
Abstract
Introduction
Foslevodopa/foscarbidopa, a soluble formulation of levodopa/carbidopa (LD/CD) prodrugs for the treatment of Parkinson’s disease (PD), is administered as a 24-hour/day continuous subcutaneous infusion (CSCI) with a single infusion site. The efficacy and safety of foslevodopa/foscarbidopa versus oral immediate-release LD/CD was previously demonstrated in patients with PD in a 12-week, randomized, double-blind, phase 3 trial (NCT04380142). We report the results of a separate 52-week, open-label, phase 3 registrational trial (NCT03781167) that evaluated the safety/tolerability and efficacy of 24-hour/day foslevodopa/foscarbidopa CSCI in patients with advanced PD.
Methods
Male and female patients with levodopa-responsive PD and ≥ 2.5 hours of “Off” time/day received 24-hour/day foslevodopa/foscarbidopa CSCI at individually optimized therapeutic doses (approximately 700–4250 mg of LD per 24 hours) for 52 weeks. The primary endpoint was safety/tolerability. Secondary endpoints included changes from baseline in normalized “Off” and “On” time, percentage of patients reporting morning akinesia, Movement Disorder Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS), Parkinson’s Disease Sleep Scale–2 (PDSS-2), 39-item Parkinson’s Disease Questionnaire (PDQ-39), and EuroQol 5-dimension questionnaire (EQ-5D-5L).
Results
Of 244 enrolled patients, 107 discontinued, and 137 completed treatment. Infusion site events were the most common adverse events (AEs). AEs were mostly nonserious (25.8% of patients reported serious AEs) and mild/moderate in severity. At week 52, “On” time without troublesome dyskinesia and “Off” time were improved from baseline (mean [standard deviation (SD)] change in normalized “On” time without troublesome dyskinesia, 3.8 [3.3] hours; normalized “Off” time, −3.5 [3.1] hours). The percentage of patients experiencing morning akinesia dropped from 77.7% at baseline to 27.8% at week 52. Sleep quality (PDSS-2) and quality of life (PDQ-39 and EQ-5D-5L) also improved.
Conclusion
Foslevodopa/foscarbidopa has the potential to provide a safe and efficacious, individualized, 24-hour/day, nonsurgical alternative for patients with PD.
Trial Registration Number
ClinicalTrials.gov identifier NCT03781167.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Foslevodopa/foscarbidopa is a soluble formulation of levodopa/carbidopa (LD/CD) prodrugs administered as a continuous (24-hour/day) subcutaneous infusion that has shown a favorable benefit/risk profile versus immediate-release LD/CD in a 12-week, randomized, double-blind, active-controlled, phase 3 study (NCT04380142). |
This open-label registrational trial (NCT03781167) evaluated the safety/tolerability and efficacy of 52 weeks of treatment with foslevodopa/foscarbidopa in patients with advanced Parkinson's disease (PD) whose symptoms were not adequately controlled with oral medication. |
What was learned from the study? |
The continuous (24-hour/day) and individualized subcutaneous infusion of foslevodopa/foscarbidopa has the potential to become an efficacious, nonsurgical treatment alternative for patients with advanced PD. |
The most common adverse events were infusion site events, the majority of which were mild/moderate in severity and nonserious. |
There were improvements in motor fluctuations, morning akinesia, sleep, and quality of life that were observed from the first post-baseline visit and persisted through 52 weeks of treatment. |
Introduction
Parkinson's disease (PD) is a neurodegenerative disorder characterized by progressive degeneration of dopaminergic neurons [1, 2]. Oral administration of levodopa (LD) with carbidopa (CD) is the gold standard pharmacological treatment for PD, often effectively controlling motor symptoms in the early stages of the disease [2, 3]. However, as disease progresses, the buffering capacity of striatal dopaminergic neurons narrows and brain dopamine concentrations become increasingly dependent on plasma LD concentrations [2]. The short plasma half-life of LD and the unreliable absorption of oral LD tablets due to impaired gastric motility are responsible for the fluctuating LD plasma concentrations, which make it more challenging to adequately control PD symptoms [3,4,5]. As a result, patients develop disabling motor fluctuations, alternating between periods of good motor system control ("On" time) and periods of poor mobility, tremor, slowness, and stiffness ("Off" time); dyskinesias, defined as involuntary choreiform movements that typically occur during the "On" time (peak-dose dyskinesia), can also develop and worsen over time [2]. To counteract motor complications, patients rely on increased doses and frequency of oral LD and/or additional concomitant medications such as dopamine agonists, anticholinergics, amantadine, selective monoamine oxidase (MAO) B inhibitors, and catechol-O-methyltransferase (COMT) inhibitors [5]. These complex drug regimens increase the risk of drug–drug interactions, prove burdensome to patients, and may promote medication nonadherence because of the number of tablets that must be taken [6,7,8].
Non-motor symptoms of PD, which include autonomic, neuropsychiatric, and sleep dysfunction, can precede or accompany motor complications [9,10,11,12] and can respond to dopaminergic therapy, but because most PD medication is administered during waking hours, a nocturnal treatment gap remains [5, 13]. In particular, sleep disturbances that may result from suboptimal nighttime treatment can negatively impact health-related quality of life (HRQoL), resulting in excessive daytime sleepiness and impaired daytime functioning for both patients and their care partners [14].
The inability to adequately control PD symptoms with oral treatment can be mitigated by delivering therapy in a continuous, rather than intermittent, fashion. Continuous therapy, including deep brain stimulation (DBS), LD/CD intestinal gel (LCIG), or continuous subcutaneous apomorphine infusion (CSAI), often necessitates the use of medical devices. While these device-aided therapies are efficacious, patients are not always amenable to considering them, either because of their invasiveness (fear of surgery and/or associated complications, cosmetic concerns) or because of possible side effects. The relegated availability to specialized centers in some areas and the regional application of international guidelines on the use of advanced therapies in PD further impact the consideration of these therapies among patients and physicians [15,16,17,18]. For example, DBS may not be recommended for elderly patients or patients with dementia or psychotic disorders [5, 18]. Similarly, the progressive loss of efficacy and the panniculitis-related reduction in apomorphine absorption may limit the use of CSAI in some patients [16, 19]. LD remains the standard of care for the treatment of PD because of its efficacy and favorable safety profile; however, the benefits of its continuous delivery (e.g., LCIG) are diminished by the surgical procedure needed to initiate treatment. These challenges represent an unmet medical need for patients with advanced PD (aPD) and underscore the need for a nonsurgical therapeutic option that can provide predictable and stable LD plasma concentrations and continuous symptomatic relief 24 hours/day.
Foslevodopa/foscarbidopa (previously known and referred to as ABBV-951) is a soluble formulation of LD and CD prodrugs developed for the treatment of motor fluctuations in patients with aPD that is administered as a 24-hour/day continuous subcutaneous infusion (CSCI) by a single infusion set connected to a portable pump [20, 21]. Upon delivery, foslevodopa/foscarbidopa undergoes rapid enzymatic conversion to the pharmacologically active forms of LD/CD. Results from a phase 1 study demonstrated that CSCI of foslevodopa/foscarbidopa quickly reached and maintained consistent and predictable steady-state plasma levels of LD across the broad range of LD doses needed to individually control symptoms in the heterogeneous aPD population [20]. In a 12-week, phase 3, double-blind, double-dummy study of foslevodopa/foscarbidopa versus oral immediate-release LD/CD, treatment with foslevodopa/foscarbidopa resulted in significantly greater improvements in motor fluctuations compared with immediate-release LD/CD and showed a favorable benefit/risk profile [22].
