
Abstract
Two gold(I) complexes of an amido-N functionalized 1,2,4-triazole derived N-heterocyclic carbene ligands have been synthesized and structurally characterized. In particular, the {[1-(R)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazol-5-ylidene]2Au}+Cl− [R = Et (1c) and i-Pr (2c)] complexes were conveniently synthesized from their respective silver(I) analogs (1–2)b by the direct reaction with (SMe2)AuCl in CH3CN at room temperature. The molecular structure determination of the gold(I) (1–2)c complexes using single crystal X-ray diffraction study revealed a syn-disposition of two N-heterocyclic carbene (NHC) ligands bound across the Au(I) center in a linear fashion with it’s two amido-N functionalized sidearms interacting with a non-coordination Cl anion through two intramolecular H-bonds.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Of late, the gold complexes have attracted attention particularly for their resurgence in catalytic applications alongside of their longstanding utility in materials and in biomedical applications research [1–3]. This has led to an unprecedented development of the gold chemistry in recent years beginning with their synthesis and leading to various application studies [4–7]. In this connection, we became interested in this new found catalytic exploits of gold, long known as an inert coinage metal, and decided to explore its catalytic potential when stabilized over an otherwise catalytically renowned ligand like the N-heterocyclic carbenes (NHCs) [8]. Towards this objective, we have reported the utility of the gold N-heterocyclic carbene complexes in the alkyne hydroamination reaction [9, 10], in the Ring-Opening Polymerization (ROP) of L-lactides [11, 12] and in the synthesis of β-enaminones from 1,3-dicarbonyl compounds and aliphatic amines [13]. In addition, we have observed an interesting and also unprecedented long-range 1,7-bromination reaction occurring in these complexes [14], the structural diversity [15] and their antimicrobial properties [16].
The other emphasis of our recent research is on broadening the horizons of the triazole derived N-heterocyclic carbene ligands [17], which thus far have remained surprisingly overlooked in comparison to their imidazole counterparts. Towards this objective, we have recently noted their utility in borylation of aryl bromides [18], and in hydroaminations of alkynes [10] and activated olefins [19]. Continuing further along the line, we decided to synthesize gold complexes of the triazole derived N-heterocyclic carbene ligands for their potential catalytic applications.
Here in this manuscript, we report the synthesis and the structural characterization of two such gold(I) complexes stabilized over 1,2,4-triazole derived N-heterocyclic carbene ligands containing an amido-N functionalized sidearm substituent (Fig. 1).

Au–NHC complexes of 1,2,4-triazole derived N-heterocyclic carbene ligands
2 Experimental Section
2.1 Materials and Methods
All manipulations were carried out using a combination of a glovebox and standard Schlenk techniques. 1-Ethyl-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazolium chloride (1a), and 1-(i-propyl)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazolium chloride (2a) were synthesized according to modified literature procedures [18]. 1H and 13C{1H} NMR spectra were recorded on a Bruker 400 MHz and Bruker 500 MHz NMR spectrometer. 1H NMR peaks are labeled as singlet (s), doublet (d), triplet (t), quartet (q), quartet of doublets (qd), doublet of doublets (dd) septet (sept) and broad (br). Infrared spectra were recorded on a Perkin Elmer Spectrum One FT-IR spectrometer. Mass spectrometry measurements were done on a Micromass Q-Tof spectrometer and Bruker maxis impact spectrometer. Elemental Analysis was carried out on Thermo Finnigan FLASH EA 1112 SERIES (CHNS) Elemental Analyzer. X-ray diffraction data for compounds 1c and 2c were collected on an Oxford Diffraction XCALIBUR-S diffractometer and Rigaku Hg 724 + diffractometer. Crystal data collection and refinement parameters are summarized in Table 1. The structures were solved using direct method and standard difference map techniques, and refined by full-matrix least-squares procedures on F 2 [21, 22]. CCDC-990570 (for 1c) and CCDC-814958 (for 2c) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data center via www.ccdc.cam.ac.uk/data_request/cif.
