Java A, Edwards A, Rossi A, Pandey R, Gaut J, Delos Santos R, et al. Cytomegalovirus-induced thrombotic microangiopathy after renal transplant successfully treated with eculizumab: case report and review of the literature. Transpl Int. 2015;28(9):1121–5.
Fraga-Rodriguez GM, Brio-Sanagustin S, Turon-Vinas E, Dixon BP, Carreras-Gonzalez E. Eculizumab in a child with atypical haemolytic uraemic syndrome and haemophagocytic lymphohistiocytosis triggered by cytomegalovirus infection. BMJ Case Rep. 2017.
Mathur P, Hollowoa B, Lala N, Thanendrarajan S, Matin A, Kothari A, et al. Enterococcus raffinosus infection with atypical hemolytic uremic syndrome in a multiple myeloma patient after autologous stem cell transplant. Hematol Rep. 2017;9(3):7094.
Inglis JM, Barbara JA, Juneja R, Milton C, Passaris G, Li JYZ. Atypical haemolytic uraemic syndrome associated with Clostridium difficile infection successfully treated with eculizumab. Case Rep Nephrol. 2018;2018:1759138.
Condom P, Mansuy JM, Decramer S, Izopet J, Mengelle C. Atypical hemolytic uremic syndrome triggered by varicella infection. IDCases. 2017;9:89–90.
Buyon JP. Systemic lupus erythematosus. In: Klipper J, Stone J, Crofford L, White P, editors. Primer on the rheumatic diseases. Springer: Springer; 2008. p. 303–38.
de Holanda MI, Porto LC, Wagner T, Christiani LF, Palma LMP. Use of eculizumab in a systemic lupus erythemathosus patient presenting thrombotic microangiopathy and heterozygous deletion in CFHR1-CFHR3. A case report and systematic review. Clin Rheumatol. 2017;36(12):2859–67.
Bermea RS, Sharma N, Cohen K, Liarski VM. Use of eculizumab in atypical hemolytic uremic syndrome, complicating systemic lupus erythematosus. Journal of clinical rheumatology : practical reports on rheumatic & musculoskeletal diseases. 2016;22(6):320–3.
El-Husseini A, Hannan S, Awad A, Jennings S, Cornea V, Sawaya BP. Thrombotic microangiopathy in systemic lupus erythematosus: efficacy of eculizumab. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2015;65(1):127–30.
Raufi AG, Scott S, Darwish O, Harley K, Kahlon K, Desai S, et al. Atypical hemolytic uremic syndrome secondary to lupus nephritis. Responsive to Eculizumab Hematology reports. 2016;8(3):6625.
Attar RZ, Ramel EI, Safdar OY, Desoky S. A case of patient with renal lupus with an initial presentation of hemolytic uremic syndrome triggered by streptococcal infection. Clinical case reports. 2018;6(4):712–8.
Ono M, Ohashi N, Namikawa A, Katahashi N, Ishigaki S, Tsuji N, et al. A rare case of lupus nephritis presenting as thrombotic microangiopathy with diffuse pseudotubulization possibly caused by atypical hemolytic uremic syndrome. Intern Med. 2018;57(11):1617–23.
Coppo R, Peruzzi L, Amore A, Martino S, Vergano L, Lastauka I, et al. Dramatic effects of eculizumab in a child with diffuse proliferative lupus nephritis resistant to conventional therapy. Pediatr Nephrol. 2015;30(1):167–72.
Weitz IC. Thrombotic microangiopathy in cancer. Thromb Res. 2018;164(Suppl 1):S103–s5.
Sussman TA, Abazeed M, McCrae K, Khorana AA. RNA sequencing approached to identify novel biomarkers for venous thromboembolism (VTE) in lung cancer. Blood: Am Soc Hematol; 2017. p. 554.
Krisinger MJ, Goebeler V, Lu Z, Meixner SC, Myles T, Pryzdial EL, et al. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood. 2012;120(8):1717–25.
Krishnappa V, Gupta M, Shah H, Das A, Tanphaichitr N, Novak R, et al. The use of eculizumab in gemcitabine induced thrombotic microangiopathy. BMC Nephrol. 2018;19(1):9.
Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358(11):1129–36.
Blake-Haskins JA, Lechleider RJ, Kreitman RJ. Thrombotic microangiopathy with targeted cancer agents. Clin Cancer Res. 2011;17(18):5858–66.
Allison SJ. VEGF–complement interactions.
Quinn CT. Minireview: clinical severity in sickle cell disease: the challenges of definition and prognostication. Exp Biol Med (Maywood). 2016;241(7):679–88.