Here we report the final results of a separate 52-week, phase 3, open-label registrational study assessing the safety, tolerability, and efficacy of 24-hour/day CSCI of foslevodopa/foscarbidopa in patients with aPD.
Methods
Study Design and Approvals
This 52-week, phase 3, open-label, single-arm, multicenter study (NCT03781167) was designed to assess the safety, tolerability, and efficacy of foslevodopa/foscarbidopa administered as a 24-hour/day CSCI in patients with PD whose motor symptoms were inadequately controlled by their current treatment. The study was conducted at 60 sites across 13 countries (Australia, Belgium, Canada, Denmark, Germany, Italy, Japan, Netherlands, Russia, Spain, Sweden, United Kingdom, and United States); patient enrollment occurred between June 6, 2019, and August 25, 2021.
The study consisted of a screening period (two study visits conducted within a 10- to 42-day timeframe) and a two-part treatment period (4-week optimization period, followed by a 48-week maintenance period) (Fig. S1 in the supplementary material). Foslevodopa/foscarbidopa dosing was initiated on study day 1, and follow-up visits were planned for day 2 and weeks 1, 2, 3, 4, 6, 13, 26, 39, and 52.
Patients
Eligible patients included adults aged 30 years or older diagnosed with LD-responsive idiopathic PD. Before initiation of foslevodopa/foscarbidopa, patients must have had symptoms that, in the investigator’s opinion, were not adequately controlled by their current therapy, and must have experienced an average of ≥ 2.5 hours of “Off” time per day, as assessed by the electronic PD diary over two consecutive days prior to day 1 (study enrollment) [23,24,25]. Patients who had received DBS therapy were eligible for this study provided they were considered to be in stable condition, remained LD-responsive, and met all other eligibility criteria. Patients were required to be receiving stable PD medications for at least 30 days before foslevodopa/foscarbidopa initiation, and those medications must have included at least one formulation of LD. Patients also had to have had recognizable “Off” and “On” states established through investigator observation and confirmed by PD diary entries recorded during a concordance test performed within the screening period (≥ 75% concordance had to be established). Low vitamin B12 (< 200 pg/mL) or low-normal B12 (< 300 pg/mL) with elevated methylmalonic acid (MMA; > 0.41 μmol/L) at screening visit 1 had to be corrected via vitamin supplementation for patients to be eligible for enrollment; subjects with normal vitamin B12 or low-normal vitamin B12 without elevated MMA plasma level at re-test during the screening period were eligible for enrollment. Patients must not have had a history of significant skin conditions (e.g., psoriasis, atopic dermatitis) or evidence of recent sunburn, acne, scar tissue, tattoo, or discoloration that, in the opinion of the investigator, could interfere with the clinical assessment of the infusion site.
Treatment
The delivery system used in this study (Canè Crono PAR Series 3 pump, Canè S.p.A., Rivoli, Italy) is a commercially available pump qualified for the delivery of subcutaneous infusions. Among the available commercial devices, this pump was considered to most closely meet the technical specifications envisioned for the delivery of this medication, while a proprietary pump specifically developed for use with foslevodopa/foscarbidopa was under development. The Canè pump was able to provide hourly continuous infusion rates of foslevodopa/foscarbidopa ranging from 0.17 to 1.04 mL/hour (approximately 700 to 4250 mg of LD per 24 hours), with an incremental precision of 0.01 mL/h (approximately 1.7 mg of LD per hour). In routine clinical use, patients will have the option of a lower hourly infusion rate (0.15 mL/hour, equivalent to a minimum of approximately 600 mg of LD per day). Foslevodopa/foscarbidopa solution is delivered though a single infusion cannula (one infusion site). It was recommended to be inserted in the periumbilical area of the abdomen, ≥ 5 cm from the navel and ≥ 2.5 cm from previous infusion sites, with infusion site rotation required at least every 3 days. In this study, the pump could be carried using a provided fabric carrying case with an associated elastic belt or collar strap.
Healthcare providers (HCPs) programmed the starting hourly infusion rate into the pump to deliver a total daily dose of foslevodopa calculated from each individual patient’s baseline daily intake of oral LD. All LD-containing medications and COMT inhibitors taken during waking time (considered to be 16 hours/day on average) were transformed to LD-equivalents leveraging published guidance [26, 27] (Tables S1 and S2 in the supplementary material) and then converted to foslevodopa and adjusted to a 24-hour treatment period using an algorithm based on LD molecular weight and pharmacokinetic data from phase 1 foslevodopa/foscarbidopa studies. Concomitant PD medications that did not contain LD or COMT inhibitors were permitted in the study and were not included in the calculation of LD equivalents used in the conversion algorithm (Table S3 in the supplementary material).
During the optimization period, concurrently with adjustments of the foslevodopa/foscarbidopa infusion rate, HCPs could increase, decrease, or even suspend concomitant PD medications to achieve the therapeutic approach that, in the HCP’s opinion, best controlled the patient’s symptoms. After the optimization period, patients were required to maintain a stable regimen of concomitant PD medications, unless modifications were considered medically necessary by the investigator. All allowed concomitant therapy and prohibited medications are listed in Table S3 in the supplementary material. Oral LD (100 mg of LD/DOPA decarboxylase inhibitor [DDCI]) or LD inhalation powder (84 mg) were permitted as rescue therapies in cases of rapid deterioration of symptom control (e.g., pump malfunction). The foslevodopa/foscarbidopa infusion could be interrupted at any time during the day for up to 1 hour (e.g., for hygiene, intimacy, swimming) with no expected clinical consequences and without the need to change infusion sets or rotate infusion sites.
In this study, patients started foslevodopa/foscarbidopa infusion from a practically defined “Off” state (i.e., no PD medications for at least 12 hour before foslevodopa/foscarbidopa initiation). To achieve quick symptom control from an “Off” state, initiation of CSCI was preceded by a loading dose corresponding to each patient’s typical first/morning dose of LD/DDCI. In this study, the loading dose was administered orally. The HCP could further modify daily doses of foslevodopa/foscarbidopa during the optimization period by adjusting the hourly infusion rate until the optimal clinical response (defined as maximizing functional “On” time during the day by minimizing “Off” episodes and “On” time with troublesome dyskinesia) for the individual patient was attained. If needed, the hourly infusion rate of foslevodopa/foscarbidopa could be adjusted by the HCP at any point during the study, based on clinical response.