2.2 Synthesis of {[1-(ethyl)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazol-5-ylidene]2Ag}+Cl− (1b)
A mixture of 1-ethyl-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazolium chloride (1a) (0.728 g, 2.68 mmol) and Ag2O (0.313 g, 1.34 mmol) was stirred in CH3CN (ca. 40 ml) at room temperature for 12 h. The reaction mixture was filtered over Celite and the filtrate was finally dried under vacuum to give the product 1b as a brown hygroscopic solid (0.585 g, 70 %) 1H NMR (CDCl3, 500 MHz, 25 °C): δ 9.00 (br, 2H, NHCO), 8.37 (s, 2H, N–C(3)H–N), 7.20 (br, 2H, C4 H 3O), 6.21 (br, 2H, C4 H 3O), 6.19 (t, 2H, 3 J HH = 2 Hz, C4 H 3O), 5.24 (s, 4H, CH 2), 4.38 (d, 4H, 3 J HH = 5 Hz, CH 2NH), 4.33 (q, 4H, 3 J HH = 7 Hz, CH 2CH3), 1.51 (t, 6H, 3 J HH = 7 Hz, CH2CH 3). 13C{1H} NMR (DMSO–d6, 100 MHz, 25 °C): δ 181.2 (Ag–NCN), 165.9 (C = O), 151.5 (C 4H3O), 144.9 (N–C(3)–N), 142.3 (C 4H3O), 110.5 (C 4H3O), 107.3 (C 4H3O), 50.1 (CH2), 48.0 (CH2CH3), 35.8 (CH2), 15.7 (CH2 CH3). IR data (KBr pellet) cm−1: 1690 (s) (νc=o). HRMS (ES): m/z 575.1299 [(NHC)2 + Ag]+, calcd. 575.1284. Anal. Calcd. for C22H28AgClN8O4·2H2O: C, 40.79; H, 4.98; N, 17.30. Found: C, 40.14; H, 4.92; N, 16.89 %.
2.3 Synthesis of {[1-(ethyl)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazol-5-ylidene]2Au}+Cl− (1c)
A mixture of {[1-(ethyl)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazol-5-ylidene]2Ag}+Cl− (1b) (0.521 g, 0.853 mmol) and (SMe2)AuCl (0.250 g, 0.850 mmol) was stirred in CH3CN (ca. 40 ml) at room temperature for 12 h, during which the formation of an off-white precipitate of AgCl was observed. The reaction mixture was filtered and filtrate was concentrated under vacuum to give the crude product as a yellow solid. The crude product was finally purified by column chromatography using silica gel as a stationary phase by elution with a mixed medium of CHCl3/MeOH (94: 6 v/v) to give the product 1c as a white solid (0.501 g, 84 %). 1H NMR (CDCl3, 500 MHz, 25 °C): δ 9.75 (br, 2H, NHCO), 8.44 (s, 2H, N–C(3)H–N), 7.11 (br, 2H, C4 H 3O), 6.21 (br, 2H, C4 H 3O), 6.14 (br, 2H, C4 H 3O), 5.27 (s, 4H, CH 2), 4.38–4.35 (m, 8H, CH 2CH3 and CH 2NH), 1.56 (t, 6H, 3 J HH = 7 Hz, CH2CH 3). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 184.8 (Au–NCN), 165.7 (C = O), 151.5 (C 4H3O), 144.5 (N–C(3)–N), 141.9 (C 4H3O), 110.4 (C 4H3O), 107.8 (C 4H3O), 50.8 (CH2), 48.9 (CH2CH3), 36.5 (CH2), 15.7 (CH2 CH3). IR data (KBr pellet) cm−1: 1685 (s) (νc=o). HRMS (ES): m/z 665.1907 [(NHC)2 + Au]+, calcd. 665.1894. Anal. Calcd. for C22H28AuClN8O4: C, 37.70; H, 4.03; N, 15.99. Found: C, 37.55; H, 4.77; N, 16.69 %.