Chudwin DS, Korenblit AD, Kingzette M, Artrip S, Rao S. Increased activation of the alternative complement pathway in sickle cell disease. Clin Immunol Immunopathol. 1985;37(1):93–7.
Gavriilaki E, Mainou M, Christodoulou I, Koravou EE, Paleta A, Touloumenidou T, et al. In vitro evidence of complement activation in patients with sickle cell disease. Haematologica. 2017;102:e481–e2.
Johnston RB Jr. Increased susceptibility to infection in sickle cell disease: review of its occurrence and possible causes. South Med J. 1974;67(11):1342–8.
Mold C, Tamerius JD, Phillips G Jr. Complement activation during painful crisis in sickle cell anemia. Clin Immunol Immunopathol. 1995;76(3 Pt 1):314–20.
Barrett-Connor E. Bacterial infection and sickle cell anemia. An analysis of 250 infections in 166 patients and a review of the literature. Medicine. 1971;50(2):97–112.
Frimat M, Tabarin F, Dimitrov JD, Poitou C, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, et al. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood. 2013;122(2):282–92.
Chonat S, Chandrakasan S, Kalinyak KA, Ingala D, Gruppo R, Kalfa TA. Atypical haemolytic uraemic syndrome in a patient with sickle cell disease, successfully treated with eculizumab. Br J Haematol. 2016;175(4):744–7.
Chen M, Zhuang J, Yang J, Wang D, Yang Q. Atypical hemolytic uremic syndrome induced by CblC subtype of methylmalonic academia: a case report and literature review. Medicine. 2017;96(43):e8284.
Adrovic A, Canpolat N, Caliskan S, Sever L, Kiykim E, Agbas A, et al. Cobalamin C defect-hemolytic uremic syndrome caused by new mutation in MMACHC. Pediatr Int. 2016;58(8):763–5.
Barlas UK, Kihtir HS, Goknar N, Ersoy M, Akcay N, Sevketoglu E. Hemolytic uremic syndrome with dual caution in an infant: cobalamin C defect and complement dysregulation successfully treated with eculizumab. Pediatr Nephrol. 2018;33(6):1093–6.
Ardissino G, Perrone M, Tel F, Testa S, Morrone A, Possenti I, et al. Late onset cobalamin disorder and hemolytic uremic syndrome: a rare cause of nephrotic syndrome. Case Rep Pediatr. 2017;2017:2794060.
Navarro D, Azevedo A, Sequeira S, Ferreira AC, Carvalho F, Fidalgo T, et al. Atypical adult-onset methylmalonic acidemia and homocystinuria presenting as hemolytic uremic syndrome. CEN Case Rep. 2018;7(1):73–6.
Zhu Z, Chen H, Gill R, Wang J, Spitalewitz S, Gotlieb V. Diabetic ketoacidosis presenting with atypical hemolytic uremic syndrome associated with a variant of complement factor B in an adult: a case report. J Med Case Rep. 2016;10:38.
Williams CR, Gooch JL. Calcineurin inhibitors and immunosuppression - a tale of two isoforms. Expert Rev Mol Med. 2012;14:e14.
Trimarchi HM, Truong LD, Brennan S, Gonzalez JM, Suki WN. FK506-associated thrombotic microangiopathy: report of two cases and review of the literature. Transplantation. 1999;67(4):539–44.
Merola J, Yoo PS, Schaub J, Smith JD, Rodriguez-Davalos MI, Tichy E, et al. Belatacept and eculizumab for treatment of calcineurin inhibitor-induced thrombotic microangiopathy after kidney transplantation: case report. Transplant Proc. 2016;48(9):3106–8.
Renner B, Klawitter J, Goldberg R, McCullough JW, Ferreira VP, Cooper JE, et al. Cyclosporine induces endothelial cell release of complement-activating microparticles. J Am Soc Nephrol. 2013;24(11):1849–62.
Ikeda T, Okumi M, Unagami K, Kanzawa T, Sawada A, Kawanishi K, et al. Two cases of kidney transplantation-associated thrombotic microangiopathy successfully treated with eculizumab. Nephrology (Carlton). 2016;21(Suppl 1):35–40.
Shochet L, Kanellis J, Simpson I, Ta J, Mulley W. De novo thrombotic microangiopathy following simultaneous pancreas and kidney transplantation managed with eculizumab. Nephrology (Carlton). 2017;22(Suppl 1):23–7.
Gray JM, Ameduri RK. Tacrolimus-associated hemolytic uremic syndrome in a pediatric heart transplant recipient. Pediatr Transplant. 2016;20(6):866–7.
Vardas PN, Hashmi ZA, Hadi MA. Identification and management of atypical hemolytic uremic syndrome immediately post-heart transplantation. J Card Surg. 2015;30(4):373–5.