To facilitate dose optimization and provide patients and their HCPs flexibility, investigators could preprogram two alternative infusion rates within the allowed range, generally one higher and one lower than the base rate. If these functionalities were enabled by the HCP, patients could choose to use the higher alternative infusion rate as needed (e.g., during intense/extended physical activity or periods of time with increased dopaminergic demand). Alternatively, patients could switch to the lower alternative infusion rate before going to sleep. Patients could also self-administer a bolus of solution as an extra dose (between 0.11 and 0.3 mL; equivalent to approximately 20–50 mg LD), at a maximum frequency (lockout time of 1–24 hours programmable in 15-minute increments) determined by the investigator. The maximum frequency and volumes of the extra dose could only be programmed/modified by the HCP.
Patients and/or care partners who could manage the delivery system as needed received in-office training on the correct use of the delivery system, infusion site selection, and infusion set application and handling before initiation of therapy. Patients and/or care partners were also instructed on appropriate infusion site rotation (rotating the infusion site at least every 3 days, using a new infusion set with each new insertion) and to pay special attention to the use of aseptic technique (including hand washing) and good skin care practices (including cleaning of the infusion site with soap and water followed by an alcohol wipe).
Additional strategies aiming to improve both the patient experience and study retention were implemented following study initiation, as reflected in an amendment to the study protocol. These strategies included providing the option of a more user-friendly infusion set and additional education related to aseptic care and device management for patients and HCPs.
Patients who completed the 52-week treatment period could enroll in a long-term extension study of foslevodopa/foscarbidopa safety/tolerability and efficacy (NCT04379050). A 30-day safety follow-up was planned for patients who prematurely discontinued or for those who completed the 52-week treatment but opted not to enroll in the extension study.
Assessments
Safety
Safety evaluations included the proportion of patients reporting AEs, changes in laboratory parameters, vital sign measurements, and electrocardiogram values from baseline. Local (skin) tolerability was additionally assessed by the Infusion Site Evaluation Scale (adapted from FDA guidance [28]).
Efficacy
Efficacy endpoints included change from baseline to the end of the study for average daily “Off” and “On” time as assessed by the PD diary (an electronic version of the PD diary was completed by patients using a hand-held device for two consecutive days immediately prior to each study visit) and normalized to a 16-hour waking day, the Movement Disorder Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) Parts I–IV (with Part III measured in the patient’s best “On” state), the Parkinson’s Disease Sleep Scale–2 (PDSS-2), the 39-item Parkinson’s Disease Questionnaire (PDQ-39) summary index, the EuroQol 5-dimension questionnaire (EQ-5D-5L), and the proportion of patients with morning akinesia (defined as reporting “Off” status in the PD diary as the predominant PD status during the first half-hour period upon awakening).
Statistical Analysis
The sample size of approximately 240 patients would provide a 70%, 91%, and 99% probability of observing an AE with an annual incidence of 0.005, 0.01, and 0.02, respectively. The safety analysis set included all patients who received at least one infusion of foslevodopa/foscarbidopa at any given time during the study. Efficacy was assessed in the full analysis set, which included all patients who received at least one infusion of foslevodopa/foscarbidopa and who had baseline and treatment observations for at least one measure of efficacy. Safety events were coded using the Medical Dictionary for Regulatory Activities (MedDRA), version 24.0, and were summarized using descriptive statistics. Efficacy endpoints were summarized using descriptive statistics, and a paired-sample t test was performed to measure the change from baseline. If no valid PD diary data were available for a visit, “Off” and “On” times were considered missing for that visit. No multiplicity adjustments were made for efficacy endpoints, so p values presented throughout this manuscript are nominal. Statistical analyses were performed using SAS® version 9.2 or later (SAS Institute Inc., Cary, NC, USA). Three interim analyses were conducted for the study prior to the final analysis.
Compliance with Ethics Guidelines
The study received institutional review board approval at all participating institutions (supplemental appendix) and was conducted in accordance with the International Council for Harmonisation, the Declaration of Helsinki of 1964 and its later amendments, and all applicable regulations and guidelines. Patients were required to provide written informed consent before undergoing any study-related procedures. This study was registered at ClinicalTrials.gov (NCT03781167). Where relevant, permissions were obtained to use the scales employed in this study (Mini-Mental State Examination [MMSE], EQ-5D-5L, MDS-UPDRS, PDQ-39, PDSS-2, PD diary).
Results
Patients
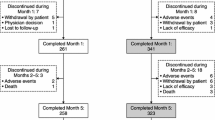
Between June 6, 2019, and August 25, 2021, 244 patients were enrolled in the study, of whom 107 prematurely discontinued treatment with foslevodopa/foscarbidopa, and 137 completed the 52-week study drug treatment (Fig. 1). Of the 137 patients who completed 52 weeks of study treatment, 94.2% (n = 129) enrolled in the subsequent long-term extension study, which remains ongoing. Primary reasons for discontinuation (patients could have multiple reasons for discontinuation) included AEs (n = 56, 23.0%), withdrawn consent (n = 30, 12.3%), and lack of efficacy (n = 11, 4.5%). The discontinuation rate was higher during the first 10 weeks after initiation of foslevodopa/foscarbidopa (66 of 107 patients) and was generally stable thereafter. Given the higher frequency of premature discontinuations during the optimization period of the study, when patients were still adjusting to the drug delivery system, a number of strategies were implemented to improve both patient experience and retention in the study. Some of these strategies, which included the introduction of an alternative, more user-friendly infusion set option and additional patient- and HCP-specific education related to aseptic care and management of devices for infusion therapies, were reflected in a study protocol amendment that was implemented during the study.

Patient disposition (all enrolled patients). aPatients could have more than one reason for discontinuation. AE adverse event
The majority of the 244 enrolled patients were male (59.8%) and White (84.8%) with a mean (standard deviation [SD]) age of 63.9 (9.2) years and PD duration of 10.7 (5.2) years since diagnosis. Patients’ mean (SD) baseline normalized “Off” time was 5.9 (2.2) hours, and their MDS-UPDRS total score was 50.4 (18.9) (Table 1). Initial optimization (no change to the base infusion rate for at least 15 days) was achieved within a mean (SD) of 3.5 (2.5) outpatient visits, and 31.2% (n = 64) of patients achieved optimization in just one visit (Table 2).
The baseline LD intake (in equivalents) was 1064.9 (584.8) mg/day. The mean (SD) study drug exposure for the 244 patients included in this analysis (including all enrolled patients who received at least one dose of study drug) was 242.9 (152.3) days, and 141 of 244 patients (57.8%) maintained a treatment duration ≥ 274 days (Table S4 in the supplementary material). The mean (SD) LD equivalent dose from foslevodopa/foscarbidopa was 1621.9 (657.3) mg at week 1, and 1859.8 (717.3) mg at week 52 (Table S5 in the supplementary material). Alternative infusion rates were common, with 87.7% (n = 214) and 87.3% (n = 213) of patients prescribed additional high and low infusion rates, respectively, at their last study prescription. As previously noted, some non-LD-containing concomitant PD medications were allowed in this study. At baseline, 13.5% (n = 33) of patients were taking only LD, 32.8% (n = 80) were taking one class of concomitant medication, and 53.3% (n = 130) were taking two or more additional classes of PD medications (includes all PD medications in addition to oral LD, which all patients were required to take before the study). The proportion of patients using no concomitant PD medication increased from 21.7% (n/N = 53/244) in week 1 to 28.0% (n/N = 60/214) by week 4, and remained at 26.7% (n/N = 55/206) during the maintenance period, while the proportion of patients using two or more classes of concomitant medications (including all permitted medications listed in Table S3 in the supplementary material) decreased from 42.6% (n/N = 104/244) in week 1 to 35.0% (n/N = 72/206) during the maintenance period (Table S6 in the supplementary material).