2.4 Synthesis of {[1-(i-propyl)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazol-5-ylidene]2Ag}+Cl− (2b)
A mixture of 1-(i-propyl)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazolium chloride (2a) (1.95 g, 6.84 mmol) and Ag2O (0.799 g, 3.45 mmol) was stirred in CH3CN (ca. 40 mL) at room temperature for 12 h. The reaction mixture was filtered over Celite and the filtrate was finally dried under vacuum to give the product 2b as a brown hygroscopic solid (0.999 g, 45 %). 1H NMR (CDCl3, 400 MHz, 25 °C): δ 9.05 (br, 2H, NHCO), 8.37 (s, 2H, N–C(3)H–N), 7.20 (br, 2H, C4 H 3O), 6.22 (br, 2H, C4 H 3O), 6.20–6.19 (m, 2H, C4 H 3O), 5.23 (s, 4H, CH 2), 4.82 (sept, 2H, 3 J HH = 7 Hz, CH(CH3)2), 4.37 (d, 4H, 3 J HH = 5 Hz, CH 2NH), 1.52 (d, 6H, 3 J HH = 7 Hz, CH(CH 3)2). 13C{1H} NMR (DMSO–d6, 100 MHz, 25 °C): δ 180.9 (Ag–NCN), 166.1 (C = O), 151.5 (C 4H3O), 144.7 (N–C(3)–N), 142.3 (C 4H3O), 110.5 (C 4H3O), 107.3 (C 4H3O), 55.2 (CH(CH3)2), 50.2 (CH2), 35.7 (CH2), 21.8 (CH(CH3)2). IR data (KBr pellet) cm−1: 1685 (s) (νc=o). HRMS (ES): m/z 603.1584 [(NHC)2 + Ag]+, calcd. 603.1597. Anal. Calcd. for C24H32AgClN8O4·3H2O: C, 41.54; H, 5.52; N, 16.15. Found: C, 40.95; H, 4.56; N, 15.21 %.
2.5 Synthesis of {[1-(i-propyl)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazol-5-ylidene]2Au}+Cl− (2c)
A mixture of {[1-(i-propyl)-4-N-(furan-2-ylmethyl)acetamido-1,2,4-triazol-5-ylidene]2Ag}+Cl− (2b) (0.778 g, 1.21 mmol) and (SMe2)AuCl (0.178 g, 0.606 mmol) was stirred in CH3CN (ca. 40 ml) at room temperature for 12 h, during which the formation of an off-white precipitate of AgCl was observed. The reaction mixture was filtered and filtrate was concentrated under vacuum to give the crude product as a yellow solid. The crude product was finally purified by column chromatography using silica gel as a stationary phase and elution with a mixed medium of CHCl3/MeOH (94: 6 v/v) to give the product 2c as a white solid (0.145 g, 34 %). 1H NMR (CDCl3, 400 MHz, 25 °C): δ 9.88 (br, 1H, NHCO), 8.39 (s, 1H, N–C(3)H–N), 7.13 (d, 1H, 3 J HH = 2 Hz, C4 H 3O), 6.32 (dd, 1H, 3 J HH = 3 Hz, C4 H 3O), 6.25 (dd, 1H, 3 J HH = 3 Hz, C4 H 3O), 5.28 (s, 2H, CH 2), 4.90 (sept, 1H, 3 J HH = 7 Hz, CH(CH3)2), 4.42 (d, 2H, 3 J HH = 6 Hz, CH 2NH), 1.60 (d, 6H, 3 J HH = 7 Hz, CH(CH 3)2). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C) δ 184.0 (Au–NCN), 165.7 (C = O), 151.7 (C 4H3O), 144.3 (N–C(3)–N), 141.9 (C 4H3O), 110.4 (C 4H3O), 107.8 (C 4H3O), 56.4 (CH(CH3)2), 51.0 (CH2), 36.5 (CH2), 22.9 (CH(CH3)2). IR data (KBr pellet) cm−1: 1686 (s) (νc=o). Anal. Calcd. for C24H32AuClN8O4·CH3CN: C, 40.55; H, 4.58; N, 16.37. Found: C, 40.46; H, 4.98; N, 17.02 %.
3 Results and Discussion
The gold(I) (1–2)c complexes were synthesized from the respective silver(I) analogs (1–2)b by the reaction with (SMe2)AuCl in CH3CN at room temperature (Scheme 1) and the workup required product purification by column chromatography. The silver(I) (1–2)b complexes were obtained by the treatment of the corresponding triazolium chloride salts (1–2)a [18] with Ag2O in moderate to good yields. It is noteworthy that despite repeated attempts the single crystals of the silver(I) (1–2)b complexes could not obtained for structural characterization studies.