Le Quintrec M, Lionet A, Kamar N, Karras A, Barbier S, Buchler M, et al. Complement mutation-associated de novo thrombotic microangiopathy following kidney transplantation. Am J Transplant. 2008;8(8):1694–701.
Garg N, Rennke HG, Pavlakis M, Zandi-Nejad K. De novo thrombotic microangiopathy after kidney transplantation. Transplant Rev (Orlando). 2018;32(1):58–68.
Jodele S, Licht C, Goebel J, Dixon BP, Zhang K, Sivakumaran TA, et al. Abnormalities in the alternative pathway of complement in children with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Blood. 2013;122(12):2003–7.
Jodele S, Fukuda T, Vinks A, Mizuno K, Laskin BL, Goebel J, et al. Eculizumab therapy in children with severe hematopoietic stem cell transplantation–associated thrombotic microangiopathy. Biol Blood Marrow Transplant. 2014;20(4):518–25.
Jodele S, Zhang K, Zou F, Laskin B, Dandoy CE, Myers KC, et al. The genetic fingerprint of susceptibility for transplant-associated thrombotic microangiopathy. Blood. 2016;127(8):989–96.
Hasegawa D, Saito A, Nino N, Uemura S, Takafuji S, Yokoi T, et al. Successful treatment of transplantation-associated atypical hemolytic uremic syndrome with eculizumab. J Pediatr Hematol Oncol. 2018;40(1):e41–e4.
Dashe JS, Ramin SM, Cunningham FG. The long-term consequences of thrombotic microangiopathy (thrombotic thrombocytopenic purpura and hemolytic uremic syndrome) in pregnancy. Obstet Gynecol. 1998;91(5 Pt 1):662–8.
Bruel A, Kavanagh D, Noris M, Delmas Y, Wong EKS, Bresin E, et al. Hemolytic uremic syndrome in pregnancy and postpartum. Clin J Am Soc Nephrol. 2017;12(8):1237–47.
Saad AF, Roman J, Wyble A, Pacheco LD. Pregnancy-associated atypical hemolytic-uremic syndrome. AJP Rep. 2016;6(1):e125–8.
Fakhouri F, Roumenina L, Provot F, Sallee M, Caillard S, Couzi L, et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol. 2010;21(5):859–67.
Huerta A, Arjona E, Portoles J, Lopez-Sanchez P, Rabasco C, Espinosa M, et al. A retrospective study of pregnancy-associated atypical hemolytic uremic syndrome. Kidney Int. 2018;93(2):450–9.
Chua J, Paizis K, He SZ, Mount P. Suspected atypical haemolytic uraemic syndrome in two post-partum patients with foetal-death in utero responding to eculizumab. Nephrology (Carlton). 2017;22(Suppl 1):18–22.
Gately R, San A, Kurtkoti J, Parnham A. Life-threatening pregnancy-associated atypical haemolytic uraemic syndrome and its response to eculizumab. Nephrology (Carlton). 2017;22(Suppl 1):32–5.
Baghli S, Abendroth C, Farooq U, Schaub JA. Atypical presentation of pregnancy-related hemolytic uremic syndrome. Am J Kidney Dis. 2018;72:451–6.
Saad AF, Roman J, Wyble A, Pacheco LD.
Andries G, Karass M, Yandrapalli S, Linder K, Liu D, Nelson J, et al. Atypical hemolytic uremic syndrome in first trimester pregnancy successfully treated with eculizumab. Exp Hematol Oncol. 2017;6:4.
Timmermans S, Abdul-Hamid MA, Vanderlocht J, Damoiseaux J, Reutelingsperger CP, van Paassen P. Patients with hypertension-associated thrombotic microangiopathy may present with complement abnormalities. Kidney Int. 2017;91(6):1420–5.
Nzerue C, Oluwole K, Adejorin D, Paueksakon P, Fremont R, Akatue R, et al. Malignant hypertension with thrombotic microangiopathy and persistent acute kidney injury (AKI). Clin Kidney J. 2014;7(6):586–9.
Zhang B, Xing C, Yu X, Sun B, Zhao X, Qian J. Renal thrombotic microangiopathies induced by severe hypertension. Hypertens Res. 2008;31(3):479–83.
Thind G, Kailasam K. Malignant hypertension as a rare cause of thrombotic microangiopathy. BMJ Case Rep. 2017.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Renal
Rights and permissions
About this article

Cite this article
Jacobs, E., Ortiz, C. & Licht, C. The Role of Complement in the Pathogenesis of HUS and the TMA Spectrum Disorders. Curr Pediatr Rep 7, 1–11 (2019). https://doi.org/10.1007/s40124-019-00186-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40124-019-00186-5