Safety
A total of 230 of 244 patients (94.3%) experienced at least one AE (all AEs, even those that would be expected for any infusion therapy, such as infusion site redness, were collected during this study). The majority of AEs were nonserious, mild or moderate in severity, and resolved. Sixty-three patients experienced at least one severe AE. Serious AEs (SAEs) were reported in 25.8% of patients. (Table 3).
The most common AEs were infusion site AEs (by MedDRA preferred terms: erythema [52.0%], nodule [28.7%], cellulitis [23.0%], edema [19.3%], pain [15.6%], reaction [12.3%], and abscess [11.1%]), followed by hallucination (17.2%), and fall (16.8%). Infusion site cellulitis (n = 10, 4.1%) and infusion site abscess (n = 8, 3.3%) were the most common SAEs (Table 3). The median time to onset for infusion site-related infections was 43.5 days, and most events resolved, with a median duration of 15.5 days. The median time to onset for infusion site reactions was 8.0 days, and most events resolved, with a median duration of 12.0 days. Fewer than one-third (n = 72, 29.5%) of patients were observed to have an Infusion Site Evaluation Scale grade ≥ 5 or letter grade ≥ D (defined a priori as notable skin reactions) at any point in the study (Table S7 in the supplementary material). Data for AEs identified as AEs of special interest (AESIs) are provided in Table S8 in the supplementary material. No fatal infusion site AEs were reported. Safety results were consistent across all interim analyses, which are reported in Table S9 in the supplementary material.
There were no clinically relevant findings in the mean changes from baseline for any laboratory parameters, vital signs, or electrocardiogram values. There were three treatment-emergent (occurring during study treatment or within 30 days of the last foslevodopa/foscarbidopa infusion) deaths during the study, including subdural hematoma (most likely related to traumatic injury from an accidental fall) and cerebral mass effect, cardiorespiratory arrest, and cerebrovascular accident. None of the treatment-emergent deaths were considered related to the study drug.
A total of 64 patients (26.2%) had AEs contributing to discontinuation of the study drug, nearly half of which (n = 26, 40.6%) occurred during the optimization period. The most common AEs leading to study drug discontinuation were hallucination (n = 10, 4.1%), infusion site erythema (n = 9, 3.7%), infusion site cellulitis (n = 9, 3.7%), infusion site nodule (n = 5, 2.0%), and dyskinesia (n = 5, 2.0%).
Efficacy
Treatment with foslevodopa/foscarbidopa resulted in a decrease in hours of “Off” time and increase in hours of “On” time without troublesome dyskinesia as early as at the first post-baseline assessment (week 1), and these improvements were sustained for the entire duration of the study (Fig. 2). At week 52, the mean (SD) change from baseline in normalized “Off” time was −3.5 (3.1) hours (p ≤ 0.001), which represents a 59% average reduction in “Off” time from baseline. Normalized “On” time without troublesome dyskinesia (the sum of “On” time without dyskinesia and “On” time with non-troublesome dyskinesia) increased from baseline to week 52 by 3.8 (3.3) hours (41% increase from baseline, p ≤ 0.001), driven entirely by increases in “On” time without dyskinesia, which, on average, increased by 58% compared to baseline (3.9 [4.2] hours; p ≤ 0.001). Hours of “On” time with non-troublesome dyskinesia decreased, resulting in a greater change from baseline in “On” time without dyskinesia than “On” time without troublesome dyskinesia.

“Off” and “On” time (full analysis set). SD standard deviation. a Mean hours of “Off” and “On” time distributed over a 16-hour day. b Change from baseline in “Off” and “On” time. “Off” and “On” times are normalized to a 16-hour waking day. “On” time without troublesome dyskinesia is the sum of “On” time without dyskinesia and “On” time with non-troublesome dyskinesia. n values were determined from valid diaries. If a patient completed a weekly visit but did not have a diary, they were not included in the analysis. ***p ≤ 0.001, **p < 0.01, *p < 0.05, calculated using a two-sided paired-sample t test
For this study, morning akinesia was defined as reporting "Off" status in the PD diary as the predominant PD status during the first half-hour period upon awakening. At week 52, approximately 50% fewer patients reported morning akinesia compared to baseline. At baseline, 77.7% (n/N = 129/166) of patients experienced morning akinesia, which decreased to 19.2% (n/N = 20/104) at week 26, and 27.8% (n/N = 25/90) at week 52. The reduction of early morning “Off” time was accompanied by a marked increase in the proportion of patients reporting “On” time without dyskinesia on awakening (62.2%; n/N = 56/90 at week 52) (Fig. 3).

Distribution of first morning state on awakening: percentage of patients (full analysis set). Symptoms assessed by 24-hour Parkinson’s disease diary. n values were determined from valid diaries. If a patient completed a weekly visit but did not have a diary, they were not included in the analysis
Treatment with foslevodopa/foscarbidopa also resulted in improvement in the PDSS-2 total score and its domain scores at all time points measured. There were also improvements in the PDQ-39 Summary Index and dimensions of mobility, activities of daily living, stigma, and bodily discomfort, and improvements in the EQ-5D-5L summary index and visual analog scale scores at all study time points (p ≤ 0.001 for all; Fig. 4 and Table S10 in the supplementary material).

Change from baseline in sleep, QoL, and HRQoL (full analysis set). EQ-5D-5L EuroQol 5-dimension questionnaire, HRQoL health-related quality of life, PDQ-39 39-item Parkinson’s Disease Questionnaire, PDSS-2 Parkinson’s Disease Sleep Scale–2, QoL quality of life, VAS visual analog scale. ***p ≤ 0.001, calculated using a two-sided paired-sample t test. aBased on the USA index value, which ranges from a worst score of −0.109 to a best score of 1
Finally, patients experienced improvements in MDS-UPDRS Part II (motor aspects of experiences of daily living) and Part IV (motor complications) at week 52 (p ≤ 0.001 for both). No significant improvements were observed for MDS-UPDRS Part III (motor examination) at week 52 (Fig. S2 in the supplementary material).
Efficacy results for week 52 were consistent across all interim analyses (Table S9 in the supplementary material).
Discussion
The results of this study demonstrate that treatment with 24-hour/day CSCI of foslevodopa/foscarbidopa for up to 52 weeks was generally safe and well tolerated and resulted in a sustained increase in “On” time without troublesome dyskinesia and reduction in “Off” time. This study builds upon the findings of a 12-week, phase 3, double-blind, double-dummy study of foslevodopa/foscarbidopa versus oral immediate-release LD/CD, which reported superior improvements for motor fluctuations in patients with aPD treated with foslevodopa/foscarbidopa compared with patients treated with immediate-release LD/CD [22].