Synthetic procedure for the Au–NHC complexes (1–2)c
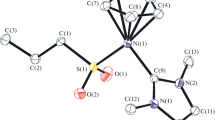
The molecular structure of the gold(I) (1–2)c complexes, as determined by the single crystal X-ray diffraction study, revealed discrete monomeric nature of these complexes (Figs. 2, 3). The Au(I) center was coordinated to the N-heterocyclic carbene ligand in a linear fashion in concurrence with the d 10 configuration of the central metal ion. The two N-heterocyclic carbene ligands are bound trans to each other displaying a syn-disposition of its substituents, with its amido-N substituent engaging with the non-coordinating Cl anion with the aid of two intramolecular H-bonding interactions. Similar syn-disposition of the amido-N substituents arising from the intramolecular H-bonding interactions of the amido-NH moieties with the non-coordinating Cl anion has been observed earlier in the silver(I) and gold(I) analogs, {[1-(R)-3-{N-(t-butylacetamido)imidazol-2-ylidene}]2M}+Cl− (M = Ag, Au; R = i-Pr, t-Bu), of the related imidazole based N-heterocyclic carbene ligands [15]. Quite interestingly, no intermolecular H-bonding interaction was present among these discrete monomeric molecules in the crystal lattice.

ORTEP of 1c with thermal ellipsoids are shown at the 50 % probability level. Selected bond lengths (Å) and angles (°): Au1–C1 2.017(7), Au1–C22 2.025(7), C22–Au1–C1 177.2(3), N1–C1–N3 103.4(6), N5–C22–N7 104.4(6)

ORTEP of 2c with thermal ellipsoids are shown at the 50 % probability level. Selected bond lengths (Å) and angles (°): Au1–C1aa 2.026(6), Au1–C0aa 2.014(6), C1aa–Au1–C0aa 177.0(2), N1–C0aa–N3 103.2(5), N4–C1aa–N6 103.8(5)
Of particular interest are the Au–Ccarbene bond distances in the 1c [2.017(7) Å, 2.025(7) Å] and the 2c [2.026(6) Å, 2.014(6) Å] complexes, that are marginally shorter than the sum of the individual covalent radii of Au and C (2.09 Å) [20] but compare well with that of the representative related imidazole analogs, {[1-(i-propyl)-3-{N-(t-butylacetamido)imidazol-2-ylidene}]2Au}+Cl− [2.017(7) Å] [15] and the {[1-(t-butyl)-3-{N-(t-butylacetamido)imidazol-2-ylidene}]2Au}+Cl− [2.028(5) Å] [15]. The angle at the gold center, ∠ Ccarbene –Au–Ccarbene, in the 1c [177.2(3)°] and in the 2c [177.0(2)°] complexes is close to linearity and is also comparable to that observed for the related representative imidazole analogs, {[1-(i-propyl)-3-{N-(t-butylacetamido)imidazol-2-ylidene}]2Au}+Cl− [178.3(4)°] [15] and the {[1-(t-butyl)-3-{N-(t-butylacetamido)imidazol-2-ylidene}]2Au}+Cl− [179.6(3)°] [15].
4 Conclusion
In summary, two gold(I) complexes of the 1,2,4-triazole derived N-heterocyclic carbene complexes (1–2)c have been synthesized. The single crystal X-ray diffraction study revealed that the (1–2)c complexes are structurally similar to their related imidazole counterparts known in the literature. Detailed evaluation of these complexes in catalytic applications is currently underway and would be a part of the future publications from the group.
5 Supporting Information Available
The 1H NMR, 13C{1H} NMR, IR, HRMS and the CHN data of the silver (1–2)b and the gold (1–2)c complexes; CIF files giving X-ray crystallographic data associated with this article can be found in the journal webpage. This material is available free of charge via the journal webpage.