While infusion site AEs were common, they were mostly nonserious, mild or moderate in severity, and generally consistent with those reported by other continuous subcutaneous therapies [29, 30]. Studies of CSCI of insulin, apomorphine, and LD/CD indicate that infusion site/skin events among continuous subcutaneous therapy users (both adults and children) are common [29,30,31,32,33,34,35,36] and occur throughout the duration of use [29]. Skin infections are also frequently reported with CSCI therapies, with most studies noting occurrences of infusion site infections in 17–41% of patients receiving subcutaneous insulin for diabetes [31,32,33]. Furthermore, results from an epidemiological investigation showed that minor skin infections occurred at least once per year among 13–15% of patients who were treated with long-term subcutaneous administration of insulin and CSAI for chronic conditions [29].
Published literature offers insight on managing infusion site skin events with subcutaneous administration including the need for AE identification and monitoring, patient/caregiver education on appropriate infusion site selection and cannula placement, regular (and possibly more frequent) rotation of infusion sites and replacement of infusion sets, use of aseptic technique and good skin care practices, and close collaboration between patients and HCPs (including physicians and nurses) to ensure optimal outcomes [37, 38]. The strategies implemented to improve the patient experience in the current study reflected these insights, and the results were generally favorable, reinforcing the importance of patient/caregiver training and education prior to initiation of foslevodopa/foscarbidopa.
The systemic safety profile of foslevodopa/foscarbidopa was generally consistent with the well-established safety profile of other LD-containing medications, including oral LD/CD [39]. The most commonly reported systemic AEs with foslevodopa/foscarbidopa were hallucinations and falls, the majority of which were nonserious, and mild/moderate in severity; their presentation was generally not different in terms of nature, severity, specificity, and outcome from that reported with oral LD-containing medications and LD-containing infusion therapies such as LCIG. Hallucination is a known manifestation seen in patients with aPD, present in up to 40% of individuals with PD, and is an established class effect for LD-containing medications [40,41,42], although hallucinations are more common with dopamine agonists than with LD [5]. The potential contribution from continuous 24-hour/day delivery of LD-containing medications on the worsening of hallucinations is debated, as there is literature to suggest a relationship between both worsening and improvement of hallucination with 24-hour continuous dosing [13, 43,44,45]. However, recent literature summarizing practical recommendations from clinics using 24-hour LCIG therapy generally recommends reducing nighttime infusion rates for the initial optimization of 24-hour therapy to avoid the risk of worsening hallucinations, psychosis, and nightmares [13]. In the current study of foslevodopa/foscarbidopa, the allowed infusion rate reduction per protocol was initially limited to within 20% of the base infusion rate, unless additional reduction was deemed medically necessary, a limitation that was removed in the long-term extension study and will likely not exist in real-world clinical practice. The ability to further individualize nighttime dosing of foslevodopa/foscarbidopa in real-world clinical practice should improve physicians’ ability to meet the specific needs of each patient.
Motor symptom improvements were observed as early as the first post-baseline assessment (week 1) after initiation of 24-hour/day CSCI of foslevodopa/foscarbidopa and persisted throughout the 52-week treatment period. Reduced “Off” time was complemented by an increase in “good On” time (“On” time without troublesome dyskinesia, defined as the sum of “On” time with no dyskinesia and “On” time with non-troublesome dyskinesia). The increase in “good On” time was driven by increases in the most desirable state of “On” time without dyskinesia rather than “On” time with non-troublesome dyskinesia. The reduction of “Off” time is particularly exemplified by the meaningful reduction in morning akinesia, with almost 50% more patients awakening in the “On” state without any dyskinesia at week 52 compared with baseline (changed from 17.5% at baseline to 62.2% at week 52). Treatment with foslevodopa/foscarbidopa also resulted in improved motor experiences of daily living.
Reduction in early morning akinesia, improved sleep outcomes, and overall improvement in HRQoL measures are among the added benefits of continuous (24-hour/day) delivery of foslevodopa/foscarbidopa. In this study, treatment with foslevodopa/foscarbidopa resulted in improved sleep, quality of life (QoL), and HRQoL. These data support mounting evidence in the literature that sustained dopaminergic stimulation may be helpful for reducing early morning “Off” symptoms [46, 47]. Improving sleep and early morning symptoms is of particular importance because sleep is one of the strongest factors associated with HRQoL [48]. Sleep disturbances are associated with an increased risk of falls and worsening of cognition in patients with PD [49], while improvements in patients’ sleep are associated with reduced caregiver burden [50]. Likewise, early morning “Off” periods occur in more than half of patients with PD, are associated with other motor and non-motor symptoms, and have a significant negative impact on patients’ QoL [46, 51].
Among the limitations to consider when interpreting results from this investigation are the open-label nature of the study and the lack of randomization and of a comparator. Because all patients received the same intervention, our ability to reach definitive conclusions regarding the relative efficacy of foslevodopa/foscarbidopa is limited. Additionally, only patients who completed each study visit were included in the efficacy analysis, and no adjustments were made to account for premature discontinuation. However, the results of this study are in line with those from the double-blind study, supporting the significant improvements in motor fluctuations with foslevodopa/foscarbidopa compared with oral immediate-release LD/CD [22].
Conclusion
In summary, individualized 24-hour/day CSCI of foslevodopa/foscarbidopa demonstrated a favorable benefit/risk profile in patients with aPD. The systemic safety profile was generally consistent with other LD-containing medications, and local tolerability was comparable to that reported by other continuous infusions that use the subcutaneous route of delivery. The majority of the AEs, including infusion site AEs, were nonserious, mild or moderate in severity, and resolved with or without treatment. It is clear from the literature that infusion site AEs are common with subcutaneous therapies and that there is no singular or universal solution for prevention. It is important that HCPs (including physicians and nurses), patients, and care partners be educated about how to recognize and respond to infusion site side effects if they occur. The prevention and management of infusion site AEs should be among the top priorities to consider when initiating foslevodopa/foscarbidopa to promote adherence to therapy and a positive experience for patients and their care partners. Strategies to improve patient outcomes will be explored in future publications that focus on clinical practice recommendations for use of foslevodopa/foscarbidopa.
Foslevodopa/foscarbidopa administered by 24-hour/day CSCI also demonstrated improvement in motor fluctuations, sleep, and QoL throughout the 52-week treatment period. Although these results are from a single-arm, open-label study, they support the benefit of a continuous, 24-hour/day infusion of LD and are consistent with the results from the double-blind study. Overall, by providing individualized, continuous, 24-hour/day, subcutaneous delivery of LD and CD prodrugs, foslevodopa/foscarbidopa has the potential to provide an efficacious and safe nonsurgical alternative therapy to other currently available treatments for controlling fluctuations and improving QoL in patients with aPD.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized individual and trial-level data (analysis datasets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the United States and Europe and after acceptance of this manuscript for publication. These data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://vivli.org/ourmember/abbvie/ then select “Home”.
Change history
10 October 2023
A Correction to this paper has been published: https://doi.org/10.1007/s40120-023-00554-w
References
Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999;56(1):33–9.
Hauser RA. Levodopa: past, present, and future. Eur Neurol. 2009;62(1):1–8.
Olanow CW, Obeso JA, Stocchi F. Continuous dopamine-receptor treatment of Parkinson’s disease: scientific rationale and clinical implications. Lancet Neurol. 2006;5(8):677–87.
Bestetti A, Capozza A, Lacerenza M, Manfredi L, Mancini F. Delayed gastric emptying in advanced Parkinson disease: correlation with therapeutic doses. Clin Nucl Med. 2017;42(2):83–7.
Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease (2009). Neurology. 2009;72(21 Suppl 4):S1-136.
Grosset D. Therapy adherence issues in Parkinson’s disease. J Neurol Sci. 2010;289(1–2):115–8.
Johnell K, Klarin I. The relationship between number of drugs and potential drug-drug interactions in the elderly: a study of over 600,000 elderly patients from the Swedish Prescribed Drug Register. Drug Saf. 2007;30(10):911–8.
Malek N, Grosset DG. Medication adherence in patients with Parkinson’s disease. CNS Drugs. 2015;29(1):47–53.
Barone P, Antonini A, Colosimo C, Marconi R, Morgante L, Avarello TP, et al. The PRIAMO study: a multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson’s disease. Mov Disord. 2009;24(11):1641–9.
Schapira AHV, Chaudhuri KR, Jenner P. Non-motor features of Parkinson disease. Nat Rev Neurosci. 2017;18(7):435–50.
Weintraub D, Aarsland D, Chaudhuri KR, Dobkin RD, Leentjens AF, Rodriguez-Violante M, et al. The neuropsychiatry of Parkinson’s disease: advances and challenges. Lancet Neurol. 2022;21(1):89–102.
van Wamelen DJ, Sauerbier A, Leta V, Rodriguez-Blazquez C, Falup-Pecurariu C, Rodriguez-Violante M, et al. Cross-sectional analysis of the Parkinson’s disease Non-motor International Longitudinal Study baseline non-motor characteristics, geographical distribution and impact on quality of life. Sci Rep. 2021;11(1):9611.
Thakkar S, Fung VSC, Merola A, Rollins M, Soileau MJ, Kovács N. 24-hour levodopa-carbidopa intestinal gel: clinical experience and practical recommendations. CNS Drugs. 2021;35(2):137–49.
Pal PK, Thennarasu K, Fleming J, Schulzer M, Brown T, Calne SM. Nocturnal sleep disturbances and daytime dysfunction in patients with Parkinson’s disease and in their caregivers. Parkinsonism Relat Disord. 2004;10(3):157–68.
Das S, Matias CM, Ramesh S, Velagapudi L, Barbera JP, Katz S, et al. Capturing initial understanding and impressions of surgical therapy for Parkinson’s disease. Front Neurol. 2021;12: 605959.
Henriksen T, Dalhoff KP, Hansen HE, Brenneche AW, Lønberg US, Danielsen EH. Access and use of device-aided therapies for Parkinson’s disease in Denmark. Mov Disord Clin Pract. 2020;7(6):656–63.
Dijk JM, Espay AJ, Katzenschlager R, de Bie RMA. The choice between advanced therapies for Parkinson’s disease patients: why, what, and when? J Parkinsons Dis. 2020;10(s1):S65-s73.
Deuschl G, Antonini A, Costa J, Śmiłowska K, Berg D, Corvol JC, et al. European Academy of Neurology/Movement Disorder Society—European Section guideline on the treatment of Parkinson’s disease: I. Invasive therapies. Eur J Neurol. 2022;29:2580–95.
Olivola E, Fasano A, Varanese S, Lena F, Santilli M, Femiano C, et al. Continuous subcutaneous apomorphine infusion in Parkinson’s disease: causes of discontinuation and subsequent treatment strategies. Neurol Sci. 2019;40(9):1917–23.
Rosebraugh M, Liu W, Neenan M, Facheris MF. Foslevodopa/foscarbidopa is well tolerated and maintains stable levodopa and carbidopa exposure following subcutaneous infusion. J Parkinsons Dis. 2021;11(4):1695–702.
Rosebraugh M, Voight EA, Moussa EM, Jameel F, Lou X, Zhang GGZ, et al. Foslevodopa/foscarbidopa: a new subcutaneous treatment for Parkinson’s disease. Ann Neurol. 2021;90(1):52–61.
Soileau MJ, Aldred J, Budur K, Fisseha N, Fung V, Jeong A, et al. Safety and efficacy of continuous subcutaneous foslevodopa-foscarbidopa in patients with advanced Parkinson’s disease: a randomised, double-blind, active-controlled, phase 3 trial. Lancet Neurol. 2022;21(12):1099–109.
Hauser RA, Deckers F, Lehert P. Parkinson’s disease home diary: further validation and implications for clinical trials. Mov Disord. 2004;19(12):1409–13.
Hauser RA, Friedlander J, Zesiewicz TA, Adler CH, Seeberger LC, O’Brien CF, et al. A home diary to assess functional status in patients with Parkinson’s disease with motor fluctuations and dyskinesia. Clin Neuropharmacol. 2000;23(2):75–81.
Hauser RA, Auinger P. Determination of minimal clinically important change in early and advanced Parkinson’s disease. Mov Disord. 2011;26(5):813–8.
Espay AJ, Pagan FL, Walter BL, Morgan JC, Elmer LW, Waters CH, et al. Optimizing extended-release carbidopa/levodopa in Parkinson disease: consensus on conversion from standard therapy. Neurol Clin Pract. 2017;7(1):86–93.
Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE. Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov Disord. 2010;25(15):2649–53.
USFDA. Assessing the irritation and sensitization potential of transdermal and topical delivery systems for ANDAs guidance for industry 2018. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/assessing-irritation-and-sensitization-potential-transdermal-and-topical-delivery-systems-andas. Accessed 25 Feb 2022.
Jick SS, Oleske DM, Persson R, Zamudio J, Facheris MF. Epidemiology of skin event rates among users of pumps for the subcutaneous administration of drugs for chronic conditions. Curr Med Res Opin. 2021;37(9):1563–71.
Katzenschlager R, Poewe W, Rascol O, Trenkwalder C, Deuschl G, Chaudhuri KR, et al. Apomorphine subcutaneous infusion in patients with Parkinson’s disease with persistent motor fluctuations (TOLEDO): a multicentre, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2018;17(9):749–59.
Pickup JC, Yemane N, Brackenridge A, Pender S. Nonmetabolic complications of continuous subcutaneous insulin infusion: a patient survey. Diabetes Technol Ther. 2014;16(3):145–9.
Ross P, Gray AR, Milburn J, Kumarasamy IM, Wu F, Farrand S, et al. Insulin pump-associated adverse events are common, but not associated with glycemic control, socio-economic status, or pump/infusion set type. Acta Diabetol. 2016;53(6):991–8.
Ross PL, Milburn J, Reith DM, Wiltshire E, Wheeler BJ. Clinical review: insulin pump-associated adverse events in adults and children. Acta Diabetol. 2015;52(6):1017–24.