References
Freakley SJ, He Q, Kiely CJ, Hutchings GJ (2015) Gold catalysis: a reflection on where we are now. Catal Lett 145(1):71–79
Gagosz F (2013) Organogold catalysis: homogeneous gold-catalyzed transformations for a golden jubilee. Wiley, New Jersey, pp 207–225
Keel T, McPherson J, (2013), Applications and future trends in gold catalysis, RSC Catal. Ser 13 Environmental Catalysis over Gold-Based Materials, 201-221
Wang YM, Lackner AD, Toste FD (2014) Development of catalysts and ligands for enantioselective gold catalysis. Acc Chem Res 47(3):889–901
Toste DF (2013) Gold catalysis for organic synthesis II. Beilstein J Org Chem 9:2040–2042
Braun I, Asiri AM, Hashmi ASK (2013) Gold catalysis 2.0. ACS Catal 3(8):1902–1907
Merino E, Nevado C (2013) On gold catalysis and beyond. Chimia 67(9):663–668
Gatineau D, Goddard JP, Mansuy MV, Fensterbank L (2013) When NHC ligands make a difference in gold catalysis. Isr J Chem 53(11–12):892–900
Katari M, Rao MN, Rajaraman G, Ghosh P (2012) Computational insight into a gold(I) N-heterocyclic carbene mediated alkyne hydroamination reaction. Inorg Chem 51(10):5593–5604
Dash C, Shaikh MM, Butcher RJ, Ghosh P (2010) Highly convenient regioselective intermolecular hydroamination of alkynes yielding ketimines catalyzed by gold(I) complexes of 1,2,4-triazole based N-heterocyclic carbenes. Inorg Chem 49(11):4972–4983
Ray L, Katiyar V, Barman S, Raihan MJ, Nanavati H, Shaikh MM, Ghosh P (2007) Gold(I) N-heterocyclic carbene based initiators for bulk ring-opening polymerization of L-lactide. J Organomet Chem 692(20):4259–4269
Ray L, Katiyar V, Raihan M, Nanavati H, Shaikh MM, Ghosh P (2006) First example of a gold(I) N-heterocyclic-carbene-based initiator for the bulk ring-opening polymerization of L-lactide. Eur J Inorg Chem 18:3724–3730
Samantray MK, Dash C, Shaikh MM, Pang K, Butcher RJ, Ghosh P (2011) Gold(III) N-heterocyclic carbene complexes mediated synthesis of β-enaminones from 1,3-dicarbonyl compounds and aliphatic amines. Inorg Chem 50(5):1840–1848
Samantaray MK, Pang K, Shaikh MM, Ghosh P (2008) Unprecedented long-range 1,7-bromination in gold complexes of N-(aryl) imino functionalized N-heterocyclic carbenes. Dalton Trans 36:4893–4902
Samantaray MK, Pang K, Shaikh MM, Ghosh P (2008) From large 12-membered macrometallacycles to ionic (NHC)(2)M + Cl-type complexes of gold and silver by modulation of the N-substituent of amido-functionalized N-heterocyclic carbene (NHC) Ligands. Inorg Chem 47(10):4153–4165
Ray S, Mohan R, Singh JK, Samantaray MK, Shaikh MM, Panda D, Ghosh P (2007) Anticancer and antimicrobial metallopharmaceutical agents based on palladium, gold, and silver N-heterocyclic carbene complexes. J Am Chem Soc 129(48):15042–15053
Dash C, Shaikh MM, Ghosh P (2011) Silver complexes of 1,2,4-triazole derived N-heterocyclic carbenes: synthesis, structure and reactivity studies. J Chem Sci 123(2):97–106
Kumar A, Bheeter LP, Gangwar MK, Sortais JB, Darcel C, Ghosh P (2015) Nickel complexes of 1,2,4-triazole derived amido-functionalized N-heterocyclic carbene ligands: synthesis, theoretical studies and catalytic application. J Organomet Chem 786:63–70
Dash C, Shaikh MM, Butcher RJ, Ghosh P (2010) A comparison between nickel and palladium precatalysts of 1,2,4-triazole based N-heterocyclic carbenes in hydroamination of activated olefins. Dalton Trans 39(10):2515–2524
Cordero B, Gomez V, Prats AEP, Reves M, Echeverria J, Cremades E, Barragan F, Alvarez S (2008) Covalent radii revisited. Dalton Trans 21:2832–2838
Sheldrick GM (1997) SHELXL-97, program for refinement of crystal structures. University of Gottingen, Germany
Sheldrick GM (1997) SHELXS-97, program for solution of crystal structures. University of Gottingen, Germany
Acknowledgments
Authors thank Department of Science and Technology (File No.: EMR/2014/000254), New Delhi, and Industrial Research and Consultancy Centre (IRCC), IIT Bombay (Grant No.: 15IRAWD006), for financial support of this research. Authors gratefully acknowledge the Single Crystal X-ray Diffraction Facility, Department of Chemistry IIT Bombay, India, for the crystallographic characterization data. AK, and MKG thank CSIR, New Delhi for research fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kumar, A., Gangwar, M.K., Shaikh, M.M. et al. Synthesis and Structural Characterization of the Gold Complexes of 1,2,4-Triazole Derived N-Heterocyclic Carbene Ligands. Proc. Natl. Acad. Sci., India, Sect. A Phys. Sci. 86, 605–609 (2016). https://doi.org/10.1007/s40010-016-0288-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40010-016-0288-7