Katzenschlager R, Poewe W, Rascol O, Trenkwalder C, Deuschl G, Chaudhuri KR, et al. Long-term safety and efficacy of apomorphine infusion in Parkinson’s disease patients with persistent motor fluctuations: results of the open-label phase of the TOLEDO study. Parkinsonism Relat Disord. 2021;83:79–85.
Isaacson S, Giladi N, Stocchi F, Salin L, Vostokova N, Poewe W. Long-term safety of continuous levodopa/carbidopa infusion with ND0612: data from patients who continued past one year in the BeyoND study [abstract]. Mov Disord. 2022;37 (suppl 1). https://www.mdsabstracts.org/abstract/long-term-safety-of-continuous-levodopa-carbidopa-infusion-with-nd0612-data-from-patients-who-continued-past-one-year-in-the-beyond-study/. Accessed 11 Nov 2022.
Poewe W, Stocchi F, Arkadir D, Ebersbach G, Ellenbogen AL, Giladi N, et al. Subcutaneous levodopa infusion for Parkinson’s disease: 1-year data from the open-label BeyoND study. Mov Disord. 2021;36(11):2687–92.
Grunberger G, Sherr J, Allende M, Blevins T, Bode B, Handelsman Y, et al. American Association of Clinical Endocrinology Clinical Practice Guideline: the use of advanced technology in the management of persons with diabetes mellitus. Endocr Pract. 2021;27(6):505–37.
Gorski LA, Hadaway L, Hagle ME, Broadhurst D, Clare S, Kleidon T, et al. Infusion therapy standards of practice, 8th edition. J Infus Nurs. 2021;44(1S Suppl 1):S1–224.
SINEMET® (Carbidopa-Levodopa) Tablets. [Prescribing Information]. Merck & Co, Inc. Whitehouse Station, NJ, USA. https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/017555s069lbl.pdf. Accessed 16 June 2022.
Ecker D, Unrath A, Kassubek J, Sabolek M. Dopamine agonists and their risk to induce psychotic episodes in Parkinson’s disease: a case-control study. BMC Neurol. 2009;9:23.
Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain. 2000;123(Pt 4):733–45.
Zahodne LB, Fernandez HH. Pathophysiology and treatment of psychosis in Parkinson’s disease: a review. Drugs Aging. 2008;25(8):665–82.
Cruse B, Morales-Briceño H, Chang FCF, Mahant N, Ha AD, Kim SD, et al. 24-hour levodopa-carbidopa intestinal gel may reduce troublesome dyskinesia in advanced Parkinson’s disease. NPJ Parkinsons Dis. 2018;4:34.
Nyholm D, Jansson R, Willows T, Remahl IN. Long-term 24-hour duodenal infusion of levodopa: outcome and dose requirements. Neurology. 2005;65(9):1506–7.
Ricciardi L, Bove F, Espay KJ, Lena F, Modugno N, Poon YY, et al. 24-Hour infusion of levodopa/carbidopa intestinal gel for nocturnal akinesia in advanced Parkinson’s disease. Mov Disord. 2016;31(4):597–8.
Rizos A, Martinez-Martin P, Odin P, Antonini A, Kessel B, Kozul TK, et al. Characterizing motor and non-motor aspects of early-morning off periods in Parkinson’s disease: an international multicenter study. Parkinsonism Relat Disord. 2014;20(11):1231–5.
De Cock VC, Dodet P, Leu-Semenescu S, Aerts C, Castelnovo G, Abril B, et al. Safety and efficacy of subcutaneous night-time only apomorphine infusion to treat insomnia in patients with Parkinson’s disease (APOMORPHEE): a multicentre, randomised, controlled, double-blind crossover study. Lancet Neurol. 2022;21(5):428–37.
Kovacs N, Bergmann L, Anca-Herschkovitsch M, Cubo E, Davis TL, Iansek R, et al. Outcomes impacting quality of life in advanced Parkinson’s disease patients treated with levodopa-carbidopa intestinal gel. J Parkinsons Dis. 2022;12(3):917–26.
Schrag A, Choudhury M, Kaski D, Gallagher DA. Why do patients with Parkinson’s disease fall? A cross-sectional analysis of possible causes of falls. NPJ Parkinsons Dis. 2015;1:15011.
Bartolomei L, Pastore A, Meligrana L, Sanson E, Bonetto N, Minicuci GM, et al. Relevance of sleep quality on caregiver burden in Parkinson’s disease. Neurol Sci. 2018;39(5):835–9.
Chapuis S, Ouchchane L, Metz O, Gerbaud L, Durif F. Impact of the motor complications of Parkinson’s disease on the quality of life. Mov Disord. 2005;20(2):224–30.
Acknowledgements
AbbVie and authors thank all the trial investigators and the patients who participated in this clinical trial.
Medical Writing and Editorial Assistance
Medical writing support was provided by Alicia Salinero, PhD, ISMPP CMPP™, of JB Ashtin, and funded by AbbVie.
Funding
AbbVie funded this study and participated in the study design, research, analysis, data collection, interpretation of data, reviewing, and approval of the publication. Funding for the Neurology and Therapy Rapid Service Fee is provided by AbbVie.
Author information
Authors and Affiliations
Contributions
Jason Aldred: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data. Eric Freire-Alvarez: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. Alexander V. Amelin: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data. Angelo Antonini: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. Bruno Bergmans: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. Filip Bergquist: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. Manon Bouchard: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data. Kumar Budur: Drafting/revision of the manuscript for content, including medical writing for content; study concept or design; analysis or interpretation of data. Camille Carroll: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. K. Ray Chaudhuri: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. Susan R. Criswell: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. Erik H. Danielsen: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data. Florin Gandor: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. Jia Jia: Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data. Thomas E. Kimber: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. Hideki Mochizuki: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data. Weining Z. Robieson: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data. Amy M. Spiegel: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data. David G. Standaert: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data. Saritha Talapala: Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data. Maurizio F. Facheris: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data. Victor S. C. Fung: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data.
Corresponding author
Ethics declarations
Conflict of interest
Jason Aldred is a study investigator for AbbVie, AC Immune, Annovis, Aptinyx, AstraZeneca, Atara, Athira, Biogen, BioVie, Boston Scientific, Celgene, Cerevance, Cerevel, Denali, EIP, Eli Lilly, Impax, Inhibikase, IRL Therapeutics, Merz, Neuraly, Neurocrine, Neuoderm, Novartis, PD Gene/PSG, Praxis, Revance, Roche/Genentech, Sage, Sanofi/Genzyme, Scion NeuroStim, Takeda, Theravance, Triplet/HSG, and UCB. He has received honorarium from AbbVie, Allergan, Boston Scientific Teva, US World Meds, Medtronic, and Abbot. Eric Freire-Alvarez has received advisory, consulting, and lecture fees from AbbVie, Almirall, Bial, Eisai, UCB Pharma, Teva, and Zambon. He is an investigator on AbbVie studies. Alexander V. Amelin has received advisory, consulting, and lecture fees from AbbVie, Pfizer, Novartis, Lundbeck, UCB Pharma, Teva, Viatris, and Stada. He is an investigator on AbbVie studies. Angelo Antonini has received compensation for consultancy and speaker-related activities from UCB, Boehringer Ingelheim, Ever Pharma, General Electric, Britannia, AbbVie, Kyowa Kirin, Zambon, Bial, Theravance Biopharma, Jazz Pharmaceuticals, Roche, and Medscape. He receives research support from Bial, Lundbeck, Roche, Angelini Pharmaceuticals, Horizon 2020 Grant 825785, Horizon 2020 Grant 101016902, Ministry of Education University and Research (MIUR) Grant ARS01_01081, Cariparo Foundation, and Movement Disorder Society for NMS Scale validation. He serves as a consultant for Boehringer Ingelheim for legal cases on pathologic gambling. He owns Patent WO2015110261-A1 and owns shares from PD Neurotechnology Limited. Bruno Bergmans has a clinical practice at AZ St-Jan Brugge-Oostende AV in Bruges and is an academic consultant at Ghent University Hospital, Ghent. He has served as an advisory board member for Allergan, Merz, and UCB, and has served as a speaker for Zambon and AbbVie. He is the principal investigator for the subcutaneous levodopa study in Bruges. Filip Bergquist has received financial compensation for lectures and advisory services as well as in-kind donations of PKG reports for clinical studies from GKC, and honorarium for an advisory board from AbbVie. He is a stock option holder of Dizlin Pharmaceuticals AB. Manon Bouchard has received honoraria for consultancy, lectures, and advisory boards from AbbVie, Allergan, Merz, Ipsen, Lilly, Lundbeck, Novartis, Paladin, Sunovion, and UCB. She is also a consultant for Société Parkinson Québec. She has received research support from AbbVie, ES-therapeutics, and Pfizer, and royalties from UptoDate. Kumar Budur is a former employee of AbbVie Inc., and is currently employed by Harmony Biosciences. Camille Carroll has received honoraria for consultancy services from AbbVie, Bial, Britannia, GKC, GSK, Kyowa Kirin, Lundbeck, Medscape, and Scient. She has received service grants from AbbVie and Bial, and research grants from Parkinson’s UK, Cure Parkinson’s, National Institute for Health Research, and the Edmond J. Safra Foundation. She is an employee of Newcastle University, Translational and Clinical Research Institute, University of Plymouth, University Hospitals Plymouth National Health Service Trust, and National Institute of Health and Care Research. K. Ray Chaudhuri is a study investigator and has served as an advisory board member for AbbVie, UCB, GKC, Bial, Cynapsus, Lobsor, Stada, Medtronic, Zambon, Profile, Sunovion, Roche, Therevance, Scion, Britannia, Acadia, and 4D. He received honoraria for lectures from AbbVie, Britannia, UCB, Zambon, Novartis, Boehringer Ingelheim, Bial, Kyowa Kirin, and SK Pharma. He has received grants (investigator initiated) from Britannia Pharmaceuticals, AbbVie, UCB, GKC, and Bial, and academic grants from EU, IMI EU, Horizon 2020, Parkinson’s UK, NIHR, PDNMG, EU (Horizon 2020), Kirby Laing Foundation, NPF, MRC, and Wellcome Trust. He receives royalties from Oxford University Press and holds intellectual property rights for the King’s Parkinson’s Pain Scale and Parkinson’s Disease Sleep Scale. Susan R. Criswell is a former employee of Washington University in St. Louis, St. Louis, MO, USA, and is currently employed by Barrow Neurological Institute, Phoenix, AZ, USA. She reports being site principal investigator for clinical trials with AbbVie, Impax, and the NIH. She receives grant funding from the NIH including grants R01 ES026891, R01ES029524, R01 OH011661, U19 AG071754-01, and R01ES021488. Erik H. Danielsen has received lecture fees from AbbVie, UCB, Nordic Infucare, Allergan, Ibsen, and Merz. Florin Gandor has served as an advisory board member for AbbVie and has received honoraria from AbbVie, Bial Pharma, Merz, and Stada. Thomas E. Kimber has received honoraria from Stada, AbbVie, UCB Pharma, Seqiris, and Teva. Hideki Mochizuki served as a consultant, lecturer, and/or scientific advisor for IQVIA, AbbVie, Insightec, Eisai Co., FP Pharmaceutical Corporation, Otsuka Pharmaceutical, Ono Pharmaceutical Corporation, Kyowa Kirin, Sumitomo Dainippon Pharma, Takeda Pharmaceutical, Mitsubishi Tanabe Pharma Corporation, Nihon Medi-Physics, and Novartis Pharma K.K. He has received scholarship grants from Kyowa Kirin, Mochida Pharmaceutical, and Japan Blood Products Organization. David G. Standaert is a member of the faculty of the University of Alabama at Birmingham and is supported by endowment and University funds. He is an investigator in studies funded by AbbVie Inc., the American Parkinson Disease Association, the Michael J. Fox Foundation for Parkinson Research, Alabama Department of Commerce, Alabama Innovation Fund, the Department of Defense, and NIH grants P50NS108675, R25NS079188, and T32NS095775. He has a clinical practice and is compensated for these activities through the University of Alabama Health Services Foundation. In addition, since January 1, 2021, he has served as a consultant for or received honoraria from AbbVie Inc., Sutter Health, Curium Pharma, Appello, Theravance, Sanofi-Aventis, Alnylam Pharmaceuticals, Coave Therapeutics, BlueRock Therapeutics and F. Hoffman-La Roche. Victor S. C. Fung receives a salary from NSW Health. He has received unrestricted research grants from the Michael J Fox Foundation, AbbVie, and Merz, and has been on advisory boards for AbbVie, Allergan, Ipsen, Merz, Praxis, Seqirus, Stada, Teva, and UCB. He receives royalties from Health Press. Jia Jia, Weining Z. Robieson, Amy M. Spiegel, Saritha Talapala, and Maurizio F. Facheris are employees of AbbVie Inc. and may hold AbbVie stock and/or stock options.
Ethical Approval
The study received institutional review board approval at all participating institutions (supplemental appendix) and was conducted in accordance with the International Council for Harmonisation, the Declaration of Helsinki of 1964 and its later amendments, and all applicable regulations and guidelines. Patients were required to provide written informed consent before undergoing any study-related procedures. This study was registered at ClinicalTrials.gov (NCT03781167). Where relevant, permissions were obtained to use the scales employed in this study (MMSE, EQ-5D-5L, MDS-UPDRS, PDQ-39, PDSS-2, PD diary).
Authorship
All authors had access to relevant data and participated in the drafting, review, and approval of this publication. No honoraria or payments were made for authorship.
Additional information
The original online version of this article was revised to correct few numerical values of some data points reported in the text.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Aldred, J., Freire-Alvarez, E., Amelin, A.V. et al. Continuous Subcutaneous Foslevodopa/Foscarbidopa in Parkinson’s Disease: Safety and Efficacy Results From a 12-Month, Single-Arm, Open-Label, Phase 3 Study. Neurol Ther 12, 1937–1958 (2023). https://doi.org/10.1007/s40120-023-00533-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-023-00533-1